
Abstract
Enterovirus 71 (EV71) is one of the main pathogens that causes hand-foot-and-mouth disease (HFMD). HFMD caused by EV71 infection is mostly self-limited; however, some infections can cause severe neurological diseases, such as aseptic meningitis, brain stem encephalitis, and even death. There are still no effective clinical drugs used for the prevention and treatment of HFMD. Studying EV71 protein function is essential for elucidating the EV71 replication process and developing anti-EV71 drugs and vaccines. In this review, we summarized the recent progress in the studies of EV71 non-coding regions (5′ UTR and 3′ UTR) and all structural and nonstructural proteins, especially the key motifs involving in viral infection, replication, and immune regulation. This review will promote our understanding of EV71 virus replication and pathogenesis, and will facilitate the development of novel drugs or vaccines to treat EV71.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Enterovirus 71 (EV71), a single-stranded RNA virus with a genome length of approximately 7.5 kb, belongs to the genus Enterovirus within the family Picornaviridae (Wang et al.2012a, b). Since it was first discovered in 1969, EV71 has caused numerous outbreaks and epidemics worldwide, particularly in Asian and Pacific regions, such as China, Korea, Singapore, Japan, and Vietnam (WHO 2018). EV71 usually infects infants and young children under the age of 5 years, and hand-foot-and-mouth disease caused by EV71 infection is usually self-limited. However, some infections can cause aseptic meningitis, brain stem encephalitis, and other nervous system diseases (Weng et al.2010; Teoh et al.2016). At present, there are no effective drugs to prevent and treat EV71 infection, and ribavirin, interferon (IFN), and other drugs are used only for symptomatic treatment (Yi et al.2011; Wang et al.2017a). Studying the structure and function of the EV71 viral genome is essential for understanding the EV71 replication process and developing anti-EV71 drugs and vaccines.
In this review, we discuss the important functions of the EV71 genome, based on recent functional genomics research.
Structural Features and Replication Process of the EV71 Genome
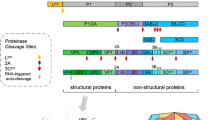
The EV71 genome contains only one open reading frame (ORF), which encodes 2193 amino acids and is flanked by 5′ and 3′ untranslated regions (UTRs). As shown in Fig. 1, the low-molecular-weight protein VPg covalently binds to the 5′ UTR, and a variable length poly-A tail is located at the terminus of the 3′ UTR. During the EV71 replication process, the polyprotein is subdivided into P1, P2, and P3 regions. P1 encodes four structural proteins (VP1–VP4), whereas P2 and P3 encode seven nonstructural proteins (2A, 2B, 2C, 3A, 3B, 3C, and 3D) (Cardosa et al.2003; McMinn 2002). These 11 proteins are closely related to EV71 infection, inflammatory responses, and host immune responses.

Structure of the enterovirus 71 (EV71) genome.
As a nonenveloped virus, the host cell entry mechanism of EV71 remains largely unknown. Recent data have indicated that the interaction between viral particles and receptors causes the spatial configuration to change and results in loss of VP4; eventually, the virus particles enter the cell through the endocytotic pathway. Then, the viral shell is removed, the viral genomic RNA is released into the cytoplasm, and the translation of the viral polymeric protein begins with the viral genomic RNA as mRNA (Hu et al.2003; Solomon et al.2010). The replication of EV71 is similar to that of other positive-stranded RNA viruses. First, polymeric precursor protein is synthesized by viral RNA as mRNA template. Then, polymer precursor protein is cut into four structural proteins and seven nonstructural proteins. The nonstructural protein 3D, an RNA-dependent RNA polymerase (RdRp), synthesizes negative-chain RNA using RNA as a template. A large number of positive-strand RNAs are further synthesized using negative-strand RNA as a template. With the accumulation of a large number of viral positive-strand RNAs in the cytoplasm, the subgeneration virus particles begin to be assembled (Bedard and Semler 2004).
Function of the Noncoding Region
The 5′ UTR
The 5′ UTR of EV71 usually folds into a specific spatial structure, including a cloverleaf structure and an internal ribosome entry site (IRES). The IRES contains six major stem loop structures associated with viral RNA replication, which regulate the initiation of viral protein translation by binding to host cell protein factors (Lin et al.2008, 2009a; Yeh et al.2011). IRES directs the initiation of translation in a cap-independent manner and can be used as a target for antiviral drugs. A single nucleotide change from cytosine to uridine at base 158 in the second stem ring of 5′ UTR reduces viral translation and EV71 virulence in mice (Yeh et al.2011). Moreover, Chang et al. found that a single nucleotide T to C mutation at nucleotide 494 of the 5′-UTR affects EV71 virulence (Chang et al.2018). During EV71 infection, the process of viral RNA translation relies mainly on translation initiation factors and IRES-specific trans-acting factors (ITAFs). These ITAFs interact with various IRES elements to regulate their activities by affecting ribosome recruitment or modifying the structure of the IRES itself. As shown in Fig. 2, several host proteins have been shown to be involved in modulating EV71 IRES function as ITAFs, including heterogeneous nuclear ribonucleoprotein A1 (hnRNP A1), hnRNP K, far upstream element binding protein 1 (FBP1), FBP2, and poly real (rC)-binding protein 1 (PCBP1) (Huang et al.2011; Lin et al.2008, 2009b, c; Luo et al.2014). During EV71 infection, hnRNPA1, hnRNP K, FBP1, FBP2, and PCBP1 are enriched in the cytoplasm, where EV71 replication occurs, whereas hnRNPA1, hnRNP K, FBP1, FBP2, and PCPB1 are localized in the nucleus in mock-infected cells. The cytoplasmic relocalization of hnRNP A1, hnRNP K, FBP1, and PCBP1 in EV71-infected cells leads to enhancement of EV71 IRES-mediated translation and directly regulates the viral protein translation process, thus promoting the replication of EV71; hnRNP A1 interacts with stem-loops II and VI, hnRNP K interacts with stem-loops I–II and IV, FBP1 interacts with the linker region downstream of the EV71 IRES (637–745 nt), and PCBP1 interacts with stem-loops I and IV (Huang et al.2011; Lin et al.2008, 2009c; Luo et al.2014). In contrast to hnRNP A1, hnRNP K, FBP1, and PCBP1, FBP2 inhibits IRES activity by directly binding to nucleotides 1–167 (stem-loops I and II), 91–228 (stem-loops II and III), and 566–745 (stem-loop VI and the spacer region) regions in the EV71 5′ UTR, thereby inhibiting EV71 replication (Lin et al.2009b). During EV71 infection, AU-rich element binding factor 1 (AUF1) accumulates in the cytoplasm where viral replication occurs, binds the IRES of EV71, and negatively regulates IRES-dependent translation (Lin et al.2014). In contrast to AUF1, the mRNA stability factor HuR and the RISC subunit Argonaute 2, as two ITAFs that bind stem-loop II, can promote EV71 IRES activity and viral replication (Fig. 2) (Lin et al.2015). EV71 promotes the expression of silent mating type information regulation 2 homolog 1 (SIRT1), stimulates SIRT1 sumoylation and deacetylase activity, and enhances SIRT1 translocation from the nucleus to the cytoplasm. After being enriched in the cytoplasm, SIRT1 interacts with the cloverleaf structure of the 5′ UTR to inhibit viral RNA transcription and binds to the IRES to attenuate viral RNA translation (Fig. 2) (Han et al.2016). As shown in Fig. 2, T cell-restricted intracellular antigen 1 (TIA-1) and TIA-1-related protein (TIAR) are translocated from the nucleus to the cytoplasm after EV71 infection. TIA-1 and TIAR can both bind to stem-loop I of the 5′ UTR and improve the stability of viral genomic RNA, facilitating EV71 replication by enhancing synthesis of the viral genome in host cells (Wang et al.2015c). Moloney leukemia virus 10 (MOV10), a highly conserved cellular protein belonging to the SF1 helicase family, positively regulates EV71 replication by binding to the EV71 cloverleaf-like structure and IRES with its C-terminus (Wang et al.2016). Based on the highly conserved 5′ UTR and its important role in EV71 replication, the 5′ UTR can be targeted to effectively inhibit EV71 replication through RNAi strategies (Deng et al.2012).

Host proteins bind to the 5′ UTR and regulate EV71 replication.
The 3′ UTR
The 3′ UTR of EV71 contains a variable poly(A) tail, which is very important for EV71 replication. The poly(A) tail in eukaryotic cells can confer mRNA stability, promote the translational efficiency of mRNA, and transport mRNA from the nucleus to the cytoplasm (Weng et al.2009). However, the specific function of the 3′ UTR and poly(A) tail for EV71 is not fully understood, and studies of PV have shown that the recombinant 3′ UTR of PV is active, although its replication rate is slower than that of the wild-type sequence (Fernandez-Miragall et al.2009; Herold and Andino 2001; Silvestri et al.2006; Todd et al.1997). Sim and others have shown that the chemical synthesis of siRNA targeting the 3′ UTR can reduce the replication of EV71. Transfection of rhabdomyosarcoma cells with siRNA targeting the 3′ UTR region significantly decreases viral RNA, viral proteins, and plaque formation; therefore, RNA interference may also be used as a method for clinical antiviral therapy (Sim et al.2005). A recent study showed that miR-23b is significantly downregulated in EV71-infected cells and that upregulation of miR-23b inhibits the replication of EV71 by targeting the EV71 3′ UTR conserved sequence through seven consecutive nucleic acids (Wen et al.2013). Feng et al. found that miR-127-5p expression was upregulated during EV71 infection and miR-127-5p can inhibit EV71 infection through downregulating the expression of SCARB2 via targeting two potential sites in 3′ UTR region (Feng et al.2017).
Protein-Coding Region
VP1 Protein
The VP1 protein of EV71 contains 297 amino acids and shows complete genetic diversity corresponding to the viral serotype, which can be used as the basis of EV71 serotype classification (McMinn 2002). Human scavenger receptor class B member 2 (SCARB2) serves as a cellular receptor for EV71 entry, and its cellular entry is through a clathrin-mediated and pH-dependent endocytic pathway (Fig. 3). EV71 binds to SCARB2 via a canyon in VP1 around residue Gln-172 (Chen et al.2012). In addition, another study found that the three VP1 mutations K98E, E145A, and L169F enhanced the combination of VP1 with the mSCARB2 protein on murine cells and permitted the virus to infect murine cells (Fujii et al.2018; Kobayashi et al.2018; Victorio et al.2016). P-selectin glycoprotein ligand-1 (PSGL-1), another receptor of EV71, enhances EV71 entry through binding to VP1, and the binding depends on sulfated tyrosine residues and the interaction between negatively charged sulfate groups and positively charged basic residues in the viral capsid (Fig. 3) (Nishimura et al.2013). PSGL-1 cannot bind to VP1 when VP1-145G/Q is replaced with E. VP1-145 is in close proximity to conserved lysine residues at VP1-242 and VP1-244. Moreover, VP1-145 controls the orientation of the lysine side-chain of VP1-244 as follows: with VP1-145Q, the lysine side chain faces outward, and VP1 combines with PSGL-1 to regulate virus entry; however, with VP1-145E, the lysine side chain is turned toward the virus surface, and VP1 cannot bind to PSGL-1 (Nishimura et al.2013). The VP1-145 amino acids vary in different strains, which may be the reason why not all EV71 strains use PSGL-1 as a receptor. In addition, a VP1 mutation (K244E) was found to be necessary for mouse-adapted EV71 virulence in adult mice using reverse genetics (Caine et al.2016). EV71 uses heparan sulfate as an attachment receptor, and both VP1-98 and VP1-145 cannot modulate heparin binding (Tan et al.2017; Tseligka et al.2018). Galectin-1, a soluble beta-galactoside binding lectin, may be associated with EV71 VP1 via carbohydrate residues and is subsequently released and bound to another cell surface along with the virus. When galectin-1 is knocked down, EV71 exhibits low infectivity in cells and less pathogenicity in mice, and galectin-1-free EV71 virus is sensitive to high temperature and loses its viability after long-term storage (Lee et al.2015). Cell surface vimentin is an attachment receptor for EV71, and vimentin expressed on the cell surface binds to VP1 via its N-terminal to promote EV71 infection (Fig. 3) (Du et al.2014). During EV71 infection, EV71 VP1 protein activates calmodulin-dependent protein kinase II, which phosphorylates the N-terminal domain of vimentin on serine 82. Vimentin phosphorylation and rearrangement may enhance EV71 replication by playing structural roles in the formation of the replication factories (Cong et al.2013). In addition, human annexin II protein can bind to EV71 VP1 via VP1 amino acids 40–100, a region different from the known receptor binding domain, thereby enhancing EV71 replication (Fig. 3) (Yang et al.2011). miR-2911, a honeysuckle-encoded atypical microRNA, can directly inhibit EV71 replication by targeting the VP1 gene (Fig. 3) (Li et al.2018a). The mutation K215A in the VP1 GH loop results in a significant increase in thermal stability, indicating that conditional thermostable mutants can be generated by altering the charge characteristics of VP1 (Yuan et al.2015). In addition, studies have found that cattle and human lactoferrin (LF) can effectively inhibit EV71 infection through binding to VP1 and that LF inhibits EV71-induced interleukin (IL)-6 production and protects mice against lethal EV71 challenge (Fig. 3) (Weng et al. 2005; Wu et al.2010a).

The protein functions of VP1 and 2A.
VP1 is the main virus-neutralizing determinant, directly determining the antigenicity of the virus, and contains the main antigen-binding site (Huang et al.2008). The N-terminal of VP1 capsid protein possesses an important antigen region, which is highly immunogenic. The peptides (amino acids 66–77 or 208–222) of the C-terminal of VP1 capsid protein may stimulate the production of neutralizing antibodies. In addition, three regions on the VP1 protein (amino acids 66–77, 145–159, and 247–261) are capable of inducing human EV-71-specific CD4+ T-cell proliferation (Foo et al.2008). The full-length VP1 is capable of self-association and forms a dimerization structure to improve the pathogenicity of the virus and the ability to adapt to the external environment. VP1 (amino acids 66–132) contains the major dimerization domain, and VP1 (amino acids 132–297) contributes largely to increasing the strength of the interaction (Lal et al.2006).
VP1, a surface antigen, is a suitable candidate for EV71 vaccines. Two peptides, SP55 (amino acids 163–177) and SP70 (amino acids 208–222) are capable of eliciting neutralizing antibodies against EV71. Immunization of mice with either SP55 or SP70 triggers an EV71-specific IgG response as high as that obtained with the whole virion as immunogen; thus, SP70 represents a promising candidate for an effective synthetic peptide-based vaccine against EV71 (Foo et al.2007). Oral immunization with recombinant VP1 protein (rVP1) induces VP1-specific IgA antibodies, serum-specific IgG, and neutralization antibodies in mice and may be a promising subunit vaccine candidate for preventing EV71 infection (Zhang et al.2014). Immunization of hamsters with an EV71 VP1 fragment (NPt-VP11-100) protein can induce good immune responses, but the high level of antibodies fails to neutralize EV71 viruses or protect vaccinated hamsters in viral challenge studies (Ch’ng et al.2012).
VP2–VP4 Proteins
The EV71 viral particle capsid composition complex and three structural proteins VP1, VP2, and VP3, which are exposed on the surface of the shell with no homology of nucleotide sequences among them, show certain similarities of the protein topology structure. The VP4 package is embedded in the inside of the virus shell, is closely connected with the virus core, and exhibits an extended spatial conformation feature, which is a bridge connecting the inside and outside (Chen et al. 2010; Rowlands et al.2010). When the virus binds to the receptors, the spatial configuration will change, and the VP4 is lost. Eventually, the viral shell is removed, the viral genomic RNA is released into the cytoplasm, and the translation of the viral polymeric protein begins with the viral genomic RNA as mRNA. The N-terminal myristoylation signal (MGXXXS) of VP4 plays an important role in EV71 replication, and different myristic acid analogs elicit differential effects on EV71 replication in vitro, suggesting that removal of the myristate moiety in the viral structural protein precursor can be an effective antiviral target for further research (Tan et al.2016). EV71 virus capsid proteins VP2 and VP3, which are important parts of the shell protein, are associated with the antigenicity of the virus. Thus, VP2 and VP3 may be potential candidates with structures similar to that of VP1, and VP2 (amino acids 142–146) contains a single, linear, non-neutralizing epitope, which is located in the E–F loop of the VP2 protein (Chen et al.2010; Kiener et al.2012). VP2 149 M mutation enhances viral binding and RNA accumulation of EV71, which promotes EV71 infectivity in vitro and mouse lethality in vivo (Huang et al.2012). T cells play an important role in the host immune response against EV71 infection. Compared with the other three capsid proteins, VP2 shows a more extensive distribution and immunogenicity in T cells (Tan et al.2013). The VP2-28 epitope containing residues 136–150 of VP2 was identified as another neutralizing epitope. Xu et al. (2015) constructed a bivalent chimeric virus-like particle (VLP) presenting the VP1 (amino acids 208–222) and VP2 (amino acids 141–155) epitopes of EV71 that could induce higher IgG titers and neutralization titers and protect neonatal mice against lethal EV71 and CA16 infections. Moreover, they found that anti-VP2 (amino acids 141–155), but not anti-VP1 (amino acids 208–222), could crossreact with normal EV71 and CA16 virions (Xu et al.2015). Kiener et al. (2014) found that the knob of EV71 VP3 encompassing residues 55–69 of VP3 is a new conformational epitope of EV71 involved in EV71 virus neutralization. The importance of this novel neutralization epitope lies in the optimization of putative EV71 vaccines because the VP3 knob can be combined with VP1 to form a bivalent subunit vaccine (Kiener et al.2014).
2A Protein
The EV71 2A protease exhibits cysteine protease activity and contains 150 amino acid residues; it is an enzyme that cleaves at its own N-terminus at the junction between VP1 and 2A of the polyprotein. A polyprotein, translated from the single ORF of EV71, is processed into mature proteins by 2A (Hellen et al.1992; Hsu et al.2007; Ventoso and Carrasco 2003). Eukaryotic translation initiation factor 4G (eIF4G) induces the synthesis of the host cell proteins by promoting the interaction between mRNA and the 40S ribosomal subunit. 2Apro inhibits host cap-dependent protein synthesis by cleaving the elongation factor eIF4G and promoting EV71 replication (Fig. 3) (Morley et al.1997). 2A can block typical stress granule formation and induce atypical stress granule formation by cleaving eIF4GI to sequester cellular mRNA but release viral mRNA, thereby facilitating viral translation (Yang et al.2018). In addition, the 2A protease of EV71 shows great transcription activity in yeast, which is independent of its protease activity. EV71 2A protease retains its transcriptional activity after truncation of 40 amino acids at the N-terminus but loses this activity after truncation of 60 amino acids at the N-terminus or deletion of 20 amino acids at the C-terminus. The acidic structure domain at the C terminal is necessary for its transcriptional activity, and deletion of amino acids 146–149 (EAME) in this acidic domain causes the loss of transcriptional activity (Yang et al.2010).
As shown in Fig. 3, 2A is capable of cleaving FBP1 at the Gly-371 residue of FBP1, which generates a functional cleavage product, FBP11−371. FBP11−371 can bind to the 5′ UTR linker region, which is different from full-length FBP1. Moreover, FBP1 and FBP11−371 act additively to promote IRES-mediated translation and enhance EV71 replication (Hung et al.2016). 2A significantly inhibits cellular endoplasmic reticulum-associated degradation (ERAD) by inhibiting the transcription of the de novo synthesis of key molecules Herp and VIMP. p97, a host factor that is distributed and co-exists with the viral protein and EV71 replication-related molecules, is hijacked from cellular ERAD by EV71 to promote viral replication (Wang et al.2017b).
Type I IFNs (IFN-Is) are key players in the innate antiviral response against viral infections. However, IFN therapy does not significantly affect EV71 infection, and induction of downstream IFN-stimulated genes is inhibited by EV71. The 2A protease can reduce IFN-I receptor protein 1 levels and suppress interferon regulatory factor 3 (IRF3) signaling by cleaving mitochondrial antiviral protein and retinoid acid-inducible gene I (RIG-I)-like receptor MDA5, thus inhibiting IFN signaling pathways and causing the virus to escape the immune response (Fig. 3) (Kuo et al.2013; Lu et al.2012; Wang et al.2013). In addition, 2A attenuates IFN-γ signaling using another mechanism by reducing the serine phosphorylation of signal transducer and activator of transcription 1 (STAT1) following the inactivation of extracellular signal-regulated kinase without affecting STAT1 expression (Fig. 3) (Wang et al.2015b). 2A protein can also induce apoptotic cell death (Kuo et al.2002a). EV71 infection induces the production of inflammasomes, whereas inflammasomes inhibit the replication of EV71. 2A protein can block the production of inflammasomes by eliminating NLRP3 protein at the G493-L494 or Q225-G226 junction (Fig. 3) (Wang et al.2015a).
2B Protein
The EV71 2B protein, a small hydrophobic ion channel protein with 99 amino acid residues, may mediate a chloride-dependent rather than calcium-dependent current in oocytes; furthermore, DIDS, an inhibitor of this current, can significantly inhibit the replication of EV71 (Xie et al.2011). Currently, little is known about the function of EV71 2B proteins. We speculate that they may have the same function as other enteroviruses. The expression of protein 2B from polioviruses shows relatively high similarity, with two transmembrane domain (TM1 and TM2) structures that are essential for the function of viroporins, such as damaging organelle membrane integrity, improving the permeability function, promoting viral RNA replication, and releasing viral particles from the host cell (Agirre et al.2002; De Jong et al. 2003; Johnson and Sarnow 1991; Madan et al.2007). The C-terminal region of 2B (amino acids 63–80) is reported to be responsible for the location of 2B in the mitochondria, and 2B induces cell apoptosis by interacting directly with and recruiting the proapoptotic protein Bax and inducing Bax conformational activation (Fig. 4). A hydrophilic region of 14 amino acids in the N-terminal region of 2B is important for Bax interaction and subsequent activation. Moreover, overexpression of the anti-apoptotic protein Bcl-XL abrogates 2B-induced release of cytochrome c and caspase activation (Cong et al.2016). Although little is known about the function of 2B protein, this protein may be a potential target for anti-EV71 drug development.

The protein functions of 2B and 2C.
2C Protein
The 2C protein of EV71 is one of the most highly conserved nonstructural proteins, containing 329 amino acid residues. 2C harbors an adenosine triphosphatase (ATPase) domain, a zinc finger structure, and an alpha helix at the end of the C-terminal region (Guan et al.2017). The 2CATPase, an RNA helicase that 3′-to-5′ unwinds RNA helices in an ATP-dependent manner and an RNA chaperone independently of ATP, facilitates EV71 RNA synthesis in vitro. 2CATPase-mediated RNA remodeling plays a critical role in the EV71 life cycle (Xia et al.2015). The N terminus of the 2C protein, which exhibits both RNA- and membrane-binding activity, interacts with reticulon 3 (RNT3) via its highly conserved reticulon homology domain and then combines with double-stranded RNA (dsRNA) viruses to form viral replication complex and participate in viral replication (Fig. 4). Reduced production of RNT3 by RNA interference markedly reduces the synthesis of EV71-encoded viral proteins and replicative dsRNA, reducing plaque formation and apoptosis (Tang et al.2007). 2C proteins can control the activity of nucleoside triphosphate and participate in the synthesis of negative-strand RNA and the capsid formation of PV subgeneration viral particles (Wu et al.2010a). By interaction with the IPT domain (amino acids 194–290) of p65, 2C can reduce the formation of the heterodimer p65/p50 and then inhibit nuclear factor (NF)-κB activation (Fig. 4) (Du et al.2015). The N-terminal of 2C (amino acids 1–125) interacts with all isoforms of the protein phosphatase 1 (PP1) catalytic subunit through PP1-docking motifs, which is efficient for EV71 2C-mediated inhibition of IKKβ phosphorylation and NF-κB activation (Fig. 4). Moreover, 2C forms a complex with PP1 and IKKβ to dephosphorylate IKKβ activation (Li et al.2016; Zheng et al.2011). Coat protein complex I (COPI) may be directed to the viral replication complex through viral 2C protein to enhance EV71 infection, whereas the inhibition of COPI activity can weaken the replication of EV71 (Fig. 4) (Wang et al.2012a).
3A Protein
The EV71 3A protein, containing 86 amino acid residues, is a membrane binding protein that regulates intracellular transport of host cells. During RNA replication, the 3A protein promotes the combination of capsule membrane and replication complex and viral RNA synthesis through its hydrophobic zone (Fujita et al.2007; Giachetti et al.1992). The 3A protein expressed in mammalian cells can lead to cleavage of the endoplasmic reticulum and the transport dysfunction of the Golgi complex protein. It may be closely related to the inhibition of host antiviral immune function and can inhibit the expression of cytokines, such as IL-1β, IL-6, and IL-8; furthermore, 3A can also reduce the expression of major histocompatibility complex I and tumor necrosis factor (TNF) receptor in the infected cells (Choe and Dodd 2005). EV71 3AB displays RNA chaperone activity. Moreover, 3B and the last seven amino acids at the C-terminal of 3A (termed 3B + 7) possess RNA chaperone activity, and five amino acids, i.e., Lys-80, Phe-82, Phe-85, Tyr-89, and Arg-103, are critical and probably the active sites of 3AB for its RNA chaperone activity (Tang et al.2014). The Golgi resident protein acyl-coenzyme A binding domain-containing 3 (ACBD3) promotes EV71 replication by interacting with the 3A protein (Fig. 5) (Lei et al.2017). EV71 infection induces the production of phosphatidylinositol-4-phosphate, and ACBD3 is required for the recruitment of phosphatidylinositol-4-kinase IIIβ (PI4 KB) to the viral RNA replication site for EV71 replication (Fig. 5). 3A stimulates the interaction of PI4 KB and ACBD3, and three proteins are all located at the viral RNA replication site to form a complex containing 3D protein to facilitate EV71 replication. However, I44A or H54Y mutation in the 3A protein can block its interaction with PI4 KB and ACBD3 (Xiao et al.2017). Qiu et al. (2017) found that 3A protein, as a viral suppressor of RNAi (VSR), effectively inhibits the production of virally derived siRNAs and antiviral RNAi immunity in vitro and in vivo.

The protein functions of 3A and 3D.
3B Protein
The 3B protein, also known as a VPg protein, is a small protein that contains 22 amino acid residues. The VPg protein forms phosphodiester bonds with pUpU at the 5′ UTR of the EV71 genome through the hydroxyl groups of the Try residues and then uses vpg-pUpU as the primer to participate in the synthesis of the negative chain and plus strand RNA (Herrero et al.2003; Liu et al.2007; McMinn 2002). VPg protein can interact with the polymerase 3D, and VPg is catalyzed by 3D polymerase to uridine acidification, which is a primer for viral RNA synthesis (Paul et al.2003).
3C Protein
The 3C protein of EV71 contains 183 amino acids and shows serine protease and cysteine protease activities. During the replication of EV71, 3C proteins catalyze the lysis of virus precursor proteins and form mature structural proteins and nonstructural proteins (Cui et al.2011). The catalytic activity site of 3C protease comprises His40, Glu71, and Cys147 (Qiu 2008). Studies have shown that 3C protease has many functions, such as promoting viral replication and enhancing apoptosis in host cells (Lee et al.2008; Li et al.2002). 3C protease can enter the nucleus through its precursor 3 CD′ and 3 CD, and its nuclear localization signal is located in the 126–129 sequence “KKKRD”. The 3C protease enters the nucleus in its precursor form, shears itself to form the 3C protease, and shears transcription factors in the nucleus, such as TATA-box binding protein, the transcription factor p53, histone H3, transcription factor C, and the pre-mRNA cleavage stimulation factor (CstF-64). 3C inhibits host gene expression by regulating host pre-mRNA processing and polyadenylation, resulting in a decline in transcription in the host cell (Lin et al.2009a, b, c; Weng et al.2009; Sharma et al.2004). 3C protease exhibits RNA binding activity, among which the “KFRDI” (amino acids 82–86) and “VGK” (amino acids 154–156) sequences are the binding sequences for RNA (Shih et al.2004). Changes in the sequence of the RNA binding region of 3C can affect the activity of 3C protease, whereas mutations in the catalytic position will not change the RNA binding capacity of 3C protein (Shih et al.2004). Apoptosis may be an important host defense mechanism, whereby virus-infected cells are eliminated, thus preventing the generation and spread of viral progeny during viral infection. A study showed that poly (ADP-ribose) polymerase, a DNA repair enzyme, is cleaved by 3C protease, which then activates caspase and induces apoptosis, and is closely related to central nervous system diseases (Fig. 6) (Kuo et al.2002b).

The protein functions of 3C.
EV71 3C protease plays an important role in viral replication, and sequence analysis showed that no homologous sequence of EV71 3C is present in mammals; therefore, the 3C protein may be a potential target for antiviral drugs (Kuo et al.2008). 3C can bind to the SUMO E2 conjugating enzyme Ubc9 after binding of the K52 amino acid and SUMO E2 ligase and be SUMO modified at residue K52 for degradation, correlating with a decrease in EV71 in virus replication and cell apoptosis (Fig. 6) (Chen et al.2011). Overexpression of the telomere binding protein PinX1 can inhibit the apoptosis induced by EV71 infection, whereas the 3C protein interacts with PinX1 to degrade PinX1, which can promote host cell apoptosis (Fig. 6) (Li et al.2017). Ubc6e, an E2 ubiquitin-conjugating enzyme, plays a key role in EV71-dependent ERAD disruption, and EV71 3C cleaves Ubc6e at Q219G, Q260S, and Q273G, which can inhibit ERAD to promote EV71 replication (Fig. 6) (Wang et al.2017b). EV71 induces the production of inflammatory cytokines, and 3C interacts with transforming growth factor-β activated kinase 1 (TAK1) and the TAK1 binding protein 1 (TAB 1), which inhibits NF-κB activation. Furthermore, 3C mediates the cleavage of TAK1/TAB 1/TAB 2/TAB 3 complexes to interfere with inflammatory responses (Fig. 6) (Lei et al.2014).
EV71 inhibits antiviral immunity by inhibiting RIG-I to downregulate IFN-β, IFN-stimulated gene 54 (ISG54), ISG56, and TNF in virus-infected cells. The 3C protein can inhibit IFN-β activation by virus and RIG-I but does not inhibit MDA5. 3C is associated with RIG-I via the caspase recruitment domain, which prevents the recruitment of an adaptor IPS-1 and subsequent nuclear translocation of IRF3 (Fig. 6) (Lei et al.2010). In addition, miR-526a positively regulates virus-triggered IFN-I production, thus suppressing viral replication. 3C can inhibit interferon production by downregulating miR-526a or inhibiting interferon regulation factor 7 to block the RIG-I signaling pathway (Lei et al.2013; Xu et al.2014). Toll-like receptor (TLR)-related pathways play an important role in antiviral immune responses. TLR3 in the endosome recognizes viral dsRNA and recruits a TIR domain-containing adaptor inducing IFN-β (TRIF) to transmit signals to IRF3 and NF-κB. 3C is capable of cleaving TRIF and impairing type I IFN production in response to TLR3 activation (Fig. 6) (Lei et al.2011). EV71 infection induces the production of inflammasomes, and the NLRP3 inflammasome plays a protective role against EV71 infection. 3C protein can block the production of the inflammasome by eliminating NLRP3 protein (Fig. 6) (Wang et al.2015a).
3D Protein
The EV71 3D protein, containing 462 amino acid residues, is an RdRp, which mainly completes the extension of the RNA chain during viral replication. The 3D polymerase is indispensable for the initiation of viral replication and plays an important role in changing viral virulence. When 3D is cleaved from 3CD, the nucleic acid location signal of the 3D protein transmits 3CD to the host cell nucleus and shuts down the transcription of the host cell (Wang et al.2010; Wu et al.2010b). The EV71 3D protein is a polymerase dependent on Mn2+, which is completely inactive in the presence of Mg2+. Studies of EV71 transcription activity in vitro have shown that 3D can use dinucleotide and 10-nucleotide RNA as a primer and initiates transcription using genomic RNA as a template (Jiang et al.2011). In addition, the EV71 3D protein attenuates STAT1 tyrosine phosphorylation independent of Janus kinase 2 inactivation, without interfering with IFN-γ receptor expression. Then, 3D blocks IRF1 activation and antagonizes the antiviral activity of IFN-γ (Fig. 5) (Agirre et al.2002). EV71 induces the production of IL-1β through activation of the NLRP3 inflammasome. 3D interacts with NLRP3 to facilitate the assembly of the inflammasome complex by forming a 3D-NLRP3-ASC ring-like structure and then stimulates activation of the NLRP3 inflammasome and the cleavage of pro-caspsase-1, which causes the release of IL-1β (Fig. 5) (Wang et al.2017c).
Summary
In summary, different viral proteins of EV71 exert various functions to guarantee the replication of the virus itself. Although some progress has been made in research on the function of the EV71 genome, further studies are still needed. For example, the function of the 3′ UTR is relatively unclear. Notably, however, EV71 infection induces the host immune response, resulting in the expression of many host factors that inhibit EV71 replication through different mechanisms. For example, the promyelocytic leukemia protein contributes to cellular antiviral effects by inhibiting autophagy (Chen et al.2018), and A3G competitively binds to the 5′ UTR to inhibit the 5′ UTR activity of EV71 and the synthesis of EV71 viral proteins and RNA (Li et al.2018b). In the future, researchers will need to focus not only on further structural and functional studies of EV71 proteins but also on how viral virulence factors interact with the human immune system, which will have a profound impact on the development of vaccines and drugs. Such studies will improve our comprehensive understanding of EV71 genome structure and function. This will also help us elucidate the pathogenesis of EV71, which will provide insights into the design of therapeutic strategies against EV71 infection.
References
Agirre A, Barco A, Carrasco L, Nieva JL (2002) Viroporin-mediated membrane permeabilization. Pore formation by nonstructural poliovirus 2B protein. Biol Chem 277:40434–40441
Bedard KM, Semler BL (2004) Regulation of picornavirus gene expression. Microbes Infect 6:702–713
Caine EA, Moncla LH, Ronderos MD, Friedrich TC, Osorio JE (2016) A single mutation in the VP1 of enterovirus 71 is responsible for increased virulence and neurotropism in adult interferon-deficient mice. J Virol 90:8592–8604
Cardosa MJ, Perera D, Brown BA, Cheon D, Chan HM, Chan KP, Cho H, McMinn P (2003) Molecular epidemiology of human enterovirus 71 strains and recent outbreaks in the Asia-Pacific region: comparative analysis of the VP1 and VP4 genes. Emerg Infect Dis 9:461–468
Chang CK, Wu SR, Chen YC, Lee KJ, Chung NH, Lu YJ, Yu SL, Liu CC, Chow YH (2018) Mutations in VP1 and 5′-UTR affect enterovirus 71 virulence. Sci Rep 8:6688
Chen XM, Zhang Q, Li JH, Cao W, Zhang JX, Zhang L, Zhang WL, Shao ZJ, Yan YP (2010) Analysis of recombination and natural selection in human enterovirus 71. Virology 398:251–261
Chen SC, Chang LY, Wang YW, Chen YC, Weng KF, Shih SR, Shih HM (2011) Sumoylation promoted enterovirus 71 3C degradation correlates with a reduction in viralreplication and cell apoptosis. J Biol Chem 286:31373–31384
Chen P, Song Z, Qi Y, Feng XF, Xu NQ, Sun YY, Wu X, Yao X, Mao QY, Li XL, Dong WJ, Wan XB, Huang N, Shen XL, Liang ZL, Li WH (2012) Molecular determinants of enterovirus 71 viral entry: cleft around GLN-172 on VP1 proteininteracts with variable region on scavenge receptor B 2. J Biol Chem 287:6406–6420
Chen DY, Feng CH, Tian XY, Zheng N, Wu ZW (2018) Promyelocytic leukemia restricts enterovirus 71 replication by inhibiting autophagy. Front Immunol 9:1268
Ch’ng WC, Stanbridge EJ, Wong KT, Ong KC, Yusoff K, Shafee N (2012) Immunization with recombinant enterovirus 71 viral capsid protein 1 fragment stimulated antibody responses in hamsters. Virol J 9:155
Choe SS, Dodd DA (2005) Kirkegaard K. Inhibition of cellular protein secretion by picornaviral 3A proteins. Virology 337:18–29
Cong HL, Ning D, Tian HC, Yang Y, Zhang W, Zhang H, Zhang WL, Song L, Tien P (2013) Enterovirus 71 VP1 Activates Calmodulin-Dependent Protein Kinase II and Results in the Rearrangement of Vimentin in Human Astrocyte Cells. PLoS ONE 8:e73900
Cong HL, Du N, Yang Y, Song L, Zhang WL, Tien P (2016) Enterovirus 71 2B induces cell apoptosis by directly inducing the conformational activation of the proapoptotic protein Bax. J Virol 90:9862–9877
Cui S, Wang J, Fan TT, Qin B, Guo L, Lei XB, Wang JW, Wang MT, Jin Q (2011) Crystal structure of human enterovirus 71 3C protease. J Mol Biol 408:449–461
De Jong AS, Wessels E, Dijkman HBPM, Galama JM, Melchers WJ, Willems PH, van Kuppeveld FJ (2003) Determinants for membrane association and permeabilization of the coxsackie virus 2B protein and the identification of the Golgi complex as the target organelle. J Biol Chem 278:1012–1021
Deng JX, Nie XJ, Lei YF, Ma CF, Xu DL, Li B, Xu ZK, Zhang GC (2012) The highly conserved 5’ untranslated region as an effective target towards the inhibition of Enterovirus 71 replication by unmodified and appropriate 2’-modified siRNAs. J Biomed Sci 19:73
Du N, Cong HL, Tian HC, Zhang H, Zhang WL, Song L, Tien P (2014) Cell surface vimentin is an attachment receptor for enterovirus 71. J Virol 88:5816–5833
Du HW, Yin PQ, Yang XJ, Zhang LL, Jin Q, Zhu GF (2015) Enterovirus 71 2C protein inhibits NF-κB activation by binding to RelA(p65). Sci Rep 5:14302
Feng CH, Fu YX, Chen DY, Wang HR, Su AR, Zhang L, Chang L, Zheng N, Wu ZW (2017) miR-127-5p negatively regulates enterovirus 71 replication by directly targeting SCARB2. FEBS Open Bio 7:747–758
Fernandez-Miragall O, Lopez de Quinto S, Martinez-Salas E (2009) Relevance of RNA structure for the activity of picornavirus IRES elements. Virus Res 139:172–182
Foo DG, Alonso S, Phoon MC, Ramachandran NP, Chow VT, Poh CL (2007) Identification of neutralizing linear epitopes from the VP1 capsid protein of Enterovirus 71 using synthetic peptides. Virus Res 125:61–68
Foo DG, Macary PA, Alonso S, Poh CL (2008) Identification of human CD4 T-cell epitopes on the P1 capsid protein of enterovirus 71. Viral Immunol 21:215–224
Fujii K, Sudaka Y, Takashino A, Kobayashi K, Kataoka C, Suzuki T, Iwata-Yoshikawa N, Kotani O, Ami Y, Shimizu H, Nagata N, Mizuta K, Matsuzaki Y, Koike S (2018) VP1 amino acid residue 145 of enterovirus 71 is a key residue for its receptor attachment and resistance to neutralizing antibody during cynomolgus monkey infection. J Virol pii:JVI.00682-18
Fujita K, Krishnakumar SS, Franco D, Paul AV, London E, Wimmer E (2007) Membrane topography of the hydrophobic anchor sequence of poliovirus 3A and 3AB proteins and the functional effect of 3A/3AB membrane association upon RNA replication. Biochemistry 46:5185–5199
Giachetti C, Hwang SS, Semler BL (1992) Cis-acting lesions targeted to the hydrophobic domain of a Poliovirus membrane-protein involved in RNA replication. J Virol 66:6045–6057
Guan HX, Tian J, Qin B, Wojdyla JA, Wang B, Zhao ZD, Wang MT, Cui S (2017) Crystal structure of 2C helicase from enterovirus 71. Sci Adv 3:e1602573
Han Y, Wang LY, Cui J, Song Y, Luo Z, Chen JB, Xiong Y, Zhang Q, Liu F, Ho WZ, Liu YL, Wu KL, Wu JG (2016) SIRT1 inhibits EV71 genome replication and RNA translation by interfering with the viral polymerase and 5′UTR RNA. J Cell Sci 129:4534–4547
Hellen CU, Lee CK, Wimmer E (1992) Determinants of substrate recognition by poliovirus 2A proteinase. J Virol 66:3330–3338
Herold J, Andino R (2001) Poliovirus RNA replication requires genome circularization through a protein-protein bridge. Mol Cell 7:581–591
Herrero LJ, Lee CS, Hurrelbrink RJ, Chua BH, Chua KB, McMinn PC (2003) Molecular epidemiology of enterovirus 71 in peninsular Malaysia, 1997-2000. Arch Virol 148:1369–1385
Hsu YY, Liu YN, Wang W, Kao FJ, Kung SH (2007) In vivo dynamics of enterovirus protease revealed by fluorescence resonance emissiontransfer (FRET) based on a novel FRET pair. Biochem Biophys Res Commun 353:939–945
Hu YC, Hsu JT, Huang JH, Ho MS, Ho YC (2003) Formation of enteroviruslike particle aggregates by recombinant baculoviruses coexpressing P1 and 3CD in insect cells. Biotechnol Lett 25:919–925
Huang SC, Hsu YW, Wang HC, Huang SW, Kiang D, Tsai HP, Wang SM, Liu CC, Lin KH, Su IJ, Wang JR (2008) Appearance of intratypic recombination of enterovirus 71 in Taiwan from 2002 to 2005. Virus Res 131:250–259
Huang PN, Lin JY, Locker N, Kung YA, Hung CT, Lin JY, Huang HI, Li ML, Shih SR (2011) Far Upstream Element Binding Protein 1 Binds the Internal Ribosomal Entry Site of Enterovirus 71 and Enhances Viral Translation and Viral Growth. Nucleic Acids Res 19:9633–9648
Huang SW, Wang YF, Yu CK, Su IJ, Wang JR (2012) Mutations in VP2 and VP1 capsid proteins increase infectivity and mouse lethality of enterovirus 71 by virus binding and RNA accumulation enhancement. Virology 422:132–143
Hung CT, Kung YA, Li ML, Brewer G, Lee KM, Liu ST, Shih SR (2016) Additive Promotion of Viral Internal Ribosome Entry Site-Mediated Translation by Far Upstream Element-Binding Protein 1 and an Enterovirus 71-Induced Cleavage Product. PLoS Pathog 12:e1005959
Jiang HB, Weng LY, Zhang N, Arita M, Li RQ, Chen LJ, Toyoda T (2011) Biochemical characterization of enterovirus 71 3D RNA polymerase. Biochim Biophys Acta 1809:211–219
Johnson KL, Sarnow P (1991) Three poliovirus 2B mutants exhibit noncomplementable defects in viral RNA amplification and display dosage-dependent dominance over wild-type poliovirus. J Virol 65:4341–4349
Kiener TK, Jia Q, Lim XF, He F, Meng T, Chow VT, Kwang J (2012) Characterization and specificity of the linear epitope of the Enterovirus 71 VP2 protein. Virol J 9:55
Kiener TK, Jia Q, Meng T, Chow VT, Kwang J (2014) A novel universal neutralizing monoclonal antibody against enterovirus 71 that targets the highlyconserved “knob” region of VP3 protein. PLoS Negl Trop Dis 8:e2895
Kobayashi K, Sudaka Y, Takashino A, Imura A, Fujii K, Koike S (2018) Amino acid variation at VP1-145 of enterovirus 71 determines attachment receptor usage and neurovirulence in human scavenger receptor B2 transgenic mice. J Virol pii:JVI.00681-18
Kuo RL, Kung SH, Hsu YY, Liu WT (2002a) Infection with enterovirus 71 or expression of its 2A protease induces apoptotic cell death. J Gen Virol 83:1367–1376
Kuo RL, Li ML, Hsu TA, Chang SC, Lee JC, Chen CC, Stollar V, Shih SR (2002b) The 3C protease activity of enterovirus 71 induces human neural cell apoptosis. Virology 293:386–395
Kuo CJ, Shie JJ, Fang JM, Yen GR, Hsu JT, Liu HG, Tseng SN, Chang SC, Lee CY, Shih SR, Liang PH (2008) Design, synthesis, and evaluation of 3C protease inhibitors as anti-enterovirus 71 agents. Bioorg Med Chem 16:7388–7398
Kuo RL, Kao LT, Lin SJ, Wang RY, Shih SR (2013) MDA5 plays a crucial role in enterovirus 71 RNA-mediated IRF3 activation. PLoS ONE 8:e63431
Lal SK, Kumar P, Yeo WM, Kar-Roy A, Chow VT (2006) The VP1 protein of human enterovirus 71 self-associates via an interaction domain spanning amino acids 66-297. J Med Virol 78:582–590
Lee JC, Shih SR, Chang TY, Tseng HY, Shih YF, Yen KJ, Chen WC, Shie JJ, Fang JM, Liang PH, Chao YS, Hsu JT (2008) A mammalian cellbased reverse two-hybrid system for functional analysis of 3C viral protease of human enterovirus 71. Anal Biochem 375:115–123
Lee PH, Liu CM, Ho TS, Tsai YC, Lin CC, Wang YF, Chen YL, Yu CK, Wang SM, Liu CC, Shiau AL, Lei HY, Chang CP (2015) Enterovirus 71 Virion- Associated Galectin-1 Facilitates Viral Replication and Stability. PLoS ONE 10:e0116278
Lei XB, Liu XL, Ma YJ, Sun ZM, Yang YW, Jin Q, He B, Wang JW (2010) The 3C protein of enterovirus 71 inhibits retinoid acid-inducible gene I-mediated interferon regulatory factor 3 activation and type i interferon responses. J Virol 84:8051–8061
Lei XB, Sun ZM, Liu XL, Jin Q, He B, Wang JW (2011) Cleavage of the adaptor protein trif by enterovirus 71 3C inhibits antiviral responses mediated by toll-like receptor 3. J Virol 85:8811–8818
Lei XB, Xiao X, Xue QH, Jin Q, He B, Wang JW (2013) Cleavage of interferon regulatory factor 7 by enterovirus 71 3C suppresses cellular responses. J Virol 87:1690–1698
Lei XB, Han N, Xiao X, Jin Q, He B, Wang JW (2014) Enterovirus 71 3C inhibits cytokine expression through cleavage of the TAK1/TAB 1/TAB 2/TAB 3complex. J Virol 88:9830–9841
Lei XB, Xiao X, Zhang ZZ, Ma YJ, Qi JL, Wu C, Xiao Y, Zhou Z, He B, Wang JW (2017) The Golgi protein ACBD3 facilitates Enterovirus 71 replication by interacting with 3A. Sci Rep 7:44592
Li ML, Hsu TA, Chen TC, Chang SC, Lee JC, Chen CC, Stollar V, Shih SR (2002) The 3C protease activity of enterovirus 71 induces human neural cell apoptosis. Virology 293:386–395
Li Q, Zheng ZH, Liu Y, Zhang ZF, Liu QS, Meng J, Ke XL, Hu QX, Wang HZ (2016) 2C proteins of enteroviruses suppress IKKα phosphorylation by recruiting protein phosphatase 1. J Virol 90:5141–5151
Li J, Yao YF, Chen Y, Xu X, Lin YQ, Yang ZL, Qiao WT, Tan J (2017) Enterovirus 71 3C promotes apoptosis through cleavage of PinX1, a telomere binding protein. J Virol 91:pii:e02016-16
Li XH, Huang Y, Sun MH, Ji H, Dou H, Hu J, Yan YF, Wang X, Chen LY (2018a) Honeysuckle-encoded microRNA2911 inhibits Enterovirus 71 replication via targeting VP1 gene. Antiviral Res 152:117–123
Li ZL, Ning SS, Su X, Liu X, Wang H, Liu Y, Zheng WW, Zheng BS, Yu XF, Zhang WY (2018b) Enterovirus 71 antagonizes the inhibition of the host intrinsic antiviral factor A3G. Nucleic Acids Res. https://doi.org/10.1093/nar/gky840
Lin JY, Li ML, Huang PN, Chien KY, Horng JT, Shih SR (2008) Heterogeneous nuclear ribonuclear protein K interacts with the enterovirus 71 5′ untranslated region and participates in virus replication. J Gen Virol 89:2540–2549
Lin JY, Chen TC, Weng KF, Chang SC, Chen LL, Shih SR (2009a) Viral and host proteins involved in picornavirus life cycle. J Biomed Sci 16:103–116
Lin JY, Li ML, Shih SR (2009b) Far upstream element binding protein 2 interacts with enterovirus 71 internal ribosomal entry site and negatively regulates viral translation. Nucleic Acids Res 37:47–59
Lin JY, Shih SR, Pan M, Li C, Lue CF, Stollar V, Li ML (2009c) hnRNP A1 interacts with the 5′ untranslated regions of enterovirus 71 and Sindbis virus RNA and is required for viral replication. J Virol 83:6106–6114
Lin JY, Li ML, Brewer G (2014) mRNA decay factor AUF1 binds the internal ribosomal entry site of enterovirus 71 and inhibits virus replication. PLoS ONE 9:e103827
Lin JY, Brewer G, Li ML (2015) HuR and Ago2 Bind the Internal Ribosome Entry Site of Enterovirus 71 and Promote Virus Translation and Replication. PLoS ONE 10:e0140291
Liu Y, Franco D, Paul AV, Wimmer E (2007) Tyrosine 3 of poliovirus terminal peptide VPg(3B) has an essential function in RNA replication in the context of its precursor protein, 3AB. J Virol 81:5669–5684
Lu J, Yi L, Zhao J, Yu J, Chen Y, Lin MC, Kung HF, He ML (2012) Enterovirus 71 disrupts interferon signaling by reducing the level of interferon receptor 1. J Virol 86:3767–3776
Luo Z, Dong XC, Li YX, Zhang Q, Kim C, Song Y, Kang L, Liu YL, Wu KL, Wu JG (2014) PolyC-Binding Protein 1 Interacts with 5,-Untranslated Region of Enterovirus 71 RNA in Membrane- Associated Complex to Facilitate Viral Replication. PLoS ONE 9:e87491
Madan V, Sanchez-Martinez S, Vedovato N, Rispoli G, Carrasco L, Nieva JL (2007) Plasma membrane-porating domain in poliovirus 2B protein. A short peptide mimics viroporin activity. J Mol Biol 374:951–964
McMinn PC (2002) An overview of the evolution of enterovirus 71 and its clinical and public health significance. FEMS Microbiol Rev 26:91–107
Morley SJ, Curtis PS, Pain VM (1997) eIF4G: translation’s mystery factor begins to yield its secrets. RNA 3:1085–1104
Nishimura Y, Lee H, Hafenstein S, Kataoka C, Wakita T, Bergelson JM, Shimizu H (2013) Enterovirus 71 Binding to PSGL-1 on Leukocytes: vP1-145 Acts as a Molecular Switch to Control Receptor Interaction. PLoS Pathog 9:e1003511
Paul AV, Yin J, Mugavero J, Rieder E, Liu Y, Wimmer E (2003) A “slide-back” mechanism for the initiation of protein-primed RNA synthesis by the RNA olymerase of poliovirus. J Biol Chem 278:43951–43960
Qiu J (2008) Enterovirus 71 infection: a new threat to global health. Lancet Neurol 7:868–869
Qiu Y, Xu Y, Zhang Y, Zhou H, Deng YQ, Li XF, Miao M, Zhang Q, Zhong B, Hu Y, Zhang FC, Wu L, Qin CF, Zhou X (2017) Human Virus-Derived Small RNAs Can Confer Antiviral Immunity in Mammals. Immunity 46:992–1004
Rowlands DJ, Tuthill TJ, Groppelli E, Rowlands DJ (2010) Picornaviruses. Curr Top Microbiol Immunol 343:43–89
Sharma R, Raychaudhuri S, Dasgupta A (2004) Nuclear entry of poliovirus protease-polymerase precursor 3CD: implications for host cell transcription shut-off. Virology 320:195–205
Shih SR, Chiang C, Chen TC, Wu CN, Hsu JT, Lee JC, Hwang MJ, Li ML, Chen GW, Ho MS (2004) Mutations at KFRDI and VGK domains of enterovirus 71 3C protease affect its RNA binding and proteolytic activities. J Biomed Sci 11:239–248
Silvestri LS, Parilla JM, Morasco BJ, Ogram SA, Flanegan JB (2006) Relationship between poliovirus negative-strand RNA synthesis and the length of the 3′ poly(A) tail. Virology 345:509–519
Sim AC, Luhur A, Tan TM, Chow VT, Poh CL (2005) RNA interference against enterovirus 71 infection. Virology 341:72–79
Solomon T, Lewthwaite P, Perera D, Cardosa MJ, McMinn P, Ooi MH (2010) Virology, epidemiology, pathogenesis, and control of enterovirus 71. Lancet Infect Dis 10:778–790
Tan S, Tan X, Sun X, Lu G, Chen CC, Yan J, Liu J, Xu W, Gao GF (2013) VP2 Dominated CD4 + T Cell Responses against Enterovirus 71 and Cross-Reactivity against Coxsackievirus A16 and Polioviruses in a Healthy Population. J Immunol 191:1637–1647
Tan YW, Hong WJ, Chu JJ (2016) Inhibition of enterovirus VP4 myristoylation is a potential antiviral strategy for hand, foot and mouth disease. Antiviral Res 133:191–195
Tan CW, Sam IC, Lee VS, Wong HV, Chan YF (2017) VP1 residues around the five-fold axis of enterovirus A71 mediate heparan sulfate interaction. Virology 501:79–87
Tang WF, Yang SY, Wu BW, Jheng JR, Chen YL, Shih CH, Lin KH, Lai HC, Tang P, Horng JT (2007) Reticulon 3 binds the 2C protein of enterovirus 71 and is required for viral replication. J Biol Chem 282:5888–5898
Tang ZF, Xia HJ, Wang PP, Yang J, Zhao TY, Zhang Q, Hu YY, Zhou X (2014) The identification and characterization of nucleicacid chaperone activity of human enterovirus 71 nonstructural protein 3AB. Virology 464–465:353–364
Teoh HL, Mohammad SS, Britton PN, Kandula T, Lorentzos MS, Booy R, Jones A, Rawlinson W, Ramachandran V, Rodriguez ML, Andrews PI, Dale RC, Farrar MA, Sampaio H (2016) Clinical Characteristics and Functional Motor Outcomes of Enterovirus 71 Neurological Diseasein Children. JAMA Neurol 73:300–307
Todd S, Towner JS, Brown DM, Semler BL (1997) Replicationcompetent picornaviruses with complete genomic RNA 3′noncoding region deletions. J Virol 71:8868–8874
Tseligka ED, Sobo K, Stoppini L, Cagno V, Abdul F, Piuz I, Meylan P, Huang S, Constant S, Tapparel C (2018) A VP1 mutation acquired during an enterovirus 71 disseminated infection confers heparan sulfatebinding ability and modulates ex vivo tropism. PLoS Pathog 14:e1007190
Ventoso I, Carrasco L (2003) A poliovirus 2A(pro) mutant unable to cleave 3CD shows inefficient viral protein synthesis and transactivation defects. J Virol 69:6280–6288
Victorio CB, Xu Y, Ng Q, Meng T, Chow VT, Chua KB (2016) Cooperative effect of the VP1 amino acids 98E, 145A and 169F in the productive infection of mouse cell lines by enterovirus 71 (BS strain). Emerging Microbes and Infections 5:e60
Wang JR, Kung YH, Huang SW, Kiang D, Ho MS, Liu CC, Yu CK, Su IJ, Wang JR (2010) Introduction of a strong temperature-sensitive phenotype into enterovirus 71 by altering an amino acid of virus 3D polymerase. Virology 396:1–9
Wang J, Wu Z, Jin Q (2012a) COPI Is Required for Enterovirus 71 Replication. PLoS ONE 7:e38035
Wang XM, Zhu CF, Bao WG, Zhao K, Niu JQ, Yu XF, Zhang WY (2012b) Characterization of full-length enterovirus 71 strains from severe and mild disease patients in northeastern China. PLoS ONE 7:e32405
Wang B, Xi XY, Lei XB, Zhang XY, Cui S, Wang JW, Jin Q, Zhao ZD (2013) Enterovirus 71 Protease 2Apro Targets MAVS to Inhibit Anti-Viral Type I Interferon Responses. PLoS Pathog 9:e1003231
Wang HB, Lei XB, Xiao X, Yang CF, Lu WL, Huang Z, Leng QB, Jin Q, He B, Meng GX, Wang JW (2015a) Reciprocal regulation between enterovirus 71 and the NLRP3 inflammasome. Cell Rep 12:42–48
Wang LC, Chen SO, Chang SP, Lee YP, Yu CK, Chen CL, Tseng PC, Hsieh CY, Chen SH, Lin CF (2015b) Enterovirus 71 Proteins 2A and 3D Antagonize the Antiviral Activity of IFN-γ via Signaling Attenuation. J Virol 89:7028–7037
Wang XH, Wang HR, Li YX, Jin Y, Chu Y, Su AR, Wu ZW (2015c) TIA-1 and TIAR interact with 5′UTR of enterovirus 71 genome and facilitate viral replication. Biochem Biophys Res Commun 466:254–259
Wang HR, Chang L, Wang XH, Su AR, Feng CH, Fu YX, Chen DY, Zheng N, Wu ZW (2016) MOV10 interacts with Enterovirus 71 genomic 5′UTR and modulates viral replication. Biochem Biophys Res Commun 479:571–577
Wang HQ, Li K, Ma LL, Wu S, Hu J, Jiang JD, Li YH (2017a) Berberine inhibits enterovirus 71 replication by downregulating the MEK/ERK signaling pathway and autophagy. Virol J 14:2
Wang T, Wang B, Huang H, Zhang CY, Zhu YM, Pei B, Cheng CF, Sun L, Wang JW, Jin Q, Zhao ZD (2017b) Enterovirus 71 protease 2Apro and 3Cpro differentially inhibit the cellular endoplasmic reticulum-associated degradation (ERAD) pathway via distinct mechanisms, and enterovirus 71 hijacks ERAD component p97 to promote its replication. PLoS Pathog 13:e1006674
Wang WB, Xiao F, Wan P, Pan P, Zhang YC, Liu F, Wu KL, Liu YL, Wu JG (2017c) EV71 3D protein binds with NLRP3 and enhances the assembly of inflammasome complex. PLoS Pathog 13:e1006123
Wen BP, Dai HJ, Yang YH, Zhuang Y, Sheng R (2013) MicroRNA-23b inhibits enterovirus 71 replication through downregulation of EV71 VPl protein. Intervirology 56:195–200
Weng TY, Chen LC, Shyu HW, Chen SH, Wang JR, Yu CK, Lei HY, Yeh TM (2005) Lactoferrin inhibits enterovirus 71 infection by binding to VP1 protein and host cells. Antivir Res 67:31–37
Weng KF, Li ML, Hung CT, Shih SR (2009) Enterovirus 71 3C protease cleaves a novel target CstF-64 and inhibits cellular polyadenylation. PLoS Pathog 5:e1000593
Weng KF, Chen LL, Huang PN, Shih SR (2010) Neural pathogenesis of enterovirus 71 infection. Microbes and infection/Institut Pasteur 12:505–510
World Health Organization. Regional Office for the Western Pacific (2018) Hand, foot and mouth disease situation update 2018. http://iris.wpro.who.int/handle/10665.1/14191
Wu KX, Ng MM, Chu JJ (2010a) Developments towards antiviral therapies against enterovirus 71. Drug Discov Today 15:1041–1051
Wu Y, Lou ZY, Miao Y, Yu Y, Dong H, Peng W, Bartlam M, Li XM, Rao ZH (2010b) Structures of EV71 RNA dependent RNA polymerase in complex with substrate and analogue provide a drug target against the hand-foot-andmouth disease pandemic in China. Protein Cell 1:491–500
Xia HJ, Wang PP, Wang GC, Yang J, Sun XL, Wu WZ, Qiu Y, Shu T, Zhao XL, Yin L, Qin CF, Hu YY, Zhou X (2015) Human Enterovirus Nonstructural Protein 2C ATPase Functions as Both an RNA Helicase and ATP-Independent RNA Chaperone. PLoS Pathog 11:e1005067
Xiao X, Lei XB, Zhang ZZ, Ma YJ, Qi JL, Wu C, Xiao Y, Li L, He B, Wang JW (2017) Enterovirus 3A facilitates viral replication by promoting PI4 KB-ACBD3 interaction. J Virol 91:e00791-17
Xie SQ, Wang K, Yu WJ, Lu W, Xu K, Wang JW, Ye B, Schwarz WG, Jin Q, Sun B (2011) DIDS blocks a chloride-dependent current that is mediated mediated by the 2B protein of enterovirus 71. Cell Res 21:1271–1275
Xu CZ, He X, Zheng ZR, Zhang Z, Wei CW, Guan K, Hou LH, Zhang BC, Zhu L, Cao Y, Zhang YH, Cao Y, Ma SL, Wang PH, Zhang PP, Xu QB, Ling YG, Yang X, Zhong H (2014) Downregulation of microRNA miR-526a by enterovirus inhibits RIG-I-dependent innate immune response. J Virol 88:11356–11368
Xu LF, He DL, Yang LS, Li ZQ, Ye XZ, Yu H, Zhao H, Li SX, Yuan LZ, Qian HL, Que YQ, Shih JW, Zhu H, Li YM, Cheng T, Xia NS (2015) A Broadly Cross-protective Vaccine Presenting the Neighboring Epitopes within the VP1 GH Loopand VP2 EF Loop of Enterovirus 71. Sci Rep 5:12973
Yang CH, Li HC, Jiang JG, Hsu CF, Wang YJ, Lai MJ, Juang YL, Lo SY (2010) Enterovirus type 71 2A protease functions as a transcriptional activator in yeast. J Biomed Sci 4:17–65
Yang SL, Chou YT, Wu CN, Ho MS (2011) Annexin II binds to capsid protein VP1 of enterovirus 71 and enhances viral infectivity. J Virol 85:11809–11820
Yang XD, Hu ZL, Fan SS, Zhang Q, Zhong Y, Guo D, Qin YL, Chen MZ (2018) Picornavirus 2A protease regulates stress granule formation to facilitate viral translation. PLoS Pathog 14:e1006901
Yeh MT, Wang SW, Yu CK, Lin KH, Lei HY, Su IJ, Wang JR (2011) A Single Nucleotide in Stem Loop II of 59-Untranslated Region Contributes to Virulence of Enterovirus 71 in Mice. PLoS ONE 6:e27082
Yi L, Lu J, Kung HF, He ML (2011) The virology and developments toward control of human enterovirus 71. Crit Rev Microbiol 37:313–327
Yuan SL, Li GM, Wang Y, Gao QQ, Wang YZ, Cui R, Altmeyer R, Zou G (2015) Identification of Positively Charged Residues in Enterovirus 71 Capsid Protein VP1 Essential for Production of Infectious Particles. J Virol 90:741–752
Zhang FH, Hao CS, Zhang S, Li AQ, Zhang QF, Wu W, Lin L, Li C, Liang MF, Li XL, Li DX (2014) Oral immunization with recombinant enterovirus 71 VP1 formulated with chitosan protects mice against lethal challenge. Virol J 11:80
Zheng ZH, Li HX, Zhang ZF, Meng J, Mao D, Bai BK, Lu BJ, Mao PY, Hu QX, Wang HZ (2011) Enterovirus 71 2C protein inhibits TNF-α–mediated activation of NF-kB by suppressing Ikβ kinase β phosphorylation. J Immunol 187:2202–2212
Acknowledgements
The work was supported by the National Natural Science Foundation of China (Grant 81503118) and CAMS Initiative for Innovative Medicine (CAMS-I2 M-1-010); The National Science and Technology Major Project of the Ministry of Science and Technology of China (2018ZX09711003-005-004).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Animal and Human Rights Statement
The authors declare that they have no conflict of interest. This article does not contain any studies with human or animal subjects performed by any of the authors.
Rights and permissions
About this article

Cite this article
Wang, H., Li, Y. Recent Progress on Functional Genomics Research of Enterovirus 71. Virol. Sin. 34, 9–21 (2019). https://doi.org/10.1007/s12250-018-0071-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12250-018-0071-9