
Abstract
We have developed a new method for the microextraction and speciation of arsenite and arsenate species. It is based on ionic liquid dispersive liquid liquid microextraction and electrothermal atomic absorption spectrometry. Arsenite is chelated with ammonium pyrrolidinedithiocarbamate at pH 2 and then extracted into the fine droplets of 1-butyl-3-methylimidazolium bis(trifluormethylsulfonyl) imide which acts as the extractant. As(V) remains in the aqueous phase and is then reduced to As(III). The concentration of As(V) can be calculated as the difference between total inorganic As and As(III). The pH values, chelating reagent concentration, types and volumes of extraction and dispersive solvent, and centrifugation time were optimized. At an enrichment factor of 255, the limit of detection and the relative standard deviation for six replicate determinations of 1.0 μg L−1 As(III) are 13 ng L−1 and 4.9 %, respectively. The method was successfully applied to the determination of As(III) and As(V) in spiked samples of natural water, with relative recoveries in the range of 93.3–102.1 % and 94.5–101.1 %, respectively.

Speciation of arsenite and arsenate by ionic liquid dispersive liquid-liquid microextraction - electrothermal atomic absorption spectrometry
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Inorganic arsenic is a known human carcinogen that can induce skin, bladder, and liver tumours and the element ranks first on the list of hazardous substances established by the Agency for Toxic Substances and Disease Registry of the US Environmental Protection Agency (EPA) [1–4]. Total arsenic concentrations in natural waters range from less than 1 μg L−1 to more than 5 mg L−1, with arsenite and arsenate generally being the most predominant species [5].
The current drinking standard set by the EPA and the World Health Organization is 10 μg L−1 [6, 7]. However, even at 10 μg L−1 the risk of arsenic-induced cancer is still 1 in 500 [8]. Compared to the EPA’s general risk management guideline for permissible skin cancer risk (1 in 10,000) and the guideline for more dangerous, more often fatal internal cancers (1 in 1,000,000), the current limit is an exceptionally high value. Arsenic is thus by far the most critical constituent in drinking water. Numerous debates have been held, especially in the US, about lowering the drinking water standard. To date, the limit has not been reduced, in part because regulatory authorities are hampered by economic reasons (with an estimated cost of 2.1 billion dollars per year for lowering it to 2 μg L−1 in the US only), but also by a lack of established analytical techniques to reliably determine arsenic at such low concentrations. An As concentration of 3 μg L−1 is currently regarded as Practical Quantitation Level and even at that concentration the cancer risk is still 1 in 1,667 [8]. There is thus an urgent need for analytical techniques capable of determining arsenic in the sub μg L−1 range.
Only a few analytical techniques, such as inductively coupled plasma- mass spectrometry, are available to measure trace concentrations of As directly, otherwise separation and pre-concentration techniques must be used prior to analysis. Several techniques have been employed to achieve a separation and pre-concentration of arsenic species. The liquid-liquid extraction is one of the oldest extraction and pre-concentration techniques, but the method is time-consuming and generally requires large quantities of organic solvent [9]. An alternative technique, which consumes much less solvent compared to liquid-liquid extraction, is solid phase extraction but it can be relatively expensive and yields a low enrichment factor [10]. Newer techniques include solid-phase microextraction [11], cloud point extraction [12, 13], liquid-phase microextraction [14], single drop microextraction [15], and dispersive liquid-liquid microextraction (DLLME) [16, 17]. Recently Han and Row reviewed newly developed liquid-phase microextraction-based techniques including their applications and limitations [18].
The DLLME method was developed in 2006, for the extraction and pre-concentration of organic compounds in water samples [19]. The DLLME method has been successfully applied for the determination of As(III) and As(V) in water samples [16, 17]. However, the reported methods were based on using a traditional organic solvent, typically carbon tetrachloride, which is known as a toxic solvent and has a high vapor pressure.
Ionic liquids (ILs) are organic salts including an organic cation with an anion. They have recently become popular as they are environmentally friendly, especially when compared with traditional organic solvents. Ionic liquids have generally negligible vapour pressures, tunable viscosities and miscibilities with water and organic solvents, and good thermal stability so are considered as viable replacements for volatile organic solvents [20]. Ionic liquids have received substantial attention in many fields of chemistry [21–23], especially for total metal and metal speciation analysis based on the different extraction techniques [20, 24–29]
The objective of the present study was to apply ionic liquid 1-butyl-3-methylimidazolium bis(trifluormethylsulfonyl) imide [BMIM][NTf2] as an extraction solvent for microextraction and speciation of As(III) and As(V) in water samples containing arsenic in concentrations lower than 10 μg L−1 followed by ETAAS as a detection method.
Experimental
Instrumentation
The arsenic measurements were performed using an atomic absorption spectrometer, Analytika Jena model ZEEnit 600 s (www.analytik-jena.de) equipped with a graphite furnace atomizer. An arsenic boosted discharge hollow cathode lamp, Photron, Australia (www.photron.com.au/) operated at 6 mA and an analytical wavelength of 193.7 nm with a spectral bandwidth of 0.8 nm was used. The temperature and time program for the graphite atomizer were given in Supplementary Material. The pH was measured with a HACH pH-meter, model HQ40d (www.hach.com). A centrifuge, Beckman USA, model Avanti™ J-25 (www.beckmancoulter.com) was used to accelerate the phase separation. A vortex model Genie 2™ (Bender and Hobein AG, Switzerland) was used for mixing the reagents.
Reagents and solutions
Standard stock solutions (1,000 mg L−1) of As(III) and As(V) were prepared by dissolving proper amounts of AsNaO2 and AsHNa2O4·7H2O (Sigma-Aldrich, Steinheim, Germany, www.sigmaaldrich.com), respectively. Working solutions were prepared daily by appropriate dilution of the standard stock solutions with Milli-Q water (18.2 MΩ cm, Millipore, France, www.millipore.com). A 0.020 g L−1 solution of ammonium pyrrolidinedithiocarbamate (APDC, Sigma-Aldrich, Steinheim, Germany, www.sigmaaldrich.com) chelating reagent was prepared daily by dissolving an appropriate amount of APDC in Milli-Q water. [BMIM][NTf2] (Merck, Germany, www.merck.de) which was kindly provided by Prof. Dr. A. Jess (University of Bayreuth, Germany). The pH adjustment was made with a 0.1 mol L−1 nitric acid (VWR, Germany, www.vwr.com) or ammonia solution (VWR international, France, www.vwr.com) for acidic and basic pH values, respectively. Methanol, acetone, and acetonitrile were purchased from (Fisher Scientific, Leicestershire, UK, www.fisher.co.uk). All water samples were filtered through 0.2 μm syringe filter (Sartorius Stedim Biotech, Germany, www.sartorius.com) prior to analysis. A 5 μL mixture of 1,000 mg L−1 Pd(NO3)2 and 100 mg L−1 Mg(NO3)2 (Merck, Darmstadt, Germany, www.merck.de/de/index.html) was used as a chemical modifier for each measurement by ETAAS.
Ionic liquid – dispersive liquid liquid microextraction procedure
A total volume of 5.0 mL of Milli-Q water spiked with 5 μg L−1 of As(III) containing 100 μL HNO3 (0.1 mol L−1) and 0.25 mg APDC chelating reagent was placed in a 10 mL polypropylene centrifuge tube with a conical bottom. This mixture was shaken well for 30 s using a vortex. A mixture of 50 μL [BMIM][NTf2] and 0.5 mL methanol was quickly injected into the sample solution. The sample solution was shaken and immersed in an ice bath for 5 min. During this step, the complex of As(III) and APDC at pH 2.0 was extracted into fine droplets of [BMIM][NTf2]. The cloudy solution was then centrifuged for 5 min at 6,000 rpm. Then, 10 μL of the sedimented phase was removed and diluted using methanol prior to ETAAS measurements. The extraction steps are illustrated in Fig. 1. For the determination of As(V) in the samples, all arsenic in solution was reduced to As(III) using a mixture of potassium iodide and sodium thiosulfate [30] and then the described method was applied for quantification of total As. As(V) was calculated by subtracting the total As concentration from As(III).

Schematic representation of different steps in IL-DLLME-ETAAS technique
Water samples collection
Tap water samples were collected from laboratory at University of Bayreuth (Upper Franconia, Germany) and four private households around the town of Gunzenhausen (Middle Franconia, Germany). The river water samples were collected from the river Main in Bayreuth (Rotmain), at the water level gauge Frauenkreuz/Gründlach, also in Upper Franconia. The river samples were filtered through a 0.2 μm syringe filter, collected in polyethylene bottles and stored at 4 °C prior to analysis.
Results and discussion
In the present work, [BMIM][NTf2] was used as an extraction solvent in DLLME combined with ETAAS for the determination of arsenite and arsenate species in water samples. To achieve the highest extraction efficiency, several parameters which influence complex formation and extraction were investigated and carefully optimized.
Selection of extractant and disperser solvent
Four ionic liquids including [BMIM][PF6], [BMIM][NTf2], [HMIM][BF4], and [OMIM][BF4] was carefully chosen in preliminary study. For selection a suitable ionic liquid as extractant in this method, some critical properties such as low solubility in water, higher density than water, and capability for arsenic extraction were considered. Furthermore, the solvent needed to be in liquid form at the selected experimental conditions. The water solubility and the density of the selected ionic decreases in the order [HMIM][BF4] > [OMIM][BF4] >> [BMIM][PF6] > [BMIM][NTf2] and [BMIM][NTf2] > [BMIM][PF6] > [HMIM][BF4] > [OMIM][BF4], respectively [31, 32]. Therefore, [BMIM][NTf2] was selected according to the above criteria in the subsequent experiments.
The disperser solvent in the DLLME method had to be miscible in both extraction solvent and the aqueous phase. Acetone, methanol, and acetonitrile were tested in the mixtures of 50 μL [BMIM][NTf2] and 0.5 mL of each disperser solvent. Acetone and acetonitrile produced very low amounts of sedimented phases and less absorbance intensity in ETAAS compared to methanol, so methanol was selected as a disperser solvent in the subsequent experiments.
Effect of amount of the extraction solvent and the disperser solvent
In order to evaluate the effect of amount of ionic liquid as extraction solvent, different volumes of IL [BMIM][NTf2] were tested (Supplementary Material). The results show that at low volume of IL, the sedimented phase was insufficient to be removed for further analysis. The highest extraction recovery and enrichment factor were obtained by using 50 μL of IL, as this volume was considered in the further experiments.
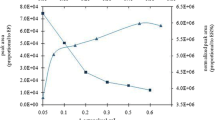
The effect of the volume of methanol as disperser solvent on the extraction recovery was studied in the range of 50 μL to 1.0 mL (Fig. 2). When applying low volumes of methanol (<0.4 mL), the cloudy state of the solution was not completely formed. At higher volumes of methanol (>0.7 mL), the solubility of the complex in the aqueous phase increased, and subsequently the extraction efficiency slightly decreased. Therefore, a volume of 0.5 mL of methanol was selected for further experiments. Influence of how and injection order of the IL and disperser solvent on extraction efficiency was discussed in Supplementary Material.

Effect of the disperser solvent (methanol) volume on the absorbance. Extraction conditions: 5 mL Milli-Q water spiked with 5 μg L−1 of arsenite containing 0.25 mg APDC, pH 2 and 50 μL of IL
Effect of pH and chelating reagent concentration
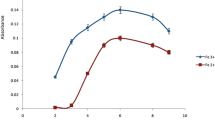
The pH of the aqueous phase plays an important role on the metal-chelate complexation and subsequent extraction. The extraction efficiency of arsenic species was investigated in the pH range from 2 to 11. As shown in Fig. 3, at acidic pH of 2–3, the maximum absorbance intensity for As(III) was obtained while the absorbance signal for As(V) was negligible at the entire pH range. This difference makes it possible to extract only As(III) at low pH. Thus, pH 2 was chosen as the optimum value to extract arsenite in further experiments.

Effect of sample solution pH on the absorbance (extraction conditions: 5 mL Milli-Q spiked with 5 μg L−1 of arsenite or arsenate containing 0.25 mg APDC, mix of 50 μL IL and 0.5 mL methanol)
The effect of the APDC chelating reagent concentration was also investigated in the range of 5–150 mg L−1. The results showed the absorbance increased by increasing the concentration of APDC up to 50 mg L−1 but did not increase any further at higher concentrations. Thus, a concentration of 50 mg L−1 of APDC was selected in the subsequent experiments.
Cooling and centrifugation time
Effect of centrifugation time and temperature on the recovery of the analyte was investigated; the experiment was described in Supplementary Material. The experimental results showed that the sedimented phase volume increased by changing the centrifugation time from 2 to 5 min but it was not increased more in the time exceeded 5 min. The temperature had no significant effect on the sedimented phase volume and the recovery of the analyte.
Effects of reducing reagents
In order to reduce As(V) completely to As(III), a mixture of potassium iodide and sodium thiosulfate solution [30] was used. Different volumes (10–500 μL) of the stock solutions of potassium iodide (2 % w/v) and sodium thiosulfate (1 % w/v) were added to the sample solution. The results showed that the absorbance signal increased as the quantities of mixed reducing agent were increased up to 100 μL. Higher volumes of reducing reagents caused a decrease in absorbance signal. Thus, 100 μL of each reducing reagent stock solution was used in the respective experiments.
Effects of potential interferences
The effect of several potentially interfering ions on the determination of arsenite at the concentration of 1.0 μg L−1 was studied under the optimized conditions. The obtained recoveries in the presence of various applied concentrations of individual interference ions are given in Table 1. The results showed that, the alkali and alkaline earth cations including Na+, K+, Mg2+, Ca2+ and Ba2+, and also common anions such as chloride and sulfate did not affect the arsenite recovery at the applied concentrations (recoveries 98.7–103 %). The lowest recovery was found in the presence of 0.5 mg L−1 Fe3+ with 90.2 %. However, in the presence of all other transition cations tested at concentrations up to at least 2,500 μg L−1, arsenite recoveries ranged from 91.3 % to 93.9 %.
Analytical figures of merit
The analytical characteristics of the method are listed in Table 2. The enrichment factor was calculated as the slope ratio of two calibration graphs with and without arsenite preconcentration. The calibration graph was linear in the range of 0.1–7.5 μg L−1, with a correlation coefficient (R2) of 0.9996. The limit of detection (LOD) calculated on the basis of three times the standard deviation for six replicate determinations of 0.1 μg L−1 arsenite and the relative standard deviations (RSDs) for six replicate determinations of 1.0 μg L−1 arsenite was 0.013 μg L−1 and 4.9 %, respectively. In order to evaluate the accuracy of the presented method, a standard reference material NASS-6 for seawater contains 1.43 ± 0.12 μg L−1 As was analysed. The amount of total As in the reference material using the presented method was 1.39 ± 0.07 μg L−1.
Analysis of natural water samples
Capability of the method for analysis of real samples was investigated by applying of the method for determination of arsenic species in some tap and river water samples (Table 3). While neither tap nor river water from Bayreuth contained any detectable traces of arsenic (<0.01 μg L−1), 2 μg L−1 of As(V) was measured in the Main river water at the water level gauge Frauenkreuz/Gründlach. The four tap water samples collected in Middle Franconia contained arsenic concentrations between 1.4 and 3.2 μg L−1 As(V), while As(III) was below detection limit. The corresponding central drinking water wells tap the Middle Keuper Blasensandstein (Late Triassic) have previously been reported to contain naturally high arsenic concentrations of several tens of μg L−1, which are probably derived from the Feuerletten Formation (Late Norian) [33]. In a field study from Freiberg University in 2000, a maximum of approximately 20 μg L−1 was determined in the wells near Büchelberg and a maximum of 90 μg L−1 in the wells close to Theilenhofen. The water is treated to remove arsenic before discharge into the public water supply network and currently meets the regulated 10 μg L−1 drinking water standard. However, with our sensitive method we have been able to show that not all arsenic is removed and the water would have to be subject to further treatment if the drinking water standard was lowered. Spiking the samples with As(III) and As(V) standards yielded relative recoveries of 93 to 102 % for As(III) and 95 to 101 % for As(V), confirming that the presented method is suitable for the determination of As(III) and As(V) in environmental samples.
The method was compared to other reported pre-concentration methods (Table 4). The presented method has a very high enrichment factor, and low sample consumption in comparison to some other reported methods. In contrast to many previous studies, the ionic liquid as extraction solvent is both safer and environmental friendly as opposed to traditional volatile organic solvents. Overall, this method is a powerful sample preparation technique and can be used as a replacement of the traditional extraction method for the routine inorganic As speciation analysis at sub μg L−1 levels in water samples.
Conclusions
A new microextraction method based on ionic liquid dispersive liquid liquid microextraction and electrothermal atomic absorption spectrometry was developed for As(III) and As(V) determination in water samples. The method is simple, fast, sensitive and requires only low quantities of sample. A high enrichment factor (255) and a low limit of detection (0.013 μg L−1) were obtained. Because the developed method is based on ionic liquid as extraction solvent instead of the traditional organic solvents, it can be considered a “green” extraction technique for inorganic arsenic speciation and pre-concentration. The method was also successfully applied to the determination of trace amounts of arsenite and arsenate species in water samples with an arsenic concentration lower than 10 μg L−1.
References
Rossman TG, Uddin AN, Burns FJ (2004) Evidence that arsenite acts as a cocarcinogen in skin cancer. Toxicol Appl Pharmacol 198:394–404
Hernandez-Zavala A, Valenzuela OL, Matousek T, Drobna Z, Dedina J, Garcia-Vargas GG, Thomas DJ, Del Razo LM, Styblo M (2008) Speciation of arsenic in exfoliated urinary bladder epithelial cells from individuals exposed to arsenic in drinking water. Environ Health Perspect 116:1656–1660
Chen YC, Guo YL, Su HJ, Hsueh YM, Smith TJ, Ryan LM, Lee MS, Chao SC, Lee JY, Christiani DC (2003) Arsenic methylation and skin cancer risk in Southwestern Taiwan. J Occup Environ Med 45:241–248
Rabieh S, Hirner AV, Matschullat J (2008) Determination of arsenic species in human urine using high performance liquid chromatography (HPLC) coupled with inductively coupled plasma mass spectrometry (ICP-MS). J Anal At Spectrom 23:544–549
Smedley PL, Kinniburgh DG (2002) A review of the source, behavior and distribution of arsenic in natural waters. Appl Geochem 17:517–568
Arsenic in Drinking Water, http://water.epa.gov/lawsregs/rulesregs/sdwa/arsenic/index.cfm
Guidelines for Drinking-water Quality, First addendum to third edition, Volume 1: http://www.who.int/water_sanitation_health/dwq/gdwq0506.pdf
Mushak P (2000) Arsenic and old laws, a scientific and public health analysis of arsenic occurrence in drinking water, its health effects, and EPA’s outdated arsenic water standard. http://www.nrdc.org/water/drinking/arsenic/chap1.asp
Dapaah AR, Ayame A (1997) Determination of arsenic in environmental samples by FI-HGAAS following solvent extraction preconcentration and back-extraction. Anal Sci 13:405–409
Yalcin S, Le XC (2001) Speciation of arsenic using solid phase extraction cartridges. J Environ Monit 3:81–85
Mester Z, Sturgeon R, Pawliszyn J (2001) Solid phase microextraction as a tool for trace element speciation. Spectrochim Acta B 56:233–260
Tang AN, Ding GS, Yan XP (2005) Cloud point extraction for the determination of As(III) in water samples by electrothermal atomic absorption spectrometry. Talanta 67:942–946
Shemirani F, Baghdadi M, Ramezani M (2005) Preconcentration and determination of ultra trace amounts of arsenic(III) and arsenic(V) in tap water and total arsenic in biological samples by cloud point extraction and electrothermal atomic absorption spectrometry. Talanta 65:882–887
Chamsaz M, Arbab-Zavar MH, Nazari S (2003) Determination of arsenic by electrothermal atomic absorption spectrometry using headspace liquid phase microextraction after in situ hydride generation. J Anal At Spectrom 18:1279–1282
Fragueiro S, Lavilla I, Bendicho C (2004) Headspace sequestration of arsine onto a Pd(II)-containing aqueous drop as a preconcentration method for electrothermal atomic absorption spectrometry. Spectrochim Acta B 59:851–855
Liang P, Peng L, Yan P (2009) Speciation of As(III) and As(V) in water samples by dispersive liquid-liquid microextraction separation and determination by graphite furnace atomic absorption spectrometry. Microchim Acta 166:47–52
Rivas RE, Lopez-Garcia I, Hernandez-Cordoba M (2009) Speciation of very low amounts of arsenic and antimony in waters using dispersive liquid–liquid microextraction and electrothermal atomic absorption spectrometry. Spectrochim Acta B 64:329–333
Han D, Row KH (2012) Trends in liquid-phase microextraction, and its application to environmental and biological samples. Microchim Acta 176:1–22
Rezaee M, Assadi Y, Millani Hosseini MR, Aghaee E, Ahmadi F, Berijani S (2006) Determination of organic compounds in water using dispersive liquid–liquid microextraction. J Chromatogr A 1116:1–9
Martinis EM, Berton P, Monasterio RP, Wuilloud RG (2010) Emerging ionic liquid-based techniques for total-metal and metal-speciation analysis. Trends Anal Chem 29:1184–1201
Safavi A, Maleki N, Bagheri M (2007) Modification of chemical performance of dopants in xerogel films with entrapped ionic liquid. J Mater Chem 17:1674–1681
Bagheri M, Rodriguez H, Swatloski RP, Spear SK, Daly DT, Rogers RD (2008) Ionic liquid-based preparation of cellulose-dendrimer films as solid supports for enzyme immobilization. Biomacromolecules 9:381–387
Wasserscheid P, Keim W (2000) Ionic liquids-new solutions for transition metal catalysis. Angew Chem Int Ed 39:3772–3789
Yousefi SR, Ahmadi SJ (2011) Development a robust ionic liquid-based dispersive liquid-liquid microextraction against high concentration of salt combined with flame atomic absorption spectrometry using microsample introduction system for preconcentration and determination of cobalt in water and saline samples. Microchim Acta 172:75–82
Majidi B, Shemirani F (2012) Salt-assisted liquid-liquid microextraction of Cr(VI) ionusing an ionic liquid for preconcentration prior to its determination by flame atomic absorption spectrometry. Microchim Acta 176:143–151
Yuan C, Liang P, Zhang Y (2011) Determination of trace silver in environmental samples by room temperature ionic liquid-based preconcentration and flame atomic absorption spectrometry. Microchim Acta 175:333–339
Berton P, Wuilloud RG (2010) Highly selective ionic liquid-based microextraction method for sensitive trace cobalt determination in environmental and biological samples. Anal Chim Acta 662:155–162
Monasterio RP, Wuillound RG (2010) Ionic liquid as ion-pairing reagent for liquid–liquid microextraction and preconcentration of arsenic species in natural waters followed by ETAAS. J Anal At Spectrom 25:1485–1490
Liu JF, Chi YG, Jiang GB (2005) Screening the extractability of some typical environmental pollutants by ionic liquids in liquid-phase microextraction. J Sep Sci 28:87–91
Zhang Q, Minami H, Inoue S, Atsuya I (2004) Differential determination of trace amounts of arsenic(III) and arsenic(V) in seawater by solid sampling atomic absorption spectrometry after preconcentration by coprecipitation with a nickel–pyrrolidine dithiocarbamate complex. Anal Chim Acta 508:99–105
Liu JF, Jönsson JA, Jiang GB (2005) Application of ionic liquids in analytical chemistry. Trends Anal Chem 24:20–27
Wang J, Pei Y, Zhao Y, Hu Z (2005) Recovery of amino acids by imidazolium based ionic liquids from aqueous media. Green Chem 7:196–202
Heinrichs G (1996) Arsenkonzentrationen in Grundwässern. In: Forschungsergebnisse aus dem Bereich Hydrogeologie und Umwelt, Heft 12, Lehr- und Forschungsbereich Hydrogeologie und Umwelt der Universität Würzburg (Hsrg.)
Huang C, Xie W, Li X, Zhang J (2011) Speciation of inorganic arsenic in environmental waters using magnetic solid phase extraction and preconcentration followed by ICP-MS. Microchim Acta 173:165–172
Acknowledgments
The authors would like to thank the German Research Foundation for base funding within the Emmy Noether program to Britta Planer-Friedrich (Grant PL 302/3-1). We would also like to thank Prof. Dr. Stefan Peiffer at University of Bayreuth for providing access to the instrumental (ETAAS) facility. Dr. Markus Bauer and Martina Rohr are gratefully acknowledged for their valuable suggestions on ETAAS analysis and Uwe Kunkel for providing river water samples.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 37 kb)
Rights and permissions
About this article
Cite this article
Rabieh, S., Bagheri, M. & Planer-Friedrich, B. Speciation of arsenite and arsenate by electrothermal AAS following ionic liquid dispersive liquid-liquid microextraction. Microchim Acta 180, 415–421 (2013). https://doi.org/10.1007/s00604-013-0946-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00604-013-0946-2