
Abstract
Liquid phase microextraction (LPME) is a popular technique for sample pretreatment before the trace determination of target compounds from complex matrices, examples being pesticides in environmental and food samples, or drug residuals in biological samples such as blood or urine. LPME is simple, affordable, easy to operate, and highly sensitive. It is a miniaturized implementation of conventional liquid-liquid extraction in which only a few microliters of solvents are used instead of several hundreds of milliliters. This review focuses on newly developed LPME-based techniques, their application to environmental and biological samples, on their limitations, and on future applications.

Liquid phase microextraction (LPME) is a popular technique for sample pretreatment before the trace determination of target compounds from complex matrices. This review focuses on newly developed LPME-based techniques, their application to environmental and biological samples, on their limitations, and on future applications.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Sample pretreatment is an important process in chemical analysis, especially for analyzing and determining the target compounds from complex matrices. A number of sample preparation methods have been developed for the separation and enrichment of analytes, such as liquid-liquid extraction (LLE) [1], supercritical fluid extraction (SFE) [2], and solid-phase extraction (SPE) [3, 4]. LLE is the most common extraction method; however, it is time-consuming, tedious, not sensitive enough for trace analysis, and requires large amounts of toxic organic solvents. SFE is a relatively rapid extraction method due to the low viscosities and high diffusivities associated with supercritical fluids. The disadvantage of SFE is that the extraction must be operated at high pressure (1,000–5,000 psi), which is required to maintain the solvent in a supercritical state, thus increasing the operating cost. Besides, due to the low polarity of CO2, SFE results in low recovery of polar components, further limiting its application. Another popular sample preparation approach is solid-phase extraction (SPE). Although this method uses much less solvent than LLE, an SPE column utilizes toxic organic solvents for pretreatment and elution. Thus, an effective enrichment method that addresses these drawbacks is necessary. Recently, solid-phase microextraction (SPME) and liquid-phase microextraction (LPME) have been developed [5–7]. However, SPME fibers are comparatively expensive and have limited lifetimes; therefore, LPME is a more attractive alternative, possessing many advantages such as simplicity, lower cost, negligible consumption of organic solvents, and high enrichment efficiency. In 1996, Liu and Dasgupta [8] were the first to report a new extraction system, wherein a micro drop of a water-immiscible organic solvent (1.3 μL) is suspended in a larger aqueous drop. Later, Jeannot and Cantwell [9] introduced a new solvent microextraction technique, wherein a micro drop (8 μL) of organic solvent was left suspended at the end of a Teflon rod immersed in a stirred aqueous sample solution. The basis of these LPME methods, called single drop microextraction (SDME), is the distribution of analytes between a small amount of water-immiscible solvent and an aqueous phase containing the analytes. Although SDME is very simple and efficient, it suffers from low stability of the hanging drop, which is easily lost into the sample during extraction. In 1999, Pedersen-Bjergaard and Rasmussen developed a new hollow fiber-based LPME technique (HF-LPME) [10]. In an HF-LPME device, the micro-extract solvent is contained within the lumen of a porous hollow fiber. In this way, the samples can be stirred or vibrated vigorously without loss of the micro-extract solvent. This technique can also provide a preconcentration of high analytes and excellent sample cleanup, and the fiber is disposable after use due to its low cost.
Another new LPME technique called dispersive liquid-liquid microextraction (DLLME) was developed recently by Rezaee and co-workers in 2006 [11]. In this method, an appropriate mixture of extraction solvent and dispenser solvent is injected into the aqueous sample by a syringe, forming a cloudy solution. The cloudy state results from the formation of fine droplets of the extraction solvent that disperse in the sample solution. The cloudy solution is then centrifuged, and the fine droplets sediment at the bottom of the conical test tube. Determination of the analytes in the remaining phase can be performed by instrumental techniques. In summary, simplicity of operation, speed, low sample volume, low cost, high recovery, and high enhancement factor are some advantages of DLLME.
LPME techniques seem to be a promising tool for trace analysis in complex samples due to easy sample cleanup and low limits of detection (LOD), which are at the level of nanograms per liter (ng L−1). Recently, the publications about LPME increase year by year (as shown in Fig. 1). This article focuses on newly developed LPME-based techniques and their application to environmental and biological samples. The advantages, limitations, and future outlook on LPME techniques are also discussed.

Evolution of number of publications concerning the combination of LPME methodologies
Classification of LPME
Single-drop LPME
Single-drop microextraction (SDME) is the simplest operational mode of the LPME technique, in which a single liquid drop is utilized as the collection phase, replacing the coated fiber. It is based on the principle of distribution of the analytes between a microdrop of extraction solvent at the tip of a micro syringe and an aqueous phase. Since the extraction medium is in the form of a single drop, this type of microextraction is called SDME. Solvent extraction is based on the principle that the equilibrium ratio of the concentration of solute between the organic phase and the aqueous phase is constant. SDME is also based on the same principle of distribution. A solvent microdrop is exposed to an aqueous sample, wherein the analyte is extracted into the drop. In this manner, high enrichment factors are obtained due to the high ratio of sample volume to organic phase volume. After extraction, the microdrop is retracted back into the microsyringe and then injected into instruments such as GC-MS, GC, and HPLC for further analysis.
Three modes of SDME, direct immersion SDME, headspace SDME, and continuous flow microextraction, have been developed for various analytical applications.
Direct immersion SDME (DI-SDME)
In 1996, Liu and Dasgupta [8] reported a drop-in-drop system to extract sodium dodecyl sulfate. In this report, a 1.3 μL microdrop of a water-immiscible organic solvent was immersed into a large flowing aqueous drop to accomplish the extraction process. At almost the same time, Jeannot and Cantwell [9] introduced a procedure that they termed solvent microextraction, in which the extraction medium is a droplet (8 μL) of 1-octanol held at the end of a Teflon rod and suspended in a stirred aqueous sample solution. After extraction for a prescribed time, the Teflon rod is withdrawn from the aqueous solution, and the organic phase is sampled with a microsyringe and injected into a GC system for analysis. In this work, the authors also proposed equilibrium and kinetic theories to explain this new mode of microextraction. A simple DI-SDME apparatus was shown in Fig. 2.

Schematic illustration of DI-SDME (reproduced with permission from Ref. [12])
To further improve the efficiency of extraction, He and Lee [13] developed dynamic-LPME. It should be noted that dynamic LPME is not strictly a SDME technique, as a drop configuration is not involved. In this approach, the aqueous sample is withdrawn into the microsyringe barrel preloaded with an organic solvent. Then, a few seconds are allowed to pass before the extraction of analytes into a thin film of organic solvent formed along the wall of the barrel as the bulk organic solvent is withdrawn back up towards the back of the barrel. Finally, the bulk solvent and organic thin film are recombined. This cycle is repeated many times within a few minutes. The enriched organic phase is then used for quantitation of analytes. In dynamic LPME, the mass transfer of analytes from the sample is faster and provides a higher enrichment factor; it is also claimed that the extraction efficiency is higher and reproducibility is improved as compared with static mode. Although SDME is a simple, low cost, and fast extraction technique, its main limitation is the instability of the droplet at high stirring speeds and in samples with a complicated matrix; consequently, careful and elaborate manual operations are required. This problem can be alleviated to some extent by using a 1 μL microsyringe instead of 10 μL one as well as by modification of the needle tip, although the organic drop is still not able to withstand a stirring speed of more than 1700 rpm. Also, when dealing with complex matrices, an extra filtration step of sample solution is imposed in order to alleviate the compromised stability of the drop. Furthermore, the sensitivity and precision of SDME methods are rather poor and require further improvement. Further, prolonged extraction times and faster stirring rate, which may result in dissolution and/or dislodgment of the drop, are not recommended.
Headspace SDME (HS-SDME)
In 2001, Jeannot and Cantwell [14] introduced a sample preparation method referred to as headspace solvent microextraction (HSME) or more commonly headspace single-drop microextraction. In this method, the extraction of analytes occurs by suspending a microliter drop of a proper non-aqueous solvent from the tip of a microsyringe located in the headspace of a sample, which is thermostated at a given temperature for a preset extraction time. The drop remains at the tip of the microsyringe throughout the extraction period, is retracted back into the microsyringe, and then used for the identification and quantification of the extracted analytes. In this mode, the analytes are distributed among three phases, the water sample, headspace, and organic drop. Aqueous phase mass transfer is the rate-determining step in this extraction, which means that a high stirring speed of the sample solution facilitates mass transfer and the extraction rate. This microextraction mode has potential for the determination of volatile compounds or volatile species produced by suitable derivatization methods in environmental, pharmaceutical, forensic, and food determinations, and it provides a high degree of cleanup of the extract as non-volatile and high molecular weight matrix interferences are reduced, if not eliminated. In this method, a solvent with a relatively low vapor pressure is preferred; thus, the practical difficulties of this technique include a limited choice of solvents due to the required viscosity and vapor pressure. Further work is needed to prove the reproducibility of these techniques.
Continuous flow microextraction (CFME)
CFME is another rapid, simple, inexpensive, and non-hazardous sample preparation technique, and it was first was first described by Liu and Lee in 2000 [15]. In this method, in a 0.5 mL glass chamber, an organic drop is held at the outlet tip of a polyetheretherketone (PEEK) connecting tube, which is immersed in a continuously flowing sample solution and acts as the fluid delivery duct and as a solvent holder. The solvent drop interacts continuously with the flowing sample solution, and extraction proceeds simultaneously. Diffusion and molecular momentum resulting from mechanical forces contribute to the effectiveness of this method. As the drop of the solvent makes full and continuous contact with the sample solution, this method could produce a higher concentration factor than the static LPME method. Xia et al. [16, 17] made some modifications to the basic CFME setup and developed a recycling-flow system in which the “waste” from the chamber returns to the sample vial. CFME has the advantage of higher performance but it still requires a peristaltic pump. Like SDME, since samples with complex matrices compromise the stability of the solvent drop during extraction, an extra filtration step of the sample solution is usually required. Later, Chen et al. [18] used the similar method determined the phenolic compounds in water samples by combined CFME with GC-FID. Practical applicability demonstrates that the method is feasible for qualitative and quantitative analysis of phenolic compounds in wastewater samples.
Hollow fiber based LPME (HF-LPME)
Pedersen-Bjergaard and Rasmussen [6] introduced an alternative concept for LPME based on the use of single, low cost, disposable, porous hollow fibers typically made of polypropylene. In this system, the microvolume of the extracting liquid is contained within the lumen of a porous hollow fiber such that the microextractant solvent is not in direct contact with the sample solution. The major advantage of this technique is that the sample can be stirred or vibrated vigorously without any loss of the extracting liquid, as it is mechanically protected. The schematic illustration of HF-LPME was shown in Fig. 3. In HF-LPME, prior to extraction, the hollow fiber is soaked into the immiscible organic solvent, which results in the immobilization of the organic solvent into the pores of the hollow fiber. The organic solvent, typically 10–20 μL in volume, forms a thin layer within the wall of the hollow fiber. The hollow fiber is then placed into a sample vial filled with the aqueous sample of interest. To speed up the extraction, the sample is extensively agitated or stirred. The analytes are then extracted from the aqueous sample through the organic phase in the pores of the hollow fiber and further into an acceptor solution inside its lumen. The disposable nature of the hollow fiber eliminates the possibility of sample carry over and ensures high reproducibility, and the pores in the walls of the hollow fiber imbue some selectivity by preventing the extraction of high molecular weight materials. HF-LPME may be accomplished both in two-phase or three-phase mode. In two-phase systems, the acceptor solution is the same organic solvent as that immobilized in the pores, and the analytes are collected in an organic phase that is compatible with GC. However, in a three-phase mode, the acceptor solution is another aqueous phase, and the analytes are extracted from an aqueous sample through the thin film of the organic solvent into an aqueous acceptor solution. Therefore, this mode is compatible with HPLC, CE, and AS.

Schematic illustration of HF-LPME (reproduced with permission from Ref. [19])
Dispersive LLME (DLLME)
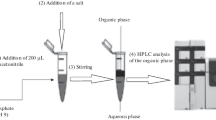
Dispersive liquid-liquid microextraction (DLLME) is another recent technique that has been successfully applied to the extraction and concentration of a wide variety of pesticides from water samples. DLLME was developed in 2006 by Rezaee and co-workers [11] and is based on a ternary solvent component system involving an aqueous phase, a non-polar water immiscible solvent (extracting solvent), and a polar water miscible solvent (disperser solvent). As shown in Fig. 4, fine droplets of the extracting solvent are dispersed into the aqueous phase when an appropriate mixture of both solvents is injected into the water samples. Following mixing, a cloudy solution is formed, followed by cooling and then centrifugation or solidification. The resulting fine particles of the extracting solvent containing the target analytes are then separated from the aqueous phase. High recoveries and high enrichment factors can be obtained, and the extraction time is relatively short. Mixing of the three components ensures equilibration within a few seconds due to the large interface between the multiple fine extractor droplets and the aqueous solution. DLLME can thus be regarded as a multiple-drop microextraction technique. In this method, water insoluble and high-density extracting solvents are mostly used. Chlorobenzene, chloroform, carbon disulfide, and carbon tetrachloride are some examples [21]. Acetone, acetonitrile, methanol, and ethanol are the main options as dispersive solvents. DLLME can be coupled with GC, HPLC, and also with atomic absorption spectrometry (AAS) [22–24].

Schematic illustration of DLLME (reproduced with permission from Ref. [20])
Directly-suspended droplet microextraction (DSDME)
This technique was developed by Lu et al. in 2006 [25]. A free microdroplet of solvent is delivered to the surface of an immiscible aqueous sample while being agitated by a stirring bar placed on the bottom of the sample cell. After some time, the microdroplet of solvent is withdrawn by a syringe and analyzed. The main disadvantage of the method is the difficult to take out the small amount of suspended droplet from the solution. Using a microsyringe, exact collection of the microdrop is impossible and some water may be transferred into the syringe.
Solidification of floating drop microextraction (SFDME)
To overcome the problem of removing a tiny amount of the suspended droplet in DSDME, a new extraction method based on solidifying the floating organic droplet was introduced by Khalili-Zanjani et al. in 2007 [26].In this microextraction mode, an appropriate volume of a suitable organic solvent (less than 20 μL) is delivered to the surface of the aqueous solution. The organic solvent must have a melting point near room temperature (in the range of 10–30 °C), such as 1-undecanol, 1-dodecanol, 2-dodecanol and n-hexadecane. After stirring a certain time, the sample is transferred into an ice bath, and the organic solvent will be solidified and can be removed from the sample matrix. The solid drop melts quickly at room temperature and can be analyzed. This novel technique has proved to be low-cost and virtually organic solvent-free and could provide a high enrichment factor. It can be easily integrated with gas chromatography (GC), high-performance liquid chromatography (HPLC), and atomic absorption spectrometry (AAS). However, its main drawback is the limited selection of extraction solvents because only a few organic solvents have melting points close to room temperature.
Influence factors on the microextraction efficiency
LPME is based on the extraction and preconcentration of analytes from the sample into a microvolume of extraction solvent. There are some factors which highly affect the microextraction efficiency.
Extraction solvent
During the HF-LPME process, the type of organic extraction solvent is essential for the efficient extraction. Firstly, the solvent must have good affinity for target compounds. Secondly, it should have a low solubility in water. Thirdly, it should be stable enough over the extraction time. Finally, the organic solvent should have excellent gas or liquid chromatographic behavior if using these detection methods. Besides, low toxicity solvents are considered to be better choice to avoid pollution. In general, water-immiscible organic solvents such as 1-octanol, toluene, di-n-hexyl ether, n-hexane, o-xylene, can be used in HF-LPME, as shown in Tables 1 and 2. For DI-SDME, DSDME and DLLME, the density of organic solvents plays a key role in the extraction process. It should be lower than water in DI-SDME and DSDME, while it should be higher than water in DLLME. As shown in Tables 1 and 3, decane, 1-butanol, n-octanol and isooctane are usually used in SDME, and chlorobenzene, tetrachloride carbon, dichlorocarbene are used in DLLME.
Extraction volume
The extraction volume of the extraction solvent directly affects the extraction efficiency since it affects the surface area of the drop and, in turn, the mass transfer of the analyte from the sample to the microdrop. In general, extraction volume in SDME is in the range of 1.0–10.0 μL, because larger drops are difficult to manipulate and lead to the microdrop’s instablility, which could result in the loss of the drop. In HF-LPME, extraction volume can be changed by using different length of hollow fiber. 2–8 cm of hollow fibers are usually used with the corresponding volume between 2.0 and 25 μL. As for the DLLME, the extraction volume is in the range of 10–300 μL. In general, as increasing the extraction volume, the extraction efficiency and enrichment factor increase, however, exceed a certain extent, the enrichment factor will decrease. Therefore, low extraction volume is used to obtain highest sensitivity.
Extraction time
LPME is not an exhaustive extraction technique, thus maximum sensitivity is attained at equilibrium conditions. On the other hand, complete equilibrium need not be attained for accurate and precise analysis. Therefore, it is vital to achieve equilibrium distribution of the extraction solvent between the aqueous phase and target compounds. In SDME and HF-LPME, it usually need long time (30–60 min) to achieve equilibrium and avoid loss of organic solvent.
Extraction time is not as important in DLLME as in the other LPME techniques. Because of the infinitely large surface area between extraction solvent and aqueous phase after the formation of cloudy solution, the target analytes diffuses quickly into the extraction solvent. Therefore, the DLLME method was time independent, which was the most important advantage of this technique.
Stirring rate
Fast stirring of the sample could be employed in LPME in order to enhance the extraction efficiency, since stirring permits the continuous exposure of the extraction surface to the aqueous sample. Stirring also reduces the time required to reach thermodynamic equilibrium and induces convection in the membrane phase.
However, in HF-LPME and SDME process, high stirring rate generate some problems such as production of air bubbles on the surface of the hollow fiber, promotion of solvent evaporation and instability of microdrop, which could decrease the extraction efficiency.
Ionic strength of sample solution
The addition of salt is widely used in microextraction to improve the analytes’ partitioning into the organic extraction phase. The addition of salt into the sample solution sometimes can improve the extraction efficiency of the analytes due to salting out effect. However, the presence of higher concentrations of salt could change the physical properties of the extraction film and thus reduce the diffusion rates of the analytes into the organic phase. In DSDME, an increase in ionic strength leads to two contrary effects. On the one hand, it enhances the extraction efficiency according to the “salting out” effect and, on the other hand, the dissolved salt may alter the physical properties of the Nernst diffusion film, then reducing the diffusion rates of analytes into the drop. Hence, depending on the analyte and the salt content of the aqueous solution, one of these two effects may be dominant, enhancing or restricting extraction. It is also possible that these two effects may cancel each other, and in this case, changes in the ionic strength of the sample solution do not affect extraction efficiencies. Furthermore, caution should be taken when high salt concentrations are used in the sample matrix, since under these conditions, in combination with the agitation of the sample, and the formation of air bubbles was promoted, increasing the incidents of drop loss and/or dislodgement of organic solvents.
Applications of LPME
SDME
In Table 1, the application of different modes of SDME to environmental and biological samples is summarized [27–51]. As mentioned earlier, the first application of SDME was reported by He and Lee [12]. They first developed LPME in a single drop of organic solvent by using a conventional microsyringe. Here, they extracted two chlorobenzenes into a single drop of toluene using a 10 μL syringe, providing higher (27-fold) enrichment within a much shorter extraction time (3 min) and relatively poorer precision (12.8%). Later, SDME was used to analyze metal ion pollution in environmental samples such as soil and water. In 2004, Fragueiro et al. [50] reported a simpler and more environmentally friendly HS-SDME-ETAAS method for the determination of As (III) and total As. This method is based on the exposure of an aqueous microdrop (3 μL) of Pd (30 mg L−1) to the headspace of a closed vial. Further, the microdrop acts as extractant for the generated hydride. Palladium is used both as an effective sorbing agent and a matrix modifier in the furnace. Total arsine is extracted from the sample in HCl medium, whereas As(III) is determined by arsine generation from the citric acid medium. Lin et al. reported SDME coupled with GC-FPD for the determination of Cr (III) in water. In this method, aqueous Cr (III) is first converted to volatile chromium trifluoroacetylacetonate (Cr(tfa)3) by reaction with 1,1,1-trifluoroacetylacetone (Htfa) under microwave irradiation. Derivatization of Cr (III) at the ng mL−1 level is completed in less than 1 min. The formed Cr (tfa)3 is then extracted into a small droplet (2 μL) of toluene suspended at the tip of a microsyringe needle. The optimal extraction time is 30 min. Mercury is an extremely toxic metal. Bagheri and Naderi used SDME combined with electrothermal vaporization atomic absorption spectroscopy for trace analysis of mercury in water samples [46]. In that study, they applied a microdrop of m-xylene as the extraction solvent. After extraction, the microdrop was introduced directly into a graphite furnace of AAS. A microdrop volume of 10 μL, a sampling temperature of 27 °C, and use of m-xylene containing dithizone as a complexing agent were found to be the major parameters to achieve a high enrichment factor of 970. Besides the determination of heavy metal content, SDME can also be used for the determination of pesticides in environmental and food samples.
Viñas et al. developed SDME combined with GC-MS for determination of seven strobilurin and six oxazole fungicides in fruits and juice samples [47]. The procedures are based on the dispersion of microvolumes of low-density organic solvents and the collection of floating organic solvent on the surfaces of the aqueous samples. Enrichment factors are between 80–1600, which implies the high sensitivity of this method. The same SDME technique was used by the group of Zhao et al. for the analysis of chloroacetanilide herbicides (alachlor, acetochlor, metolachlor, pretilachlor, and butachlor) residues in water [42]. The optimum experimental conditions were found to be 1.6 μL toluene microdrop, 5 mL water sample, 400 rpm stirring rate, 15 min extraction time, and no salt addition. Sharma et al. reported solid-phase extraction (SPE) of phenol and chlorophenols, their derivatization to methyl ethers, headspace single-drop microextraction (HS-SDME) of methyl ethers using 1-butanol as extraction solvent, and direct transfer of the drop into the injector for high performance liquid chromatography with diode array detection (HPLC-DAD) [28]. A rectilinear relationship was obtained between the amount of chlorophenols and peak area ratio of their methyl ethers/internal standard (4-methoxyacetophenone) in the range 0.01–10 mgL−1, correlation coefficient in the range 0.9956–0.9996, and limit of detection in the range 1.5–3.9 μg L−1 when HS-SDME alone was used for sample preparation. When using coupled SPE and HS-SDME, the linear range obtained was 0.1–500 μg L−1, correlation coefficient in the range 0.9974–0.9998, and the limit of detection in the range 0.04–0.08 μg L−1, which implied higher sensitivity.
Lambropoulou et al. extracted 10 organophosphorous insecticides from water samples coupling SPME with GC-MS [30]. In this method, extraction was achieved by suspending a 1.5 μL toluene drop to the tip of a microsyringe immersed in a 5 ml donor aqueous solution containing 2.5% NaCl (w/v) and stirred at 800 rpm. After microextraction, the organic drop is then drawn back into the syringe, after which the needle is removed from the vial and transferred immediately into the GC injection port for analysis. Under selected ion monitoring mode, the limits of detection were found to be in the range between 0.010 and 0.073 μg L-1.
SDME is also a trace analysis tool for drug residue in biological samples. In 2006, He and Kang used SDME for determination of the popular drug methamphetamine and its major metabolite, amphetamine, in a urine sample [31]. The enrichment factor was above 500−fold, providing an effective detection method for abuse drugs. Amongst recent developments, a simple SDME coupled with capillary electrophoresis was developed to determine six fluoroquinolones in human urine [38]. The limits of detection (LODs) varied from 7.4 to 31.5 μg L−1 at a signal-to-noise (S/N) ratio of 3. The recoveries at two spiking levels were found to be 81.8–104.9% with relative standard deviations of <8.3%. Fentanyl was a potent synthetic narcotic analgesic administered in the form of a transdermal patch for the management of chronic pain. Ebrahimzadeh et al. determined fentanyl in biological (plasma and urine) and wastewater samples using SDME-HPLC [41]. Fentanyl was extracted from 0.01 M NaOH solution (donor phase) into a thin layer of organic phase (100 μL), then back-extracted into 5 μL of the acidic acceptor microdrop (1 × 10−3 M HClO4) immersed in the organic membrane from the tip of a 25 μL HPLC syringe. At the most appropriate conditions (100 μL of noctane, 3.6 mL of the donor phase maintained at 0.01 mol L−1NaOH, 5 μL of 1 × 10−3 M HClO4 as the acceptor microdrop, stirring rate of 1000 rpm for pre-extraction and 700 rpm for backextraction, 30 °C, no salt addition, 30 min for pre-extraction and 20 min for back-extraction), an enrichment factor (EF) of 355 was obtained.
HF-LPME
In Table 2, current applications of HF-LPME reported in the scientific literature are summarized [52–99]. As seen from the table, most applications have been reported within the fields of environmental analysis and drug/pharmaceutical analysis. In addition, a few papers have focused on food, beverages, and peptides. Typically, two-phase LPME involves the use of either toluene or n-octanol as the organic phase, whereas three-phase LPME in most cases has been conducted with n-octanol or dihexyl ether as the SLM. In three-phase LPME, HCl and NaOH are used to make appropriate pH adjustments in the sample and acceptor solution.
As seen in Table 2, GC or GC-MS are mostly used for the final analysis of extracts from two-phase LPME, whereas HPLC, LC-MS, or CE are used in combination with three-phase LPME. Since LPME often provides very clean extracts, some papers have reported the direct coupling of LPME with different spectroscopic techniques in which the chromatographic or electrophoretic step has been eliminated. Varanusupakul and co-workers et al. [52] analyzed haloacetic acids (HAAs) in water using HF-LPME. The HAAs were derivatized with acidic methanol into their methyl esters and simultaneously extracted with supported liquid hollow fiber membrane in headspace mode. The derivatization was attempted directly in water sample without sample evaporation. The HF-LPME was performed using 1-octanol as the extracting solvent at 55 ◦C for 60 min with 20% Na2SO4. The method detection limits of most analytes were below 1 μg L−1.
Later, Payán et al. [97] explored the extraction and preconcentration of acidic pharmaceuticals in wastewaters using HF-LPME. A full factorial design for three factors and two levels was used to determine the effects and importance of donor pH, acceptor pH, and stirring time. Detection limits were found to be 20, 100, and 300 ng L−1 for salicylic acid, diclofenac, and ibuprofen, respectively. The same group also carried out the determination of sulfonamides and their main metabolites in environmental water [98]. Here, a Q3/2 Accurel KM polypropylene hollow fiber supporting 1-octanol was used with 2 M Na2SO4 aqueous solution (pH 4) as the donor phase and aqueous solution (pH 12) as the acceptor phase. The procedure produced very low detection and quantitation limits of 0.3–33 ng L−1 and 0.9–100 ng L−1, respectively.
Yang et al. used HF-LPME coupled with HPLC-UV to simultaneously determine three Aconitum alkaloids in human urine [55]. Analytes were extracted from a 5 mL urine sample containing 1.0 mmol L−1 of NaOH in 1-octanol membrane phase impregnated in the pores of a hollow fiber wall and then back into acidified aqueous solution in the lumen of the hollow fiber. After extraction, 10 μL of the acceptor phase was analyzed directly by HPLC. Another innovative approach involves the use of dynamic LPME based on hollow-fiber supported liquid membrane (SLM) extraction for extracting ionisable xenobiotics from human plasma [72]. The system is non-expensive, convenient, requires minimal manual handling, and enables samples with volumes as small as 0.2 mL to be extracted. For plasma samples, extraction efficiencies between 30 and 58% were achieved within 20 min. This method also produced 98- to 288-fold enrichment factors within 60 min of extraction and good repeatability with RSDs of 0.99–7.22%.
Since LPME can provide very high enrichments, combinations with CE may be used to determine drugs and drug metabolites in human plasma, although CE with UV detection is known to provide relatively poor detection limits. This may be especially interesting for chiral applications since CE is well known for its excellent chiral selectivity. Previously, three-phase LPME of both [69] was followed by chiral CE to monitor chiral metabolism in humans. Although CE analysis was conducted with UV detection, therapeutically relevant concentrations were easily measured due to the high enrichment obtained by LPME. Lin et al. [70] used microemulsion electrokinetic chromatography (MEEKC) coupled with HF-LPME for the determination of six aromatic amines. The obtained enrichment factors ranged between 70 and 157 in a 30 min extraction time, and the LODs ranged between 0.0021 and 0.0048 μg mL−1. Compared to conventional sample preparation procedures, this environmentally friendly method certainly provides better sensitivity and has the distinct advantage of simplicity. HF-LPME can also be combined with GC-MS. Ghasemi et al. [65] developed using a headspace HF-LPME combined with capillary gas chromatography–mass spectrometry for determination of volatile organic compounds of selenium (dimethylselenide (DMSe) and dimethyldiselenide (DMDSe).
DLLME
Besides HF-LPME, DLLME also has been widely used in environmental and biological analyses (Table 2) [22–24, 100–128]. Rezaee et al. used DLPME for extraction and determination of polycyclic aromatic hydrocarbons (PAHs) in water samples [124]. Specifically, 1 mL of acetone (as disperser solvent) containing 8.0 μL of C2Cl4 (as extracting solvent) was rapidly injected into 5 mL of the sample solution using a 1 mL syringe, after which the mixture was gently shaken. Then, the mixture was centrifuged and 2 μL of the sedimented phase was injected into the GC for analysis. Under optimal conditions, the obtained PFs were found to range from 603 to 1113. The linear range was 0.02–200 μg L−1 and the LOD was 0.007–0.030 μg L−1 for most of the analytes. Yan et al. preconcentrated and determined six pyrethroids in river water samples using ultrasound-assisted dispersive liquid-liquid microextraction [118]. In this method, a suitable mixture of extraction solvent (20 μL of tetrachloromethane) and dispersive solvent (1.00 mL of acetone) are injected into the aqueous samples (10 mL), resulting in a cloudy solution. After centrifugation, the enriched analytes in the sediment phase are determined by HPLC-UV. Under optimal conditions, the enrichment factors for the six pyrethroids were ranged from 767 to 1033 folds.
Leong et al. [117] developed a technique based on solidification of a floating organic drop (DLLME-SFO) for the determination of six organochlorine pesticides in water. The experimental procedure consisted of adding disperser and extraction solvents (acetonitrile and hexane, respectively) to the aqueous solution, such that small hexane droplets formed. Then, the tube was deposited in crushed ice, and the solidified organic solvent drop was transferred to another receptacle, where it melted. Enrichment factors were found to be in the order of 37–872.
Another example is the work of Du et al. [121], who developed a similar methodology for the extraction of cypermethrin and permethrin from pear juice. In this work, the DLLME disperser-acting solvent (methanol) and the extraction solvent (C2Cl4) had already been used to extract the pesticides from the matrix. Under the optimum condition, the enrichment factors for cypermethrin and permethrin were 344 and 351 fold respectively.
Another important group of analytes that have been the focus of DLLME procedures are pharmaceuticals. Liu et al. combined SPE with DLLME for the determination of clenbuterol (CLB) in porcine tissue samples [111]. The recoveries at three spiked levels were ranged from 87.9% to 103.6% with RSD less than 3.9% and the EF of 62 folds could be obtained. Recently, Lv et al. developed a new DLLME method combined with floating organic droplet for the determination of volatile aldehyde biomarkers (hexanal and heptanal) in human blood [126]. In this method, 1-dodecanol is used as an extraction solvent and its density is lighter than water, such that it forms a floating drop and can easily be removed for analysis.
Chen et al. [127] applied DLLME to determine chloramphenicol, a broad-spectrum antibiotic banned internationally, in honey. To develop the method, it was necessary to dilute the honey with water and vortex the solution until it became homogeneous. The high viscosity of the initial sample precluded the formation of the droplets, leaving dilution as the best approach. A mixture of acetonitrile and 1, 1, 2, 2-tetrachloroethane (as extraction solvent) was injected into an aliquot of 5 μL of the homogeneous diluted honey sample. After centrifugation, the final extraction droplet was analyzed by HPLC-UV. Mean relative recoveries were in the range 89.5–91.7% with RSD less than 5.1%.
Tsai et al. [128] used a modified DLLME method combined with HPLC-DAD to determine quinolones in pig muscle. About 5 g of tissue was extracted with acetonitrile (containing 70 μL of 70–72% perchloric acid), which was used as a disperser solvent. In this case, 300 μL of dichloromethane were added, and the mixture was quickly introduced into 7.5 μL of deionized water. DLLME was used as more of a cleaning step than an extraction procedure. The effects of both extraction solvent volume and pH of water were investigated. Increased dichloromethane volume resulted in higher extraction recoveries, but the cloudy suspension of droplets was not well formed, and the ternary component system should have been vortexed. However, larger volumes of dichloromethane resulted in larger volumes of the settled phase as well as decreases in the enrichment factor.
DSDME
As mentioned above, DSDME was introduced by by Lu et al. in 2006 [25] using, 8-dioxyanthraquinone as research subject. Later, Sarafraz-Yazdi et al. developed a similar methodology for determining two tricyclic antidepressant drugs (amitriptyline and nortriptyline) in urine sample[129]. In this technique, an aqueous sample is agitated with a stirring bar, creating a mild vortex at the center of the vial. A droplet of toluene is placed at the bottom of the vortex. After 20 min, toluene is withdrawn with a syringe and injected into the GC. Under the optimum condition, typical enrichment factors were 167 and 179 for amitriptyline and nortriptyline, respectively. The same author combined HPLC with DSDME for determining the diclofenac, 3-nitroaniline, chlorophenols and volatile organic compounds such as benzene, toluene, ethylbenzene and o-xylene in environmental water samples [130–132], which provided a fast, effective trace detection method for environmental monitor. In 2009, Farahani et al. examined the potential of DSDME for pretreatment complicate sample matrix (human plasma) [133]. An enrichment factor of 187 along with substantial sample clean up was obtained under the optimized conditions. Excellent extraction efficiency is achieved almost independent of the matrix in the actual application. Subsequently, Viñas et al. developed DSDME with injection-port derivatization coupled to GC-MS for the analysis of polyphenols in herbal infusions, fruits and functional foods [134]. The procedure uses undecanone as extraction solvent. The sensitivity and detection limits for polyphenols using the DSDME sample pretreatment method were very low. Enrichment factors are between 413 and 578. DSDME also can be combined with CE for analyzing three alkaloids [135]. Microliters of n-octanol were dripped on the top of the aqueous sample and the mixture was agitated for 8 min at 1150 rpm for DSDME. Afterwards, the stirring rate was adjusted at 800 rpm and the larger droplet of organic phase kept steady. Then, the microsyringe filled with 1 μL of 20 mmol L−1 HCl was inserted into the vial by piercing the septa. The needle tip was fixed in the center of the larger droplet of organic extractive phase and the plunger of the syringe was depressed completely to suspend the droplet in it for back-extraction. Under the optimum conditions, the enrichment factors ranged from 231 to 524.
SFDME
As a novel sample preparation method, SFDME can be used in combination with HPLC, GC, and AAS. It has been widely applied in the fields such as the analysis of pesticide residues and heavy metals. The typical applications are listed in Table 4 [136–149].
It was first introduced by Khalili Zanjani et al. [26] for analyzing polycyclic aromatic hydrocarbons in water samples. 1-undecanolwas delivered to the surface of solution containing analytes and solution was stirred for a desired time. Then sample vial was cooled by inserting it into an ice bath for 5 min. The solidified 1-undecanol was transferred into a suitable vial and immediately melted; then, 2 μL of it was injected into a gas chromatograph for analysis. Under the optimized conditions, enrichment factors were in the range of 594–1940, which greatly improved the sensitivity of the trace amount of pollutes in environmental water [136]. Later, Sobhi et al.was first to employ orthogonal array designs (OADs) to screen the liquid-phase microextraction (LPME) method in which few microliters of 1-undecanol were delivered to the surface of the aqueous sample to analysis fat-soluble vitamins [137]. Besides, another important application of SFDME is to detect the heavy metals pollution in environment and food [138–146]. After SFDME, the trace amount of heavy metals could be detected by ETAAS or other instruments. Combined with GC or HPLC, SDME also have been used to analysis the pesticide residual [147–149].
Development of novel microextraction solvent “ionic liquid”
Extraction mechanisms
Ionic liquids (Ils) are gaining widespread recognition as novel solvents in chemistry. Compared to classical organic solvents, Ils generally consist of bulky, nonsymmetrical organic cations, such as imidazolium, pyrrolidinium, pyridinium, ammonium, or phosphonium, as well as numerous different inorganic or organic anions, such as tetrafluoroborate and bromide anions. The unique properties of Ils, such as a negligible vapor pressure, good thermal stability, tunable viscosity, and miscibility with water and organic solvents, as well as their good extractability for various organic compounds and metal ions mainly depend on their special structures. Some Ils are suitable for conventional liquid-liquid extraction due to their immiscibility with water (which allows formation of biphasic systems) as well as the high solubility of their organic species. The design of safe and environmentally benign separation processes plays an increasingly important role in the development of extraction technology.
Huddleston et al. [150] studied the partition coefficients between Ils and water and compared the results with the octanol-water partition coefficient. They found that these two coefficients exhibit a good linear relationship, and the distribution coefficient is higher for the uncharged form. Later, Armstrong’s group measured the ionic liquid/water and ionic liquid/anthene distribution coefficients of a set of 40 compounds with various functionalities, including organic acids, organic bases, amino acids, antioxidants, and neutral compounds, using liquid chromatography [151]. Marked differences in the partitioning behavior of basic, acidic, and neutral compounds were observed. This study indicated a lower basicity of the IL phase compared to octanol. Acidic solutes have distribution coefficients lower than their distribution coefficients in octanol. The opposite is true for aminoaromatic compounds. These results can probably be attributed to the lower basicity of the Ils compared to octanol. In general, for ionizable compounds, transfer from aqueous phase to an IL at room temperature is more efficient for the neutral form of the compound. Changing the pH of the aqueous phase is an effective means of adjusting selectivity for extraction by Ils, as is the case for non-ionic solvents.
Application of ionic liquid-based LPME
Table 5 summarizes some representative examples of IL-based extraction of metal ions and organic contaminants in environmental samples, such as water, soil, and so on [152–170].
Extraction of metal ions from polluted environmental samples is another main use of Ils. The ions Ag+, Hg2+, Cu2+, Pb2+, Cd2+, and Zn2+ have successfully been extracted into [BMIM][PF6] by employing dithizone as a chelator to form neutral metal-dithizone complexes [152, 153, 155, 158, 161, 162, 168–170]. It was found that the extraction efficiency of Ils is higher than that of chloroform at low Ph. Furthermore, metal ions can be extracted from aqueous phase into [BMIM][PF6] and then back into aqueous phase with high recovery by manipulating the Ph value of the extraction system.
Manzoori et al. [152] used IL ([C4MIM][PF6])-based SDME for preconcentration of lead in environmental water. Lead was complexed with ammonium pyrroldinedithiocarbamate (APDC) and extracted into a 7 μL ionic liquid drop. The extracted complex was directly injected into the graphite furnace. In the optimum experimental conditions, the limit of detection (3 s) and the enhancement factor were 0.015 μgL−1 and 76, respectively. And later, the similar result was reported for the extraction of manganese using the same IL [155].
DLLME takes advantage of the low solubility of the extraction solvent, in this case Ils such as [HMIM][PF6] that are dispersed throughout a larger sample (aqueous) volume assisted by a disperser solvent and subsequently recovered from solution as a discrete drop. Liu et al. [168] first performed IL-based dispersive liquid-phase microextraction (IL-DLPME) of polycyclic aromatic hydrocarbons (PAHs) from water. A mixture of 0.052 g of [HMIM][PF6] and 0.50 μL of methanol (disperser solvent) was quickly injected into a sample solution using a 1 μL syringe. A cloudy solution quickly formed as fine droplets of the immiscible extraction solvent dispersed in the aqueous sample, which greatly enlarged the contact area between the extraction solvent and aqueous phase. The analytes in the aqueous sample were extracted into the fine IL droplets at this step. Then, the water–methanol-[HMIM][PF6] mixture was centrifuged at 4,000 rpm for 10 min. After this, the dispersive particles of the IL phase sedimented at the bottom of a conical test tube. The upper aqueous phase was then removed with a syringe, and the IL phase (about 19 μL) was dissolved in 50 μL of methanol, after which 10 μL was injected into the HPLC system for analysis. The enrichment factor ranged from 10 to 200, which is about three times that obtained with 1-octanol. Recently, Zhou et al. [163] developed a temperature-controlled IL dispersive liquid-phase microextraction technique for the extraction of pyrethroid pesticides from water samples. A homogeneous phase of IL and water was obtained by heating, whereas cooling of the homogeneous liquid mixture produced phase separation due to decreased solubility. Briefly, 45 μL of [HMIM] [PF6] was added to about 10 μL of the water sample, and this was then heated to 70 °C, allowing the IL to completely dissolve in water. After the extraction, the system was cooled down, and phase separation of the IL from water was obtained by centrifugation.
Using a similar protocol, chlorobenzenes, phenols, dichlorodiphenyltrichloroethane, and its metabolites in a water sample were preconcentrated. A later study showed that many organic pollutants, including chlorobenzenes, anthraquinones, organophosphorus pesticides, herbicides, and Triazines herbicides can be preconcentrated using this method [164–167]. The advantage of dispersive methods is the greater surface area provided by the dispersed or dissolved extraction solvent, which enhances the rate of analyte transfer to the extraction solvent.
HF-LPME using ILs has also been used in extraction. In this method, the extraction solvent is immobilized within the pores of the membrane and forms a liquid barrier between the donor phase (sample solution) and acceptor phase (injection solvent). For aqueous donor phases, room temperature ILs [OMIM][PF6] are suitable solvents for the extraction of chlorophenols into basic buffer. The LODs for chlorophenols are in the range from 0.5–2.5 μg/L, which is suitable for analysis of typical environmental samples [154].
Limitation and outlook
LPME-based techniques are potential extraction procedures for the analysis of pesticide residuals in environmental and food samples as well as drug residuals in biological samples. In general, LPME can provide high extraction recoveries, high enrichment, and excellent sample cleanup within a short extraction time (1–45 min). However, the implementation of LPME is currently limited by the unavailability of commercial equipment. Work in this area is in progress, and the near future should produce some commercial equipment for LPME. This equipment should be fully automated and compatible with common laboratory robotics and auto-samplers. Based on the strong advantages of LPME, it is expected to be an important future sample preparation technique complementing existing techniques such as liquid-liquid extraction, solid-phase extraction, and solid-phase microextraction.
Abbreviations
- LPME:
-
liquid phase microextraction
- LLE:
-
liquid-liquid extraction
- SFE:
-
supercritical fluid extraction
- SPE:
-
solid-phase extraction
- SPME:
-
solid-phase microextraction
- SDME:
-
single drop microextraction
- HF-LPME:
-
hollow fiber-based LPME
- DLLME:
-
dispersive liquid-liquid microextraction
- DSDME:
-
directly-suspended droplet microextraction
- SFDME:
-
Solidification of floating drop microextraction
- LOD:
-
low limits of detection
- DI-SDME:
-
direct immersion SDME
- HS-SDME:
-
headspace SDME
- CFME:
-
continuous flow microextraction
- GC-MS:
-
gas chromatography–mass spectrometry
- ETAAS:
-
electrothermal atomic absorption spectrometry
- GC-ICP-MS:
-
gas chromatography-inductively coupled plasma mass spectrometry
- ETV-ICP-MS:
-
electrothermal vaporization inductively coupled plasma mass spectrometry
- MEKC:
-
micellar electrokinetic chromatography
- GF-AAS:
-
graphite furnace atomic absorption spectrometry
- ESI-IMS:
-
electrospray ionizationion mobility spectrometry, AFS, Atomic fluorescence spectrometry
- FAAS:
-
Flame atomic absorption Spectrometry
- EAAS:
-
electrothermal-atomic absorption spectrometry
- ICP-OES:
-
inductively coupled plasma-optical emission spectrometry
- [C4MIM][PF6]:
-
1-butyl-3- methylimidazolium hexafluorophosphate
References
Agrawal YK (2002) Liquid-liquid extraction, separation recovery and transport of tantalum by crown-ether. Talanta 58:875–882
Rissato SR, Galhiane MS, Knoll FRN, Apon BM (2004) Supercritical fluid extraction for pesticide multiresidue analysis in honey: Determination by gas chromatography with electron-capture and mass spectrometry detection. J Chromatogr A 1048:153–159
Suárez B, Santos B, Simonet BM, Cárdenas S, Valcárcel M (2007) Solid-phase extraction-capillary electrophoresis-mass spectrometry for the determination of tetracyclines residues in surface water by using carbon nanotubes as sorbent material. J Chromatogr A 1175:127–132
Chen L, Yu A, Zhuang X, Zhang K, Wang X, Ding L, Zhang H (2007) Determination of andrographolide and dehydroandrographolide in rabbit plasma by on-line solid phase extraction of high-performance liquid chromatography. Talanta 74:146–152
Vuckovic D, Zhang X, Cudjoe E, Pawliszyn J (2010) In-tube solid-phase microextraction coupled by in valve mode to capillary LC-DAD: Improving detectability to multiresidue organic pollutants analysis in several whole waters. J Chromatogr A 1217:2695–2702
Pena-Pereira F, Lavilla I, Bendicho C (2010) Liquid-phase microextraction techniques within the framework of green chemistry. Trac-Trends Anal Chem 29:617–628
Dadfarnia S, Haji Shabani AM (2010) Recent development in liquid phase microextraction for determination of trace level concentration of metals—A review. Anal Chim Acta 658:107–119
Liu H, Dasgupta PK (1996) Analytical chemistry in a drop solvent extraction in a microdrop. Anal Chem 68:1817–1821
Jeannot MA, Cantwell FF (1996) Solvent microextraction into a single drop. Anal Chem 68:2236–2240
Pedersen-Bjergaard S, Rasmussen KE (1999) Liquid–liquid–liquid microextraction for sample preparation of biological fluids prior to capillary electrophoresis. Anal Chem 71:2650–2656
Rezaee M, Assadi Y, Milani Hosseini MR, Aghaee E, Ahmadi F, Berijani S (2006) Determination of organic compounds in water using dispersive liquid-liquid microextraction. J Chromatogr A 1116:1–9
Xu L, Basheer C, Lee HK (2007) Developments in single-drop microextraction. J Chromatogr A 1152:184–192
He Y, Lee HK (1997) Liquid-phase microextraction in a single drop of organic solvent by using a conventional microsyringe. Anal Chem 69:4634–4640
Theis AL, Waldack AJ, Hansen SM, Jeannot MA (2001) Headspace solvent microextraction. Anal Chem 73:5651–5654
Liu W, Lee HK (2000) Continuous-flow microextraction exceeding 1000-fold concentration of dilute analytes. Anal Chem 72:4462–4467
Xia L, Hu B, Jiang Z, Wu Y, Li L, Chen R (2005) 8-Hydroxyquinoline-chloroform single drop microextraction and electrothermal vaporization ICP-MS for the fractionation of aluminium in natural waters and drinks. J Anal Atom Spectrom 20:441–446
Xia L, Hu B, Jiang Z, Wu Y, Liang Y (2004) (2004) Single-drop microextraction combined with low-temperature electrothermal vaporization ICPMS for the determination of trace Be, Co, Pd, and Cd in biological samples. Anal Chem 76:2910–2915
Chen X, Zhang T, Liang P, Li Y (2006) Application of continuous-flow liquid phase microextraction to the analysis of phenolic compounds in wastewater samples. Microchim Acta 155:415–420
Lee J, Lee HK, Rasmussen KE, Pedersen-Bjergaard S (2008) Environmental and bioanalytical applications of hollow fiber membrane liquid-phase microextraction: A review. Anala Chim Acta 624:253–268
Bosch Ojeda C, Sánchez Rojas F (2009) Separation and preconcentration by dispersive liquid-liquid microextraction procedure: a review. Chromatographia 69:1149–1159
Anthemidis AN, Ioannou KIG (2009) Recent developments in homogeneous and dispersive liquid-liquid extraction for inorganic elements determination. A review Talanta 80:413–421
Zarei AR, Gholamian F (2011) Development of a dispersive liquid-liquid microextraction method for spectrophotometric determination of barbituric acid in pharmaceutical formulation and biological samples. Anal Biochem 412:224–228
Bidari A, Ganjali MR, Norouzi P, Hosseini MRM, Assadi Y (2011) Sample preparation method for the analysis of some organophosphorus pesticides residues in tomato by ultrasound-assisted solvent extraction followed by dispersive liquid-liquid microextraction. Food Chem 126:1840–1844
Wu CX, Wu QH, Wang C, Wang Z (2011) A novel method for the determination of trace copper in cereals by dispersive liquid-liquid microextraction based on solidification of floating organic drop coupled with flame atomic absorption, spectrometry. Chin Chem Lett 22:473–476
Lu Y, Lin Q, Luo G, Dai Y (2006) Directly suspended droplet microextraction. Anal Chim Acta 566:259–264
Khalili Zanjani MR, Yamini Y, Shariati S, Jönsson JA (2007) A new liquid-phase microextraction method based on solidification of floating organic drop. Anal Chim Acta 585:286–293
Xiao Q, Hu B, He M (2008) Speciation of butyltin compounds in environmental and biological samples using headspace single drop microextraction coupled with gas chromatography-inductively coupled plasma mass spectrometry. J Chromatogr A 1211:135–141
Sharma N, Jain A, Singh VK, Verma KK (2011) Solid-phase extraction combined with headspace single-drop microextraction of chlorophenols as their methyl ethers and analysis by high-performance liquid chromatography-diode array detection. Talanta 83:994–999
Saraji M, Bidgoli AAH (2009) Single-drop microextraction with in-microvial derivatization for the determination of haloacetic acids in water sample by gas chromatography-mass spectrometry. J Chromatogr A 1216:1059–1066
Lambropoulou DA, Psillakis E, Albanis TA, Kalogerakis N (2004) Single-drop microextraction for the analysis of organophosphorous insecticides in water. Anal Chim Acta 516:205–211
He Y, Kang YJ (2006) Single drop liquid-liquid-liquid microextraction of methamphetamine and amphetamine in urine. J Chromatogr A 1133:35–40
Garbi A, Sakkas V, Fiamegos YC, Stalikas CD, Albanis T (2010) Sensitive determination of pesticides residues in wine samples with the aid of single-drop microextraction and response surface methodology. Talanta 82:1286–1291
Park YK, Choi K, Ahmed AYBH, AlOthman ZA, Chung DS (2010) Selective preconcentration of amino acids and peptides using single drop microextraction in-line coupled with capillary electrophoresis. J Chromatogr A 1217:3357–3361
Fang C, Xiong Y, Liang Q, Li Y, Peng P (2011) Optimization of headspace single-drop microextraction technique for extraction of light hydrocarbons (C6-C12) and its potential applications. Org Geochem. Article in Press
Gil S, de Loos-Vollebregt MTC, Bendicho C (2009) Optimization of a single-drop microextraction method for multielemental determination by electrothermal vaporization inductively coupled plasma mass spectrometry following in situ vapor generation. Spectroc Acta Pt B-Atom Spectr 64:208–214
Wang Q, Qiu H, Li J, Liu X, Jiang S (2010) On-line coupling of ionic liquid-based single-drop microextraction with capillary electrophoresis for sensitive detection of phenols. J Chromatogr A 1217:5434–5439
Zhao E, Han L, Jiang S, Wang Q, Zhou Z (2006) Application of a single-drop microextraction for the analysis of organophosphorus pesticides in juice. J Chromatogr A 1114:269–273
Gao W, Chen G, Chen Y, Zhang X, Yin Y, Hu Z (2011) Application of single drop liquid-liquid-liquid microextraction for the determination of fluoroquinolones in human urine by capillary electrophoresis. J Chromatogr B 879:291–295
Saraji M, Farajmand B (2008) Application of single-drop microextraction combined with in-microvial derivatization for determination of acidic herbicides in water samples by gas chromatography-mass spectrometry. J Chromatogr A 1178:17–23
Amvrazi EG, Tsiropoulos NG (2009) Application of single-drop microextraction coupled with gas chromatography for the determination of multiclass pesticides in vegetables with nitrogen phosphorus and electron capture detection. J Chromatogr A 1216:2789–2797
Ebrahimzadeh H, Yamini Y, Gholizade A, Sedighi A, Kasraee S (2008) Determination of fentanyl in biological and water samples using single-drop liquid–liquid–liquid microextraction coupled with high-performance liquid chromatography. Anal Chim Acta 626:193–199
Zhao EC, Shan WL, Jiang SR, Liu Y, Zhou ZQ (2006) Determination of the chloroacetanilide herbicides in waters using single-drop microextraction and gas chromatography. Microchem J 83:105–110
Li L, Hu B, Xia L, Jiang Z (2006) Determination of trace Cd and Pb in environmental and biological samples by ETV-ICP-MS after single-drop microextraction. Talanta 70:468–473
Deng C, Yao N, Wang B, Zhang X (2006) Development of microwave-assisted extraction followed by headspace single-drop microextraction for fast determination of paeonol in traditional Chinese medicines. J Chromatogr A 1103:15–21
Wu Y, Xia L, Chen R, Hu B (2008) Headspace single drop microextraction combined with HPLC for the determination of trace polycyclic aromatic hydrocarbons in environmental samples. Talanta 74:470–477
Bagheri H, Naderi M (2009) Immersed single-drop microextraction–electrothermal vaporization atomic absorption spectroscopy for the trace determination of mercury in water samples. J Hazard Mater 165:353–358
Viñas P, Martínez-Castillo N, Campillo N, Hernández-Córdoba M (2010) Liquid-liquid microextraction methods based on ultrasound-assisted emulsification and single-drop coupled to gas chromatography-mass spectrometry for determining strobilurin and oxazole fungicides in juices and fruits. J Chromatogr A 1217:6569–6577
Lin MY, Whang CW (2007) Microwave-assisted derivatization and single-drop microextraction for gas chromatographic determination of chromium(III) in water. J Chromatogr A 1160:336–339
Yu Y, Chen B, Shen C, Cai Y, Xie M, Zhou W, Chen Y, Li Y, Duan G (2010) Multiple headspace single-drop microextraction coupled with gas chromatography for direct determination of residual solvents in solid drug product. J Chromatogr A 1217:5158–5164
Fragueiro S, Lavilla I, Bendicho C (2004) Headspace sequestration of arsine onto a Pd(II)-containing aqueous drop as a preconcentration method for electrothermal atomic absorption spectrometry. Spectroc Acta Pt B-Atom Spectr 59:851–855
Wu W, Zhou Z (2008) Application of liquid-phase microextraction and gas chromatography to the determination of chlorfenapyr in water samples. Microchim Acta 162:161–165
Varanusupakul P, Vora-adisak N, Pulpoka B (2007) In situ derivatization and hollow fiber membrane microextraction for gas chromatographic determination of haloacetic acids in water. Anal Chim Acta 598:82–86
Ratola N, Alves A, Kalogerakis N, Psillakis E (2008) Hollow-fibre liquid-phase microextraction A simple and fast cleanup step used for PAHs determination in pine needles. Anal Chim Acta 618:70–78
Ito R, Kawaguchi M, Honda H, Koganei Y, Okanouchi N, Sakui N, Saito K, Nakazawa H (2008) Hollow-fiber-supported liquid phase microextraction with in situ derivatization and gas chromatography-mass spectrometry for determination of chlorophenols in human urine samples. J Chromatogr B 872:63–67
Yang Y, Chen J, Shi YP (2010) Determination of aconitine, hypaconitine and mesaconitine in urine using hollow fiber liquid-phase microextraction combined with high-performance liquid chromatography. J Chromatogr B 878:2811–2816
Kawaguchi M, Takatsu A (2009) Miniaturized hollow fiber assisted liquid-phase microextraction and gas chromatography-mass spectrometry for the measurement of progesterone in human serum. J Chromatogr B 877:343–346
Jiang H, Hu B, Chen B, Xia L (2009) Hollow fiber liquid phase microextraction combined with electrothermal atomic absorption spectrometry for the speciation of arsenic (III) and arsenic (V) in fresh waters and human hair extracts. Anal Chim Acta 634:15–21
Saleh A, Yamini Y, Faraji M, Shariati S, Rezaee M (2009) Hollow fiber liquid phase microextraction followed by high performance liquid chromatography for determination of ultra-trace levels of Se(IV) after derivatization in urine, plasma and natural water samples. J Chromatogr B 877:1758–1764
Chen HC, Chen WT, Ding WH (2009) Determination of perchlorate in river by ion-pair hollow-fiber liquid-phase microextraction coupled with electrospray ionization tandem mass spectrometry. Talanta 79:442–445
Li Y, Xiong Y, Fang J, Wang L, Liang Q (2009) Application of hollow fiber liquid-phase microextraction in identification of oil spill sources. J Chromatogr A 1216:6155–6161
Xiao Q, Hu B (2010) Hollow fiber-liquid phase microextraction combined with gas chromatography for the determination of phenothiazine drugs in urine. J Chromatogr B 878:1599–1604
Romero-González R, Frenich AG, Vidal JLM, Aguilera-Luiz MM (2010) Determination of ochratoxin A and T-2 toxin in alcoholic beverages by hollow fiber liquid phase microextraction and ultra high-pressure liquid chromatography coupled to tandem mass spectrometry. Talanta 82:171–176
Emídio ES, de Menezes PV, de Santana FJM, Dórea HS (2010) Hollow fiber-based liquid phase microextraction with factorial design optimization and gas chromatography-tandem mass spectrometry for determination of cannabinoids in human hair. J Chromatogr B 878:2175–2183
Xiong J, Chen J, He M, Hu B (2010) Simultaneous quantification of amphetamines, caffeine and ketamine in urine by hollow fiber liquid phase microextraction combined with gas chromatography-flame ionization detector. Talanta 82:969–975
Ghasemi E, Najafi NM, Raofie F, Ghassempour A (2010) Simultaneous speciation and preconcentration of ultra traces of inorganic tellurium and selenium in environmental samples by hollow fiber liquid phase microextraction prior to electrothermal atomic absorption spectroscopy determination. J Hazard Mater 181:491–496
Yang Q, Guo Y, Wang L, Liang S, Liu X (2010) Simultaneous determination of trace benzene and toluene in beverage by ultrasound-enhanced hollow-fiber liquid-phase microextraction coupled with GC. Chromatographia 72:1157–1161
Siang GH, Makahleh A, Saad B, Lim BP (2010) Hollow fiber liquid-phase microextraction coupled with gas chromatography-flame ionization detection for the profiling of fatty acids in vegetable oils. J Chromatogr A 1217:8073–8078
Shrivas K, Patel DK (2011) Ultrasound assisted-hollow fibre liquid-phase microextraction for the determination of selenium in vegetable and fruit samples by using GF-AAS. Food Chem 124:1673–1677
Simões RA, De Oliveira ARM, Bonato PS (2011) Hollow fiber-based liquid-phase microextraction (HF-LPME) of isradipine and its main metabolite followed by chiral HPLC analysis: Application to an in vitro biotransformation study. Anal Bioanal Chem 399:2435–2443
Lin Z, Zhang J, Cui H, Zhang L, Chen G (2010) Determination of aromatic amines in environmental water sample by hollow fiber-liquid phase microextraction and microemulsion electrokinetic chromatography. J Chromatogr A 1217:4507–4510
Wang C, Li C, Zang X, Han D, Liu Z, Wang Z (2007) Hollow fiber-based liquid-phase microextraction combined with on-line sweeping for trace analysis of Strychnos alkaloids in urine by micellar electrokinetic chromatography. J Chromatogr A 1143:270–275
Peng J, Lü J, Hu X, Liu J, Jiang G (2007) (2007) Determination of atrazine, desethyl atrazine and desisopropyl atrazine in environmental water samples using hollow fiber-protected liquid-phase microextraction and high performance liquid chromatography. Microchim Acta 158:181–186
Xia L, Hu B, Wu Y (2007) Hollow fiber-based liquid-liquid-liquid microextraction combined with high-performance liquid chromatography for the speciation of organomercury. J Chromatogr A 1173:44–51
Esrafili A, Yamini Y, Shariati S (2007) Hollow fiber-based liquid phase microextraction combined with high-performance liquid chromatography for extraction and determination of some antidepressant drugs in biological fluids. Anal Chim Acta 604:127–133
Sobhi HR, Yamini Y, Abadi RHHB (2007) Extraction and determination of trace amounts of chlorpromazine in biological fluids using hollow fiber liquid phase microextraction followed by high-performance liquid chromatography. J Pharm Biomed Anal 45:769–774
Liu M, Qiu B, Jin X, Zhang L, Chen X, Chen G (2008) Determination of estrogens in wastewater using three-phase hollow fiber-mediated liquid-phase microextraction followed by HPLC. J Sep Sci 31:622–628
Jiang H, Hu B, Chen B, Zu W (2008) (2008) Hollow fiber liquid phase microextraction combined with graphite furnace atomic absorption spectrometry for the determination of methylmercury in human hair and sludge samples. Spectroc Acta Pt B-Atom Spectr 63:770–776
Shah FU, Barri T, Jönsson JA, Skog K (2008) Determination of heterocyclic aromatic amines in human urine by using hollow-fibre supported liquid membrane extraction and liquid chromatography-ultraviolet detection system. J Chromatogr B 870:203–208
Shariati S, Yamini Y, Esrafili A (2009) Carrier mediated hollow fiber liquid phase microextraction combined with HPLC-UV for preconcentration and determination of some tetracycline antibiotics. J Chromatogr B 877:393–400
Tahmasebi E, Yamini Y, Saleh A (2009) Extraction of trace amounts of pioglitazone as an anti-diabetic drug with hollow fiber liquid phase microextraction and determination by high-performance liquid in biological fluids. J Chromatogr B 877:1923–1929
Payán MR, López MAB, Fernández-Torres R, Bernal JLP, Mochón MC (2009) HPLC determination of ibuprofen, diclofenac and salicylic acid using hollow fiber-based liquid phase microextraction (HF-LPME). Anal Chim Acta 653:184–190
Wu Y, Hu B (2009) Simultaneous determination of several phytohormones in natural coconut juice by hollow fiber-based liquid-liquid-liquid microextraction-high performance liquid chromatography. J Chromatogr A 1216:7657–7663
Barahona F, Gjelstad A, Pedersen-Bjergaard S, Rasmussen KE (2010) Hollow fiber-liquid-phase microextraction of fungicides from orange juices. J Chromatogr A 1217:1989–1994
Ebrahimzadeh H, Yamini Y, Firozjaei HA, Kamarei F, Tavassoli N, Rouini MR (2010) Hollow fiber-based liquid phase microextraction combined with high-performance liquid chromatography for the analysis of gabapentin in biological samples. Anal Chim Acta 665:221–226
Al Azzam KM, Makahleah A, Saad B, Mansor SM (2010) Hollow fiber liquid-phase microextraction for the determination of trace amounts of rosiglitazone (anti-diabetic drug) in biological fluids using capillary electrophoresis and high performance liquid chromatographic methods. J Chromatogr A 1217:3654–3659
Saraji M, Jafari MT, Sherafatmand H (2010) Hollow fiber-based liquid-liquid-liquid microextraction combined with electrospray ionization-ion mobility spectrometry for the determination of pentazocine in biological samples. J Chromatogr A 1217:5173–5178
Wang J, Hu S, Bai X (2010) Determination of trace amounts of chlorogenic acid and three of its metabolites using time-resolved LPME and LC-UV detection in biological specimens. Chromatographia 72:453–458
Calixto LA, Bonato PS (2010) Simultaneous determination of rosiglitazone and its metabolites in rat liver microsomal fraction using hollow-fiber liquid-phase microextraction for sample preparation. J Sep Sci 33:2872–2880
Sagristà E, Larsson E, Ezoddin M, Hidalgo M, Salvadó V, Jönsson JÅ (2010) Determination of non-steroidal anti-inflammatory drugs in sewage sludge by direct hollow fiber supported liquid membrane extraction and liquid chromatography-mass spectrometry. J Chromatogr A 1217:6153–6158
Yang Y, Chen J, Shi YP (2010) Determination of aconitine, hypaconitine and mesaconitine in urine using hollow fiber liquid-phase microextraction combined with high-performance liquid chromatography. J Chromatogr B 878:2811–2816
Saraji M, Mousavi F (2010) Use of hollow fibre-based liquid-liquid-liquid microextraction and high-performance liquid chromatography-diode array detection for the determination of phenolic acids in fruit juices. Food Chem 123:1310–1317
Yang Y, Chen J, Shi YP (2010) Determination of aristolochic acid in urine using hollow fiber liquid-phase microextraction combined with high-performance liquid chromatography. Biomed Chromatogr 24:1350–1355
Yudthavorasit S, Chiaochan C, Leepipatpiboon N (2011) Simultaneous determination of multi-class antibiotic residues in water using carrier-mediated hollow-fiber liquid-phase microextraction coupled with ultra-high performance liquid chromatography. Microchim Acta 172:39–49
Jafari MT, Saraji M, Sherafatmand H (2011) (2011) Electrospray ionization-ion mobility spectrometry as a detection system for three-phase hollow fiber microextraction technique and simultaneous determination of trimipramine and desipramine in urine and plasma samples. Anal Bioanal Chem 399:3555–3564
Zhang C, Ye L, Xu L (2011) Orthogonal array design for the optimization of hollow fiber protected liquid-phase microextraction of salicylates from environmental waters. Anal Chim Acta 689:219–225
Ramos Payán M, Bello López MÁ, Fernández-Torres R, González JAO, Callejón Mochón M (2011) Hollow fiber-based liquid phase microextraction (HF-LPME) as a new approach for the HPLC determination of fluoroquinolones in biological and environmental matrices. J Pharm Biomed Anal 55:332–341
Payán MR, López MAB, Fernández-Torres R, Mochón MC, Ariza JLG (2010) Application of hollow fiber-based liquid-phase microextraction (HF-LPME) for the determination of acidic pharmaceuticals in wastewaters. Talanta 82:854–858
Ramos Payán M, López MTB, Fernández-Torres R, Navarro MV, Mochón MC (2011) Hollow fiber-based liquid phase microextraction (HF-LPME) for a highly sensitive HPLC determination of sulfonamides and their main metabolites. J Chromatogr B 879:197–204
Hu X, Huang Y, Tao D, Yin D, Liu J (2010) Hollow fiber membrane supported thin liquid film extraction for determination of trace phenoxy acid herbicides and phenols in environmental water samples. Microchim Acta 168:23–29
Zhou Q, Zhao N, Xie G (2011) Determination of lead in environmental waters with dispersive liquid–liquid microextraction prior to atomic fluorescence spectrometry. J Hazard Mater 189:48–53
Yan H, Wang H, Qiao J, Yang G (2011) Molecularly imprinted matrix solid-phase dispersion combined with dispersive liquid–liquid microextraction for the determination of four Sudan dyes in egg yolk. J Chromatogr A 1218:2182–2188
Campillo N, Viñas P, Martínez-Castillo N, Hernández-Córdoba M (2011) Determination of volatile nitrosamines in meat products by microwave-assisted extraction and dispersive liquid–liquid microextraction coupled to gas chromatography–mass spectrometry. J Chromatogr A 1218:1815–1821
Wang WX, Yang TJ, Li ZG, Jong TT, Lee MR (2011) A novel method of ultrasound-assisted dispersive liquid–liquid microextraction coupled to liquid chromatography–mass spectrometry for the determination of trace organoarsenic compounds in edible oil. Anal Chim Acta 690:221–227
Pizarro C, Sáenz-González C, Pérez-del-Notario N, González-Sáiz JM (2011) Development of a dispersive liquid–liquid microextraction method for the simultaneous determination of the main compounds causing cork taint and Brett character in wines mass spectrometry. J Chromatogr A 1218:1576–1584
De La Calle I, Pena-Pereira F, Cabaleiro N, Lavilla I, Bendicho C (2011) Ion pair-based dispersive liquid–liquid microextraction for gold determination at ppb level in absorption spectrometry. Talanta 84:109–115
Wen X, Yang Q, Yan Z, Deng Q (2011) Determination of cadmium and copper in water and food samples by dispersive liquid–liquid microextraction combined with UV–vis spectrophotometry. Microchem J 97:249–254
Afzali D, Mohadesi AR, Jahromi BB, Falahnejad M (2011) Separation of trace amount of silver using dispersive liquid-liquid based on solidification of floating organic drop microextraction. Anal Chim Acta 684:45–49
Zgoła-Grześkowiak A, Kaczorek E (2011) Isolation, preconcentration and determination of rhamnolipids in aqueous samples by dispersive liquid–liquid microextraction and liquid chromatography with tandem mass spectrometry. Talanta 83:744–750
Cortada C, Vidal L, Canals A (2011) Determination of geosmin and 2-methylisoborneol in water and wine samples by ultrasound-assisted dispersive liquid–liquid microextraction coupled to gas chromatography-mass spectrometry. J Chromatogr A 1218:17–22
Yan H, Wang H, Qin X, Liu B, Du J (2011) Ultrasound-assisted dispersive liquid–liquid microextraction for determination of fluoroquinolones in pharmaceutical wastewater. J Pharm Biomed Anal 54:53–57
Liu B, Yan H, Qiao F, Geng Y (2011) Determination of clenbuterol in porcine tissues using solid-phase extraction combined with ultrasound-assisted dispersive liquid-liquid microextraction and HPLC-UV detection. J Chromatogr B 879:90–94
Jia X, Han Y, Liu X, Duan T, Chen H (2011) Speciation of mercury in water samples by dispersive liquid–liquid microextraction combined with high performance liquid chromatography-inductively coupled plasma mass spectrometry. Spectroc Acta Pt B-Atom Spectr 66:88–92
Jofré VP, Assof MV, Fanzone ML, Goicoechea HC, Martínez LD, Silva MF (2010) Optimization of ultrasound assisted-emulsification-dispersive liquid–liquid microextraction by experimental design methodologies for the determination of sulfur compounds in wines by gas chromatography-mass spectrometry. Anal Chim Acta 683:126–135
Pizarro C, Sáenz-González C, Perez-del-Notario N, González-Sáiz JM (2010) Optimisation of a dispersive liquid–liquid microextraction method for the simultaneous determination of halophenols and haloanisoles in wines. J Chromatogr A 1217:7630–7637
Xiong C, Ruan J, Cai Y, Tang Y (2009) Extraction and determination of some psychotropic drugs in urine samples using dispersive liquid–liquid microextraction followed by high-performance liquid chromatography. J Pharm Biomed Anal 49:572–578
Moradi M, Yamini Y, Esrafili A, Seidi S (2010) Application of surfactant assisted dispersive liquid–liquid microextraction for sample preparation of chlorophenols in water samples. Talanta 82:1864–1869
Leong MI, Chang CC, Fuh MR, Huang SD (2010) Low toxic dispersive liquid–liquid microextraction using halosolvents for extraction of polycyclic aromatic hydrocarbons in water samples. J Chromatogr A 1217:5455–5461
Yan H, Liu B, Du J, Yang G, Row KH (2010) Ultrasound-assisted dispersive liquid–liquid microextraction for the determination of six pyrethroids in river water. J Chromatogr A 1217:5152–5157
Dai L, Cheng J, Matsadiq G, Liu L, Li JK (2010) Dispersive liquid–liquid microextraction based on the solidification of floating organic droplet for the determination of polychlorinated biphenyls in aqueous samples. Anal Chim Acta 674:201–205
Biparva P, Ranjbari E, Hadjmohammadi MR (2010) Application of dispersive liquid–liquid microextraction and spectrophotometric detection to the rapid determination of rhodamine 6 G in industrial effluents. Anal Chim Acta 674:206–210
Du J, Yan H, She D, Liu B, Yang G (2010) Simultaneous determination of cypermethrin and permethrin in pear juice by ultrasound-assisted dispersive liquid-liquid microextraction combined with gas chromatography. Talanta 82:698–703
Fontana AR, Lana NB, Martinez LD, Altamirano JC (2010) Ultrasound-assisted leaching-dispersive solid-phase extraction followed by liquid-liquid microextraction for the determination of polybrominated diphenyl ethers in sediment samples by gas chromatography-tandem mass spectrometry. Talanta 82:359–366
Najafi NM, Tavakoli H, Alizadeh R, Seidi S (2010) Speciation and determination of ultra trace amounts of inorganic tellurium in environmental water samples by dispersive liquid–liquid microextraction and electrothermal atomic absorption spectrometry. Anal Chim Acta 670:18–23
Rezaee M, Yamini Y, Khanchi A, Faraji M, Saleh A (2010) A simple and rapid new dispersive liquid–liquid microextraction based on solidification of floating organic drop combined with and determination of aluminium in water samples. J Hazardous Materials 178(1–3):766–770
Bernardo M, Gonçalves M, Lapa N, Mendes B (2010) Determination of alkylphenols in eluates from pyrolysis solid residues using dispersive liquid–liquid microextraction. Chemosphere 79(11):1026–1032
Lv LL, Xu H, Song D, Cui Y, Hu S, Zhang G (2010) Analysis of volatile aldehyde biomarkers in human blood by derivatization and dispersive liquid-liquid microextraction based on solidification of floating organic droplet method by high performance liquid chromatography. J Chromatogr A 1217:2365–2370
Chen H, Chen H, Ying J, Huang J, Liao L (2009) Dispersive liquid-liquid microextraction followed by high-performance liquid chromatography as an efficient and sensitive technique for simultaneous determination of chloramphenicol and thiamphenicol in honey. Anal Chim Acta 632:80–85
Tsai WH, Chuang HY, Chen HH, Huang JJ, Chen HC, Cheng SH, Huang TC (2009) Application of dispersive liquid-liquid microextraction and dispersive micro-solid-phase extraction for the determination of quinolones in swine muscle by high-performance liquid chromatography with diode-array detection. Anal Chim Acta 656:56–62
Sarafraz-Yazdi A, Raouf-Yazdinejad S, Es’haghi Z (2007) Directly suspended droplet microextraction and analysis of amitriptyline and nortriptyline by GC. Chromatographia 66:613–617
Sarafraz-Yazdi A, Mofazzeli F, Es’haghi Z (2008) Directly suspended droplet three liquid phase microextraction of diclofenac prior to LC. Chromatographia 67:49–53
Sarafraz-Yazdi A, Amiri AH, Es’haghi Z (2009) Separation and determination of benzene, toluene, ethylbenzene and o-xylene compounds in water using directly suspended droplet microextraction coupled with gas chromatography-flame ionization detector. Talanta 78:936–941
Sarafraz-Yazdi A, Mofazzeli F, Es’haghi Z (2010) Determination of chlorophenols in environmental water samples using directly suspended droplet liquid-liquid-liquid phase microextraction prior to high-performance liquid chromatography. Int J Environ Anal Chem 90:1108–1118
Farahani H, Norouzi P, Beheshti A, Sobhi HR, Dinarvand R, Ganjali MR (2009) Quantitation of atorvastatin in human plasma using directly suspended acceptor droplet in liquid-liquid-liquid microextraction and high-performance liquid chromatography-ultraviolet detection. Talanta 80:1001–1006
Viñas P, Martínez-Castillo N, Campillo N, Hernández-Córdoba M (2011) Directly suspended droplet microextraction with in injection-port derivatization coupled to gas chromatography-mass spectrometry for the analysis of polyphenols in herbal infusions, fruits and functional foods. J Chromatogr A 1218:639–646
Gao W, Chen G, Chen T, Zhang X, Chen Y, Hu Z (2011) Directly suspended droplet microextraction combined with single drop back-extraction as a new approach for sample preparation compatible with capillary electrophoresis. Talanta 83:1673–1679
Khalili Zanjani MR, Yamini Y, Shariati S, Jönsson JA (2007) A new liquid-phase microextraction method based on solidification of floating organic drop. Anal Chim Acta 585:286–293
Sobhi HR, Yamini Y, Esrafili A, Abadi RHHB (2008) (2008) Suitable conditions for liquid-phase microextraction using solidification of a floating drop for extraction of fat-soluble vitamins established using an orthogonal array experimental design. J Chromatogr A 1196–1197:28–32
Dadfarnia S, Salmanzadeh AM, Shabani AMH (2008) A novel separation/preconcentration system based on solidification of floating organic drop microextraction for determination of lead by graphite furnace atomic absorption spectrometry. Anal Chim Acta 623:163–167
Dadfarnia S, Haji Shabani AM, Kamranzadeh E (2009) Separation/preconcentration and determination of cadmium ions by solidification of floating organic drop microextraction and FI-AAS. Talanta 79:1061–1065
Yamini Y, Rezaee M, Khanchi A, Faraji M, Saleh A (2010) Dispersive liquid-liquid microextraction based on the solidification of floating organic drop followed by inductively coupled plasma-optical emission spectrometry as a fast technique for the simultaneous determination of heavy metals. J Chromatogr A 1217:2358–2364
López-García I, Rivas RE, Hernández-Córdoba M (2010) Liquid-phase microextraction with solidification of the organic floating drop for the preconcentration and determination of mercury traces by electrothermal atomic absorption spectrometry. Anal Bioanal Chem 396:3097–3102
Ghambarian M, Khalili-Zanjani MR, Yamini Y, Esrafili A, Yazdanfar N (2010) Preconcentration and speciation of arsenic in water specimens by the combination of solidification of floating drop microextraction and electrothermal atomic absorption spectrometry. Talanta 81:197–201
Rezaee M, Yamini Y, Khanchi A, Faraji M, Saleh A (2010) A simple and rapid new dispersive liquid-liquid microextraction based on solidification of floating organic drop combined with inductively coupled plasma-optical emission spectrometry for preconcentration and determination of aluminium in water samples. J Hazard Mater 178:766–770
Asadollahi T, Dadfarnia S, Shabani AMH (2010) Separation/preconcentration and determination of vanadium with dispersive liquid-liquid microextraction based on solidification of floating organic drop (DLLME-SFO) and electrothermal atomic absorption spectrometry. Talanta 82:208–212
Afzali D, Mohadesi AR, Jahromi BB, Falahnejad M (2011) Separation of trace amount of silver using dispersive liquid-liquid based on solidification of floating organic drop microextraction. Anal Chim Acta 684:45–49
Mirzaei M, Behzadi M, Abadi NM, Beizaei A (2011) Simultaneous separation/preconcentration of ultra trace heavy metals in industrial wastewaters by dispersive liquid-liquid microextraction based on solidification of floating organic drop prior to determination by graphite furnace atomic absorption spectrometry. J Hazard Mater 186:1739–1743
Gao Y, Zeng Y, Zheng L, Li L (2009) Determination of triazine herbicides in aqueous samples using solidification of a floating drop for liquid-phase microextraction with liquid chromatography. Anal Lett 42:1620–1631
Leong MI, Huang SD (2009) Dispersive liquid-liquid microextraction method based on solidification of floating organic drop for extraction of organochlorine pesticides in water samples. J Chromatogr A 1216:7645–7650
Cheng J, Xiao J, Zhou Y, Xia Y, Guo F, Li J (2011) Dispersive liquid-liquid microextraction based on solidification of floating organic droplet method for the determination of diethofencarb and pyrimethanil in aqueous samples. Microchim Acta 172:51–55
Huddleston JG, Visser AE, Reichert WM, Willauer HD, Broker GA, Rogers RD (2001) Characterization and comparison of hydrophilic and hydrophobic room temperature ionic liquids incorporating the imidazolium cation. Green Chem 3:156–164
Carda-Broch S, Berthod A, Armstrong DW (2003) Solvent properties of the 1-butyl-3-methylimidazolium hexafluorophosphate ionic liquid. Anal Bioanal Chem 375:191–199
Manzoori JL, Amjadi M, Abulhassani J (2009) Ultra-trace determination of lead in water and food samples by using ionic liquid-based single drop microextraction-electrothermal atomic absorption spectrometry. Anal Chim Acta 644:48–52
Chisvert A, Román IP, Vidal L, Canals A (2009) Simple and commercial readily-available approach for the direct use of ionic liquid-based single-drop microextraction prior to gas chromatography Determination of chlorobenzenes. J Chromatogr A 1216:1290–1295
Aguilera-Herrador E, Lucena R, Cárdenas S, Valcárcel M (2008) Determination of trihalomethanes in waters by ionic liquid-based single drop microextraction/gas chromatographic/mass spectrometry. J Chromatogr A 1209:76–82
Manzoori JL, Amjadi M, Abulhassani J (2009) Ionic liquid-based single drop microextraction combined with electrothermal atomic absorption spectrometry for the determination of manganese in water samples. Talanta 77:1539–1544
Vidal L, Chisvert A, Canals A, Salvador A (2010) Ionic liquid-based single-drop microextraction followed by liquid chromatography-ultraviolet spectrophotometry detection to determine typical UV filters in surface water samples. Talanta 81:549–555
Peng JF, Liu JF, Hu XL, Jiang GB (2007) Direct determination of chlorophenols in environmental water samples by hollow fiber supported ionic liquid membrane extraction coupled with high-performance liquid chromatography. J Chromatogr A 1139:165–170
Abulhassani J, Manzoori JL, Amjadi M (2010) Hollow fiber based-liquid phase microextraction using ionic liquid solvent for preconcentration of lead and nickel from environmental and biological samples prior to determination by electrothermal atomic absorption spectrometry. J Hazard Mater 176:481–486
Tao Y, Liu JF, Hu XL, Li HC, Wang T, Jiang GB (2009) Hollow fiber supported ionic liquid membrane microextraction for determination of sulfonamides in environmental water samples by high-performance liquid chromatography. J Chromatogr A 1216:6259–6266
Yao C, Li T, Twu P, Pitner WR, Anderson JL (2011) Selective extraction of emerging contaminants from water samples by dispersive liquid–liquid microextraction using functionalized ionic liquids. J Chromatogr A 1218:1556–1566
Gharehbaghi M, Shemirani F (2011) Ionic liquid-based dispersive liquid-liquid microextraction and enhanced spectrophotometric determination of molybdenum (VI) in water and plant leaves samples by FO-LADS. Food Chem Toxicol 49:423–428
Molaakbari E, Mostafavi A, Afzali D (2011) Ionic liquid ultrasound assisted dispersive liquid–liquid microextraction method for preconcentration of trace amounts of rhodium prior to flame atomic absorption spectrometry determination. J Hazard Mater 185:647–652
Zhou Q, Bai H, Xie G, Xiao G (2008) Temperature-controlled ionic liquid dispersive liquid phase micro-extraction. J Chromatogr A 1177:43–49
Kamarei F, Ebrahimzadeh H, Yamini Y (2010) Optimization of temperature-controlled ionic liquid dispersive liquid phase microextraction combined with high performance liquid chromatography for analysis of chlorobenzenes in water samples. Talanta 83:36–41
Zhang HF, Shi YP (2010) Temperature-assisted ionic liquid dispersive liquid-liquid microextraction combined with high performance liquid chromatography for the determination of anthraquinones in Radix et Rhizoma Rhei samples. Talanta 82:1010–1016
He L, Luo X, Jiang X, Qu L (2010) A new 1,3-dibutylimidazolium hexafluorophosphate ionic liquid-based dispersive liquid-liquid microextraction to determine organophosphorus pesticides in water and fruit samples by high-performance liquid chromatography. J Chromatogr A 1217:5013–5020
Wang Y, You J, Ren R, Xiao Y, Gao S, Zhang H, Yu A (2010) Determination of triazines in honey by dispersive liquid–liquid microextraction high-performance liquid chromatography. J Chromatogr A 1217:4241–4246
Yousefi SR, Shemirani F (2010) Development of a robust ionic liquid-based dispersive liquid-liquid microextraction against high concentration of salt for preconcentration of trace metals in saline aqueous samples: application to the determination of Pb and Cd. Anal Chim Acta 669:25–31
Berton P, Martinis EM, Wuilloud RG (2010) Development of an on-line temperature-assisted ionic liquid dispersive microextraction system for sensitive determination of vanadium in environmental and biological samples. J Hazard Mater 176:721–728
Chen H, Du P, Chen J, Hu S, Li S, Liu H (2010) Separation and preconcentration system based on ultrasonic probe-assisted ionic liquid dispersive liquid-liquid microextraction for determination trace amount of chromium(VI) by electrothermal atomic absorption spectrometry. Talanta 81:176–179
Acknowledgements
This research was supported by Basic Science Research Program through the National Research Foundation (NRF) of Korea funded by the Ministry of Education, Science and Technology (2011–0010673)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Han, D., Row, K.H. Trends in liquid-phase microextraction, and its application to environmental and biological samples. Microchim Acta 176, 1–22 (2012). https://doi.org/10.1007/s00604-011-0678-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00604-011-0678-0