
Abstract
Transcription factors (TF) control gene expression by binding to specific regions of DNA sequence. TF play an important role in various disease processes, and their identification helps in understanding underlying gene regulation leading to disease risk. Currently, the most powerful models used for the predicting binding sites between TF and DNA sequence from ChIP-Seq dataset are lagging in terms of good feature extraction capabilities. We propose two models named PCLAtt and TranAtt for the prediction of 690 TF-cell line pairs from DNA sequence data. PCLAtt consists of two sets of convolutional neural networks—bidirectional long short-term memory (CNN-BiLSTM) layers in parallel followed by a multi-head attention layer and weight-shared dense layer which all contribute towards extracting efficient features from DNA sequence. TranAtt consists of convolution layers of a pre-trained model along with a BiLSTM layer and attention layer. The convolutional layers of the model act as a motif scanner and the BiLSTM layer learns the regulatory grammar of the motifs. Further, the attention mechanism is applied to give more importance to those sequence regions of DNA that consist of transcription factor binding motifs thus resulting in better performance of the proposed models. PCLAtt outperformed other state-of-the-art methods like DeepSEA, DanQ, TBiNet and DeepATT in prediction of binding sites between TF and the DNA sequence.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Transcription factors control the rate of transcription from DNA to messenger RNA by binding to a particular DNA sequence. In other words, it controls the copying of a particular segment of DNA into messenger RNA using RNA polymerase. Transcription factors can be activators or repressors. The activators increase the rate of transcription from DNA to messenger RNA while repressors decrease or block the rate of gene transcription. Transcription factors turn genes on or off so that the right genes are expressed in the right cells of the body at the right time and in the right amount. Some genes need to be expressed in more than one body part or cell, that means there may be situations where a gene needs to be turned on in the skull, spine and fingertips but not required to be turned on in other parts of the body at that time. Groups of transcription factors coordinate to direct cell division, cell growth throughout life. Improper coordination may lead to mutations that results in a large number of human diseases such as cancer and diabetes. For understanding the gene regulatory mechanism of various diseases, accurate prediction of transcription factor binding sites is required.
Nowadays, deep learning methods have become popular and are being used in various fields. They have shown better performance than traditional machine learning methods. Convolutional neural networks, a variant of deep neural networks are popular for their automatic feature extraction capabilities from raw input sequences. It has been applied in various fields like computational biology, bioinformatics and medical informatics [3, 4, 9, 23, 24] and has shown better performance than traditional machine learning methods. The weight sharing strategy used by the CNNs are useful to capture local patterns from the input data. Using this property, the kernels of the CNN can capture sequence motifs from DNA sequences required for predicting biological functions. The deep neural networks consisting of convolutional layers have also been applied in various models used for the prediction of the relationship between transcription factors with the DNA sequence. Some of the models like DeepSEA [34], DanQ [26], DeepATT [20] have been developed, that use deep learning methods for prediction of 919 non-coding DNA regulatory functions. DeepBind [1], a deep learning method, generates a mutation map that depicts the binding affinity inside a sequence. Statistical methods were combined with deep learning to predict DNA-protein binding using a technique known as expectation pooling [22].
TBiNet [25] is another deep learning model developed for prediction of 690 TF-cell line pairs from ChIP-Seq dataset. DeepSEA, which employs deep convolutional neural networks for predicting the functions of DNA sequences outperformed gkmSVM [12] that used a support vector machine for prediction. After DeepSEA, DanQ was built that extended DeepSEA architecture by adding BiLSTM to the CNN layer. LSTM [16] is a variant of recurrent neural networks that learns long-term dependencies which were developed to solve the vanishing gradient problem of basic recurrent neural network (RNN). Another variant of DNNs is the attention mechanism that has been used in many machine learning tasks such as machine translation tasks [5], computer vision, speech processing and many more. It has also been used in various bioinformatics tasks [2, 17, 18, 29, 31]. DeepATT and TBiNet both are attention-based neural networks. DeepATT uses a category multi-head attention layer (modified version of self-attention mechanism) and a category dense layer (weight-shared mode) along with CNN and BiLSTM layer for prediction of 919 regulatory functions from DNA sequence data. Another attention-based neural network, TBiNet [25] is developed for predicting TF-DNA binding which is trained on the ChIP-Seq dataset that consists of 690 ChIP-Seq experiments from ENCODE [7] that yield better results than DeepSEA and DanQ. PBVPP-Hybrid [8], a more recent state-of-the-art approach, is a combination of CNN and RNN that can extract vital features from large-scale genomic sequences obtained by high throughput technology to predict the occurrence of TFBS and RBP sites. However, still there is a need of a model with better feature extraction capabilities for more accurate TF-DNA binding prediction task.
In this paper, we propose two models, TranAtt and PCLAtt. TranAtt consists of two convolution layers and two max-pooling layers used alternatively which were transferred from a pre-trained model (an implementation of DeepSEA model that was trained for predicting DNA functions consisting of transcription factors, histone modification, and DNase I hypersensitivity sites) and then followed by bidirectional LSTM layer and an attention layer. PCLAtt consists of two sets of CNN-BiLSTM layers, such that one set of CNN-BiLSTM layer is in parallel with the another set of CNN (uses small kernel size) with two BiLSTM layers, which are followed by a multi-head attention layer and weight-shared dense layer. To increase the feature extraction capabilities of the model so as to get better performance than the existing models we used ensemble of two different sets of CNN-BiLSTM layers in parallel. Since both CNN and RNN are good feature extractors, so by combining the features extracted from two different sets of CNN-BiLSTM layers, the feature extraction capabilities of the proposed model are increased. The convolution layer captures the regulatory motifs and the bidirectional LSTM layer learns the regulatory grammar of the sequence motifs obtained from the CNN layer. We used multi-head attention layer and weight-shared dense layer [20] in PCLAtt. The multi-head attention layer selects the important features required for the prediction of transcription factor binding sites and the weight-shared dense layer classifies transcription factors with features selected by the attention layer.
2 Related work
Earlier, traditional machine learning methods were used for the prediction problem from biological sequences (e.g. DNA sequences) that needed manual feature extraction, which was both tedious and ignored important information. To overcome these problems, deep learning methods were used and have shown better performance than traditional machine learning methods. Deep learning methods have been used in many bioinformatics problems. The automatic feature extraction capabilities of deep convolutional neural networks have been successfully applied in the prediction of functions from biological sequences. DeepSEA [34] which is based on CNN architecture was used for predicting DNA functions consisting of transcription factors, histone modification and DNase I hypersensitivity sites. It consists of three convolution layers and two max-pooling layers arranged alternatively followed by a fully connected layer and an output layer. The kernels of the CNN layer are used to capture the sequence motifs. DeepSEA is trained in a multi-task way to predict DNA functions. It showed better performance than gkm-SVM [12] that uses a support vector machine to predict regulatory functions from DNA sequences. The classifiers that were trained on DeepSEA output showed better classification performance than representative variant scoring methods like CADD [27], GWAVA [28] and FunSeq2 [10]. Later, the DanQ model was proposed as an improvement over the DeepSEA model.
DanQ [26] is designed using CNN-RNN architecture. It consists of a CNN layer followed by a BiLSTM layer, a fully connected layer and an output layer. The CNN layer of DanQ uses kernels that act as a motif scanner for given input DNA sequence and the BiLSTM learns the regulatory grammar from the motif information obtained from the CNN layer. DanQ has a hybrid architecture that helps it to simultaneously learn motif features and the regulatory grammar of the sequence motifs. Both DeepSEA and DanQ models have given equal importance to all sequence regions of input DNA when predicting TF-DNA binding and other biological functions. Although, to get a better prediction of TF-DNA binding, the regions of the DNA sequence that contain TF-binding motifs should be given more importance than other sequence regions, that can be attained using an attention mechanism. Later, TBiNet was proposed as an improvement over DeepSEA and DanQ in the prediction of binding sites of TF with the DNA sequence.
TBiNet [25] used an attention mechanism in addition to the CNN layer and BiLSTM layer. It consists of a CNN layer along with an attention layer which is further followed by a BiLSTM layer, a fully connected layer and an output layer. Attention mechanism has shown good performance in machine translation tasks, computer vision and many other applications [11, 32]. In TBiNet, the attention layer gives more importance to those sequence regions of DNA that contain TF-binding motifs by giving different weights known as attention scores to different parts of input sequence thus selecting important features required for predicting transcription factor binding sites. DeepATT [20] is another attention-based neural network for multi-class prediction of 919 regulatory functions from DNA sequence data. It consists of a CNN layer followed by a Bidirectional-LSTM, multi-head attention layer and weight-shared dense layer. The multi-head attention layer designed in DeepATT is modified version of self-attention mechanism. The multi-head attention layer and weight-shared dense layer were used in DeepATT in order to select important features required for predicting different DNA regulatory functions.
Although, state-of-the-art deep learning models have shown noticeable performance, still there is a room for improvement in TF-DNA binding prediction task. In this paper, we built Parallel convolutional-LSTM with Attention (PCLAtt) consisting of two sets of CNN-BiLSTM layers such that one set of CNN-BiLSTM layer is in parallel with a second set of CNN with two BiLSTM layers, followed by a multi-head attention layer and weight-shared dense layer. We also designed another model, Transfer learning with Attention (TranAtt) that proved to be an improvement over TBiNet for more accurate prediction of TF-DNA binding. TranAtt consists of two convolution layers and two max-pooling layers used alternatively which were transferred from a pre-trained model (an implementation of DeepSEA model) used for predicting DNA functions consisting of transcription factors, histone modification and DNase I hypersensitivity sites and then added a bidirectional LSTM layer and an attention layer to it. Transfer learning has been used in different bioinformatics tasks [21, 33] and has yield good performance results. The two convolutional layers which were transferred from a pre-trained model were kept freezed in TranAtt.
3 Materials and methods
In this section the dataset used and details of the design of TranAtt and PCLAtt are described.
3.1 Dataset collection
In our work, we used a dataset obtained from ENCODE (Encyclopedia of DNA elements) [7] which is same as that used by DeepSEA, DanQ, TBiNet and DeepATT, consisting of 4,863,024 total samples that includes training, validation and test samples. Each sample consists of a DNA sequence represented as 1000 X 4 one-hot encoding matrix and the target binary vector of size 690 X 1. The dataset was preprocessed by the authors of DeepSEA and this preprocessed data was further used by DanQ, TBiNet and DeepATT and then in our proposed models. Each column of the one-hot encoded matrix of a DNA sequence consists of DNA bases A, C, G and T and each element of the target binary vector corresponds to one of the 690 TF-cell line pairs (e.g., K562-Pol2). Each sample is labelled based on the middle 200bp of the DNA sequence and the remaining 400bp on each side of the 200bp is added to give additional context, thus making the DNA sequence of 1000bp length. For each sample, the target binary vector consists of 690 tasks such that for each task if more than half of the middle 200bp bins of an input sequence belong to the peak, then that task of the target vector is labelled positive otherwise negative. The GRCh37 reference genome sequence was used to generate the input DNA sequence. The dataset consisting of a total of 4,863,024 samples includes 4,400,000 training samples, 8,000 validation samples and 455,024 test samples. Reverse complement of sequences doubled the size of the dataset.
3.2 Proposed methods
We have proposed two models in this paper. The first model TranAtt is an attention-based neural network with transfer learning and can outperform DeepSEA, DanQ and TBiNet in TF-DNA binding prediction task. Our second model PCLAtt is built using Convolutional Neural Network, Bidirectional Long short-term memory, multi-head attention layer and weight-shared dense layer. PCLAtt outperforms all the current best performing models (DeepSEA, DanQ, TBiNet and DeepATT) in TF-DNA binding prediction task.
3.2.1 Convolutional neural networks
Convolutional Neural Network is a multi-layer perceptron with automatic feature extraction capabilities. The convolution layer uses kernels to extract features from the input data and a non-linear activation function, ReLU as described by Eq. (1) is applied between the convolution and pooling layer to increase non-linearity. The pooling layer used after the convolutional layer reduces the spatial size of the output generated by the convolution layer. Various models such as DeepBind [1] and DeepSEA [34] are built using CNN architecture. Here one-hot encoded matrix of DNA sequences is treated as an input image and uses filters to extract motif features from it.
3.2.2 Recurrent neural networks
Recurrent Neural Network is a kind of Deep Neural Network (DNNs) that learns sequential data and are capable of processing variable lengths data. Basic Recurrent Neural Network has vanishing gradient problem and is unable to learn long-term dependencies. To solve the vanishing gradient problem of RNN, long short-term memory (LSTM) was used. DeeperBind [14] and DanQ [26] are models that used CNN-LSTM architecture for better performance. DanQ uses Bidirectional LSTM which is a variant of RNN that learns sequence from both the direction. BiLSTM has been used to learn long-term dependencies in bioinformatics applications [15] and has been used in various other machine learning applications [13, 30].
3.2.3 Attention neural network
The attention mechanism is known to give more importance to those regions of input sequence which are important for generating output. It gives different weights to different parts of the input sequence, these weights are known as attention scores. Attention mechanism has been used in various machine learning tasks like computer vision but it gained popularity from its application in Natural Language Processing (NLP) [5, 11, 32]. The multi-head attention layer used in PCLAtt is based on that used in DeepATT [20], which is modified version of the self-attention mechanism [32]. The query, key and value vectors used in self-attention layer can be expressed as shown below:
Here q, k and v are the query, key and value vectors respectively. The symbol \(\mathbf {\alpha }\) is used to represent the scaled dot-product attention. The output is then normalized using softmax function. Finally the context vector c is obtained as shown in Eq. (7).
For our model, PCLAtt, we create category query code with 690 X 690 diagonal matrix that represents first stage query vector of 690 transcription factor-cell line pairs while the key vectors and value vectors both are same containing the resultant output obtained after concatenation of the output from the two final BiLSTM layers in parallel.

Fig (a)Architecture of the TranAtt: An input of 1000 X 4 one-hot encoded matrix representation of a DNA sequence is passed through pre-trained convolution layers followed by a BiLSTM layer. The output is then passed through attention layer, fully connected layer and output layer thus resulting into 690 TF-cell line pairs (b) pre-trained model architecture: Freezed convolutional layers of the model
3.3 Architecture of TranAtt
TranAtt consists of an input layer, convolution layers of a pre-trained model followed by a BiLSTM layer, attention layer, fully connected layer, output layer. Convolution layer extracts TF-binding motif related features. After each convolution, a rectifier activation function was applied to increase non-linearity. To reduce the spatial size of the output obtained from the convolution layer, the max pooling layer is applied next to it. The Bidirectional LSTM is used to get information about the regulatory grammar of the sequence motifs. Then an attention layer was applied to give more importance to those regions of input DNA sequence that contains TF binding motifs.
We used transfer learning in TranAtt by using the convolution layers of a pre-trained model. The pre-trained model consists of 3 convolutional layers, 2 max-pooling layers, a fully connected layer and an output layer. Each of the first two convolution layers is followed by a max-pooling layer. This pre-trained model was designed for predicting DNA functions, consisting of histone-mark profile , transcription factor bindings and DNase I sensitivity. We first freezed all the layers of the pre-trained model and took the first two convolutional layers up to the dropout layer of the pre-trained model and then added a BiLSTM layer followed by an attention layer to it for more accurate prediction of TF-DNA binding. Thus the two convolutional layers of TranAtt were freezed such that their weights cannot be updated and the features learned by the convolutional layers in the pre-trained model can be used in TranAtt.

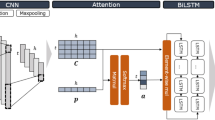
Architecture of PCLAtt model. The 1000 X 4 one-hot encoded matrix representation of a DNA sequence is an input of the model. The kernels of the convolution layer act as motif scanner. The BiLSTM layer added after each convolution layer learns the regulatory grammar of the sequence motifs. The output of two BiLSTM layers in parallel are concatenated and passed through the multi-head attention layer and the resultant is then passed through weight-shared dense layer. The multi-head attention layer selects the important features and the weight-shared dense layer predicts transcription factor binding sites with the selected features
The architecture of TranAtt is as shown in Fig. 1. The 1000 X 4 one-hot encoded matrix is passed through the two freezed convolutional layers of TranAtt and the output obtained from the convolution layers is then passed through bidirectional LSTM layer. The Bidirectional LSTM concatenates the output of forward and backward LSTM thus producing 60 RNN vectors with 960 dimensions. The output matrix of BiLSTM layer is then fed to the attention layer. In an attention layer, a key vector of size 2 X hidden units of LSTM is randomly initialized. Then a dot product is performed between each row of the output matrix of BiLSTM layer and the key vector of the attention layer. The softmax activation function is further applied to the resultant output to get normalized output known as attention vector. Later, an element-wise multiplication is performed between the attention vector and each column of the output matrix obtained from the BiLSTM layer. The resultant matrix obtained is finally passed through the fully connected layer, which is a dense layer with ReLU as an activation function. The output of the fully connected layer is finally fed to the output layer to get an output vector of size 690 representing 690 TF-cell line pairs. The output layer uses sigmoid as an activation function.
3.4 Architecture of PCLAtt
Our second proposed model PCLAtt consists of an input layer, two sets of CNN-BiLSTM layers in parallel followed by a multi-head attention layer and weight-shared dense layer as shown in Fig. 2. The 1000 X 4 one-hot encoded matrix is passed through the two parallel convolutional layers which pass through their respective connected bidirectional LSTM layers. The two convolution layer is used to capture TF-binding motif related features. The set consisting of a CNN with two BiLSTM layers with a max-pooling layer in between the two BiLSTM layers produces the output consisting of 64 RNN vectors with 400 dimensions. The another set consisting of one CNN followed by one BiLSTM layer produces output consisting of 64 RNN vectors with 1024 dimensions. The bidirectional LSTM learns the regulatory grammar of the sequence motifs obtained from their respective connected CNN layers. The output from the two BiLSTM layers is then concatenated and passed through the attention layer. In the multi-head attention layer, we create category query code with 690 X 690 diagonal matrix for first stage query vector of 690 transcription factor-cell line pairs, then the second stage query vector is generated by the linear combination from that obtained from the first stage. The query vector is splitted among 4 heads. The key and value vector matrix is the same as the output obtained after the concatenation of the output from the two BiLSTM layers. The output of the attention layer is 690 ATT vectors with 400 dimensions which is then passed through the weight-shared dense layer [20]. The 690 dense layers are assigned to 690 attention vectors respectively such that different dense layers have the same weight. The resultant is then passed through the sigmoid output layer.
3.5 Loss function
Both of the models use binary cross-entropy as their loss function which is defined as given in e Eq. (8).
Here \(y_n\) represents the target label, \({\hat{y}}_n\) represents the predicted output by the model and N represents the number of samples.
3.6 Model training
Proposed models are trained using Adam optimizer [19] and 0.0005 as the learning rate with varying batch size. It was trained for 20 epochs. Due to limited RAM, the training set consisting of 4,400,000 samples has been split into 10 chunks, each chunk consists of 440,000 samples while the validation and test set remained the same as used by the existing models (DeepSEA, DanQ, TBiNet and DeepATT).
4 Results and discussion
4.1 Evaluation metrics
We evaluated PCLAtt and TranAtt, used for predicting TF-DNA binding with various state-of-the-art models: DeepBind, Expectation Pooling, DeepSEA, DanQ, TBiNet, DeepATT and PBVPP-Hybrid. All these models were trained on 690 ChIP-Seq dataset. The evaluation metrics, AUROC and AUPR were used for evaluating the prediction of TF-DNA binding. AUROC is the area under receiver operating characteristic curve which is the area under true-positive rate and false-positive rate. True-positive rate is given by Eq. (9) and False-positive rate is as shown by Eq. (10) respectively. AUPR is the area under the precision-recall curve. Precision and recall are defined by Eqs. (11) and (12) respectively. AUPR is the better metric for evaluation in case of imbalanced dataset [6] as compared to the AUROC evaluation metric.
4.2 Hyper parameter tuning
We considered various hyperparameters during training of the proposed models. The hyper-parameters that are tuned during training are batch-size and learning rate (0.0005). We used a random search to find the best hyper-parameter values, which are listed in Table 1. The tuned hyper-parameters include the size of the CNN filters, learning rate, number of filters and LSTM units, dropout ratio, activation functions, optimizers, and so on.

(a) Area under precision-recall (AUPR) curve for TranAtt (b) Area under receiver operating characteristics (AUROC) curves of TranAtt

(a) Scatter-plots showing the comparison of AUPR scores of PCLAtt with DeepATT (b) Scatter-plots showing the comparison of AUROC scores of PCLAtt with DeepATT

(a) Area under precision-recall (AUPR) curve for PCLAtt (b) Area under receiver operating characteristics (AUROC) curves of PCLAtt
4.3 Discriminative ability of the proposed models
We compared both proposed models with various existing models: DeepBind, Expectation Pooling, DeepSEA, DanQ, TBiNet, DeepATT and PBVPP-Hybrid, and found that TranAtt and PCLAtt, achieved higher average AUROC and AUPR scores. TranAtt has an average AUROC value of 0.9564 and an average AUPR value of 0.3616. PCLAtt has an average AUROC value of 0.9630 and an average AUPR score of 0.3987. Results of the models shown in Table 2 are trained in our system on 690 ChIP-Seq dataset. TBiNet achieved an average AUROC score of 0.9454 and an average AUPR score of 0.3253 and DeepATT achieved an average AUROC score of 0.9582 and an average AUPR score of 0.3771 as shown in Table 2. Table 3 shows the scores of the proposed models compared with various state-of-the-art models trained on 690 ChIP-Seq dataset including our two proposed models TranAtt and PCLAtt. The percentage of improvement in the average AUC-ROC in PCLAtt when compare with recent models TBiNet [25] and PBVPP-Hybrid [8] are 1.65%, 1.42% respectively.
Figure 3 shows the curve plotted between precision-recall (AUPR curve) and the curve plotted between true-positive rate and false-positive rate (AUROC curve) of TranAtt. We plotted scatter plots for comparing AUPR and AUROC scores between PCLAtt and DeepATT as shown in Fig. 4. For most TF-cell line prediction, PCLAtt showed better performance than DeepATT in terms of AUPR scores and AUROC scores (Fig. 5).
5 Conclusion
Identification of transcription factor binding sites is necessary as it helps in understanding the gene regulation. Deep neural networks along with attention mechanisms have been used in various applications. We proposed two models TranAtt and PCLAtt focusing more on increasing the feature extraction capabilities that results in more accurate TF-DNA binding prediction task. In TranAtt, the transfer learning mechanism plays a key role in efficient feature extraction from the DNA sequence thus improving the performance of the model. The model PCLAtt is built using two sets of CNN-BiLSTM layers in parallel such that one set consists of one CNN layer with two BiLSTM layers and the another set consists of one CNN and one BiLSTM layer, followed by multi-head attention layer and weight-shared dense layer, all contributing towards efficient feature extraction from DNA sequence and selecting the valid features required for prediction of transcription factor binding sites. We trained all the models with the 690 ChIP-Seq dataset (contains data from 690 ChIP-Seq experiments) obtained from ENCODE Project. We evaluated both models based on average AUROC and AUPR scores. Finally, PCLAtt achieved higher AUPR and AUROC scores on average and outperformed state-of-the-art models (DeepSEA, DanQ, TBiNet and DeepATT) in TF-DNA binding prediction task.
References
Alipanahi B, Delong A, Weirauch MT, Frey BJ (2015) Predicting the sequence specificities of dna-and rna-binding proteins by deep learning. Nat Biotechnol 33(8):831–838
Almagro Armenteros JJ, Sønderby CK, Sønderby SK, Nielsen H, Winther O (2017) Deeploc: prediction of protein subcellular localization using deep learning. Bioinformatics 33(21):3387–3395
Amilpur S, Bhukya R (2020) Edeepssp: explainable deep neural networks for exact splice sites prediction. J Bioinform Comput Biol 18:2050024
Angermueller C, Pärnamaa T, Parts L, Stegle O (2016) Deep learning for computational biology. Mol Syst Biol 12(7):878
Bahdanau D, Cho K, Bengio Y (2014) Neural machine translation by jointly learning to align and translate. arXiv preprint arXiv:1409.0473
Chicco D (2017) Ten quick tips for machine learning in computational biology. BioData Min 10(1):1–17
Consortium EP et al (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489(7414):57
Dasari CM, Amilpur S, Bhukya R (2021) Exploring variable-length features (motifs) for predicting binding sites through interpretable deep neural networks. Eng Appl Artif Intell 106:104485
Dasari CM, Bhukya R (2020) Intersspp: investigating patterns through interpretable deep neural networks for accurate splice signal prediction. Chemom Intell Lab Syst 206:104144
Fu Y, Liu Z, Lou S, Bedford J, Mu XJ, Yip KY, Khurana E, Gerstein M (2014) Funseq2: a framework for prioritizing noncoding regulatory variants in cancer. Genome Biol 15(10):1–15
Galassi A, Lippi M, Torroni P (2020) Attention in natural language processing. IEEE Trans Neural Netw Learn Syst 32(10):4291–308
Ghandi M, Lee D, Mohammad-Noori M, Beer MA (2014) Enhanced regulatory sequence prediction using gapped k-mer features. PLoS Comput Biol 10(7):e1003711
Graves A, Jaitly N, Mohamed Ar (2013) Hybrid speech recognition with deep bidirectional lstm. In: 2013 IEEE Workshop on Automatic Speech Recognition and Understanding, pp 273–278
Hassanzadeh HR, Wang MD (2016) Deeperbind: enhancing prediction of sequence specificities of dna binding proteins. In: 2016 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), pp 178–183
He J, Pu X, Li M, Li C, Guo Y (2020) Deep convolutional neural networks for predicting leukemia-related transcription factor binding sites from DNA sequence data. Chemom Intell Lab Syst 199:103976
Hochreiter S, Schmidhuber J (1997) Long short-term memory. Neural Comput 9(8):1735–1780
Hu H, Xiao A, Zhang S, Li Y, Shi X, Jiang T, Zhang L, Zhang L, Zeng J (2019) Deephint: understanding hiv-1 integration via deep learning with attention. Bioinformatics 35(10):1660–1667
Hu Y, Wang Z, Hu H, Wan F, Chen L, Xiong Y, Wang X, Zhao D, Huang W, Zeng J (2019) Acme: pan-specific peptide-mhc class i binding prediction through attention-based deep neural networks. Bioinformatics 35(23):4946–4954
Kingma DP, Ba J (2014) Adam: a method for stochastic optimization. arXiv:1412.6980
Li J, Pu Y, Tang J, Zou Q, Guo F (2020) Deepatt: a hybrid category attention neural network for identifying functional effects of DNA sequences. Brief Bioinform 22(3):bbaa159
López-García G, Jerez JM, Franco L, Veredas FJ (2020) Transfer learning with convolutional neural networks for cancer survival prediction using gene-expression data. PloS one 15(3):e0230536
Luo X, Tu X, Ding Y, Gao G, Deng M (2020) Expectation pooling: an effective and interpretable pooling method for predicting DNA-protein binding. Bioinformatics 36(5):1405–1412
Min S, Lee B, Yoon S (2017) Deep learning in bioinformatics. Brief Bioinform 18(5):851–869
Miotto R, Wang F, Wang S, Jiang X, Dudley JT (2018) Deep learning for healthcare: review, opportunities and challenges. Brief Bioinform 19(6):1236–1246
Park S, Koh Y, Jeon H, Kim H, Yeo Y, Kang J (2020) Enhancing the interpretability of transcription factor binding site prediction using attention mechanism. Sci Rep 10(1):1–10
Quang D, Xie X (2016) Danq: a hybrid convolutional and recurrent deep neural network for quantifying the function of dna sequences. Nucl Acids Res 44(11):e107–e107
Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M (2019) Cadd: predicting the deleteriousness of variants throughout the human genome. Nucl Acids Res 47(D1):D886–D894
Ritchie GR, Dunham I, Zeggini E, Flicek P (2014) Functional annotation of noncoding sequence variants. Nat Methods 11(3):294–296
Sekhon A, Singh R, Qi Y (2018) Deepdiff: deep-learning for predicting differential gene expression from histone modifications. Bioinformatics 34(17):i891–i900
Sundermeyer M, Alkhouli T, Wuebker J, Ney H (2014) Translation modeling with bidirectional recurrent neural networks. In: Proceedings of the 2014 Conference on Empirical Methods in Natural Language Processing (EMNLP), pp 14–25
Tsubaki M, Tomii K, Sese J (2019) Compound-protein interaction prediction with end-to-end learning of neural networks for graphs and sequences. Bioinformatics 35(2):309–318
Vaswani A, Shazeer N, Parmar N, Uszkoreit J, Jones L, Gomez AN, Kaiser L, Polosukhin I (2017) Attention is all you need. arXiv preprint arXiv:1706.03762
Zheng A, Lamkin M, Wu C, Su H, Gymrek M (2020) Deep neural networks identify context-specific determinants of transcription factor binding affinity. BioRxiv
Zhou J, Troyanskaya OG (2015) Predicting effects of noncoding variants with deep learning-based sequence model. Nature Methods 12(10):931–934
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bhukya, R., Kumari, A., Dasari, C.M. et al. An attention-based hybrid deep neural networks for accurate identification of transcription factor binding sites. Neural Comput & Applic 34, 19051–19060 (2022). https://doi.org/10.1007/s00521-022-07502-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00521-022-07502-z