
Abstract
Within the domain of molecular biology research, the intricate regulation of transcription continues to present a challenging yet imperative area of study. According to recent scientific studies, the nucleotide double helix shape is a major factor in improving the accuracy and comprehensibility of Transcription Factor Binding Sites (TFBSs). Despite the significant growth in computational methods aiming to concurrently incorporate both DNA sequence and DNA shape features, devising an effective model remains a challenging and unresolved issue. In this paper, we proposed a deep learning prediction model for TFBSs using attention mechanism, convolutional, and RNN-based networks by incorporating the DNA sequence and shape data. Attention mechanisms recognise the long-range dependencies but encounter challenges in focusing on local feature details. On the other hand, convolutional operations are proficient at extracting local features but may inadvertently neglect global information. Recurrent Neural Networks (RNNs) capture long-term dependencies within sequences. We demonstrate that the ability to predict TFBSs is greatly improved by our proposed technique, DeepCTF, using 12 in-vitro datasets collected from Protein Binding Microarray (PBMs) compared to the other state-of-the-art models.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Understanding how proteins and DNA interact is crucial for controlling gene transcription, splicing, translation, replication, and degradation. These interactions significantly influence the complex systems of genetic regulation [1,2,3]. To annotate and investigate the activity of cis-regulatory elements, modelling of Transcription Factor (TF) binding affinity and predictions of TF binding locations are of key importance. Transcription factor binding sites (TFBSs), also known as motifs [4], are a particular class of functional DNA sites that typically range in size from a few to around 20 base pairs (bps). Recognising the many processes involved in gene expression and gaining knowledge about in vitro cellular processes and the design of medicinal treatments [5] depend on precisely finding the TFBS within the DNA sequence. In the past ten years, improvements in techniques such as Protein Binding Microarrays (PBMs) [6, 7], Chromatin Immunoprecipitation coupled with high-throughput sequencing (ChIP-seq) [8, 9], and Systematic Evolution of Ligands by Exponential Enrichment coupled with high-throughput sequencing (SELEXseq) have produced detailed datasets of TFBSs, encompassing both in vivo and in vitro contexts. Nevertheless, regardless of the steadily growing variety of these datasets, we can still not predict the genomic regions where a certain TF binds with total accuracy.
Numerous precise techniques for analysing PBM data have been suggested to predict TFBSs accurately [10,11,12]. Due to the availability of this data, computational methods performance is enhanced for the prediction of specific expression of transcription factor binding [13,14,15]. Thus, computational technologies have replaced biological experimentation as the principal strategy for answering critical biological questions [12, 16]. Plus, computational technologies have inherent simplicity, speed, and cost-effectiveness advantages that are responsible for replacing conventional biological experiments.
Computational technology like Deep learning (DL) [17,18,19,20,21] has experienced rapid advancements and showcased remarkable performance in diverse fields [22,23,24], and similarly in predicting functional genomics [25,26,27,28]. Input data of high-dimension can be processed and automatically identified by this technology. Encoding input data in DL models like Convolutional Neural Networks (CNN) or Recurrent Neural Networks (RNN) have demonstrated favorable results in the identification of TFBSs [29,30,31,32,33] by producing a probability value representing the binding or no binding of TFBSs. Thus, these DL models greatly outperform conventional methods.
Identifying particular sequences (TFBS) within DNA sequences is regarded as a Natural Language Processing (NLP) task, and we know that advancements in NLP have been propelled by the emergence of the self-attention mechanism [34, 35]. Ullah et al. [36] introduced a Deep Learning (DL) model based on CNN and self-attention layers to capture interactions among regulatory elements within genomic sequences. This model incorporates attention mechanisms to enhance the network’s learning capability by inferring a global view of interactions in the genomic dataset. Shen et al. [37] introduced SAResNet, a model that merges the self-attention mechanism with a residual network structure, enhancing the network’s learning capability by capturing positional information in biological sequences using the self-attention mechanism and with residual connections to extract high-level features, enabling accurate prediction of DNA-protein binding interactions. These notable studies demonstrate the self-attention layer’s significant utility in detecting potential motifs and its capacity to accurately picture the relationships between regulatory components inside a particular sequence.
An expanding volume of research suggests that the shape of DNA in specific targeted locations may provide insight into a critical aspect of TF binding. The reason for this lies in the 3D structure of DNA, formed by the stacking of physical interactions among adjacent base pairs, which inherently contains the dependencies among nucleotides [38]. Studies have demonstrated that TF binding is notably affected by four separate shape characteristics derived from Monte Carlo (MC) simulations: Minor Groove Width (MGW), Roll, Propeller Twist (ProT), and Helix Twist (HelT) [39]. In [40], a kernel-based framework was introduced to identify TF-DNA binding similarities precisely. In this approach, the spectrum + shape kernel and the di-mismatch + shape kernel were employed for modeling TF binding without requiring sequence alignment and potentially offering better scalability for large datasets. Unlike Ullah et al. [36] and Shen et al. [37], Ma et al. [40] rely on kernel-based methods, which may have limitations in capturing complex interactions and long-range dependencies within genomic sequences, potentially leading to lower predictive performance.
Yang et al. [41] employed the DEep Sequence and Shape mOtif (DESSO) model, a straightforward DL model that incorporated DNA shape to predict TFBSs using human ChIP-seq datasets. They discovered that the shape of DNA holds significant predictive capability for TF-DNA binding, offering novel potential shape motifs for human TFs. However, DESSO may lack the advanced attention mechanisms utilized by Ullah et al. [36] and Shen et al. [37], potentially limiting its ability to capture intricate interactions and long-range dependencies within genomic sequences. Additionally, it may not offer the flexibility and scalability of kernel-based methods like Ma et al. [40] for handling diverse datasets. Zhang et al. [42] introduced a sequence + shape framework called DLBSS, and Wang et al. [43] introduced a hybrid convolutional recurrent neural network framework named CRPTS both predicted TFBS using DNA sequence and shape features. Thus, the conclusion was that including DNA shape significantly enhances the results of TFBS prediction.
Although promising results were obtained by using primary DNA sequences and shape features as input, like in DLBSS and CRPTS models, these models lack the lack the advanced attention mechanisms presented in Ullah et al. [36] and Shen et al. [37], potentially limiting their ability to capture complex dependencies and interactions within genomic sequences effectively. Moreover, the approach encounters challenges like prioritizing key features over comprehensively considering all features and working with the continuous nature of shape features, which differs from the discrete nature of sequence features. So, there is still room for advancement in DL models. Thus, we present an improved shared DL architecture incorporating an attention mechanism, drawing inspiration from Wang et al. [43]. Our approach in the DeepCTF model combines attention mechanisms with CNN and recurrent neural networks (RNNs) to adapt DNA sequences and their associated local DNA shape features, enabling an enhanced predictive model for TFBS identification. The improved performance of our proposed model, DeepCTF, stems from two important advances: (1) the strategic incorporation of a self-attention mechanism into CNN and RNN, which effectively allows for the extraction of complex features from DNA sequences acquired from high-throughput technologies; (2) DeepCTF’s exceptional ability to extract hidden local structural information from DNA sequences, which reduces the need for solely depending on DNA shape data. This combination highlights the model’s adaptability to complex genomic contexts while improving performance.
2 Approach
This study employed kernel methods to construct quantitative TF binding prediction models [40]. We have considered a set of triples \((s_1, x_1, y_1)\) up to \((s_n, x_n, y_n)\). Each \(s_i\) represents a DNA sequence of a specific length, denoted as w. The information in \(x_i\) pertains to the DNA shape conformation of \(s_i\). Meanwhile, \(y_i\) is a binary indicator, signifying whether a TF binds to the sequence. We aim to construct a predictive model, denoted as f(.), with the objective that when given \(s_i\) and \(x_i\) as inputs, the model \(f(s_i, x_i)\) accurately predicts \(y_i\). As we have integrated DNA shape local features within our prediction framework, we examined four DNA Shapes: MGW, Roll, ProT, and HelT.
2.1 Attention mechanism
Figure 1 depicts the attention mechanism’s framework. To allow the layer after batch normalisation to learn from earlier layers and concentrate on achieving its objective of expediting the training process, we first applied the batch normalisation technique on the input features to reduce the internal covariate shift. Then, we utilise the Rectified Linear Unit (ReLU) activation, computed by the formula described below:
Subsequently, X is transformed linearly to yield three vectors which are Query \(Q_{r}\in R^{T \times d_k}\), Key \(K_{Y} \in R^{T \times d_k}\), and Value \(V_{V} \in R^{T \times d_v}\). T denotes the sequence length, while the hidden dimensionality for query or key and value is indicated by \(d_k\) and \(d_v\), respectively. These three vectors are formulated as follows:
Where the learned weight metrics of the query, key, and value vectors are denoted by the variables \(W_{Q_{r}}\), \(W_{K_{Y}}\), and \(W_{V_{V}}\). We have selected the scaled dot-product attention; in other words, the attention value from x to y is determined by the similarity between \((Q_{r})_x\) and \((K_{Y})_y\), and it is then normalized and multiplied by \(V_{V}\) to provide the final attention weight \( A(Q_{r},K_{Y},V_{V})\), which may be expressed as follows:
\(\frac{1}{\sqrt{d_k}}\), is essential to manage the attention values with an appropriate variance. It primarily prevents the softmax function’s input from becoming overly large. For each possible combination of Queries and Keys, \(Q_{r}K_{Y}^T\) provides the dot product, producing a matrix with the shape \(T \times T\). The model can obtain long-term relationships among residues by dynamically focusing on the residues that comprise the sequences and capturing the global properties of the input DNA sequences because of the self-attention mechanism.

Illustrative diagram of self-attention module
2.2 Convolutional neural network (CNN)
As widely recognised, the convolutional layer, usually followed by the ReLU unit, is a motif scanner that calculates a score for all possible motifs. Thus, this stage is in charge of detecting motif features. Prior CNN-based prediction techniques have shown that CNNs can pick up complex features. Nonetheless, different CNN architectures will result in various network efficiency levels [44, 45]. Increasing the number of convolution kernels makes it easier to identify motif variants while stacking convolutional layers deepens the model and improves feature identification/extraction. The single convolution layer focuses more on extracting local features in the absence of the stacking step. The multilayer convolutional neural network is frequently employed to create layered representations of the input sequence, facilitating the extraction of meaningful features at different levels of abstraction [45] and more thoroughly detect TFBSs [44]. By collaborating among convolution layers, the network achieves its goals. However, this makes it challenging to train with excessive parameters, and the global information produced is typically incomplete and lossy. As a result, our model only employs one CNN layer to extract local features, and the 2D convolution at every location i is as follows:
Where \(W_k\) denotes convolutional filters corresponding to S, the input sequence, m, is the location of the convolutional operation. The sequence motif detector is a \(l \times \gamma \) weight matrix, where \(\gamma \) is the channel number of S and l is the filter’s length. \(\tau \) is the filter’s index.
2.3 Recurrent neural network (RNN)
Long Short-Term Memory (LSTM) (one of the types of RNN) [46] addresses the issue that regular RNN is unable to handle with long-term dependency. We used LSTM to extract long-term characteristics from the DNA sequence, considering its double-stranded structure. The cell state is crucial to LSTM and is carefully monitored by structures known as gates, which include output, forget, and input gates. In the first phase, the "forget gate" determines which data should be saved or deleted. The next step is to choose the appropriate amount of new data to be added to the cell state. The output value is decided in the last stage.
Where \(fg_{t}\), \(ig_{t}\), and \(Og_{t}\) stand for the forget, input, and output gates weight values; W is the weight matrix, and b is the bias; the input vector, the memory representation, and the hidden layer state are denoted, respectively, by the variables \(x_{t}\), \(Cm_{t}\), and \(hl_{t}\) at time t; and \(\odot \) used to represent element-wise multiplication. \(\sigma \) stands for sigmoid function. For clarity, the summary of notations used here is described in the following Table 1.
3 Material and method
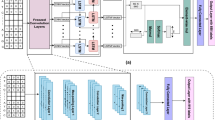
We develop a two-path deep learning sequence plus shape kernels (DeepCTF) framework: one for DNA sequences computation with attention mechanism and the other regarding DNA shape-related data processing. The specifics of DeepCTF are explained as follows, as illustrated in Fig. 2.
3.1 Dataset and processing
The data processing technique for the proposed DeepCTF model is depicted in Fig. 2a.
3.1.1 DNA sequence data
The PBM approach provides biological understanding regarding the regulatory roles and in vivo activities of protein-DNA interactions. We extracted 12 uPBM data [47], which originates from a range of protein families, to assess the efficiency of the proposed model. Every input DNA sequence was first converted by one-hot encoding into a matrix \(n \times l\), suitable for a DL model. Here, n denotes the four nucleotides (A, T, C and G), indicated by the binary vectors written as follows, and l is the sequence length, i.e.35, in the uPBM we utilised.
3.1.2 DNA shape data
The binding patterns are significantly influenced by the 3D structure of DNA of TFs [48]. The four DNA shape features have distinct pentamers utilising a sliding-window method and a query table-which were identified in earlier work [49]. Table S3 [50] included the preliminary DNA shape data. The efficient contribution of the four DNA shapes is determined by averaging the two roll and HelT values, one MGW value, and one ProT value contributed by each pentamer. As in [43], to generate a DNA sequence of length \(l + 4\), it is padded with two zeros at both sides of the sequence. Next, we utilise sliding window a to produce an input shape feature matrix \(n \times l\), with a size similar to the DNA sequence. Zero-mean normalisation was applied to each feature to remove the bias resulting from varying value ranges for distinct shapes.

A visual representation of the suggested model DeepCTF, where m is the number of shape features (m=4), n is the length of DNA sequences, and \(B_{m}\) is the size of the mini-batch
3.2 Architecture of DeepCTF
The overall layout of the DeepCTF model is seen in Fig. 2. On the left side of Fig. 2b, our model DeepCTF begins by encoding the DNA sequences into one-hot form and fed to a self-attention module. Then, we move on to a convolutional layer that performs the convolution function and a 2D max pooling layer to obtain an initial sense of local and global attributes. The purpose of the max pooling layer is to shorten the lengthy sequences to minimise the number of parameters and avoid overfitting. An LSTM layer is placed after the max pooling layer to record long-term relationships between the motifs and the orientations and spatial separations of DNA sequences. To mitigate overfitting, a dropout layer was implemented following the LSTM layer. Left-sided module configuration of Proposed DeepCTF model is presented in Table 2.
Implementing the DNA shape feature data in our model DeepCTF as shown on the right side of Fig. 2b, we used the same method as CRPTS [43] i.e. a convolution layer for processing DNA shape features to match the size of the DNA sequence feature. After the convolution layer, the output from this layer is fed into the activation function ReLU, which is used in our model. It improves convergence performance and addresses gradient disappearing issues during back-propagation training. Right-sided module configuration of Proposed DeepCTF model is presented in Table 3.
In the end, outputs from both the left and right sides of the DeepCTF model are concatenated and processed through the dense layer. The dense layer consists of two Fully Connected (FC) layers: batch normalisation and dropout (Table 4).
Batch normalisation was used at the output stage to simplify the network parameter initialisation process and reduce gradient problems during back-propagation. The previous layer’s outputs were fed into an FC layer to enable feature integration. A dropout layer containing a single neuron followed the output layer, which was utilised to predict the binding/no binding probability of TF-DNA binding. Table 4 presents a comprehensive configuration of this dense layer.
4 Experimental results
Conducting several comparative experiments in this section to show how well the proposed model DeepCTF performed.
4.1 Experimental setup and hyper-parameter settings

Performance comparison of DeepCTF model with state-of-the-art models using \(R^2\) evaluation metric

Performance comparison of DeepCTF model with state-of-the-art models using PCC evaluation metric

Boxplot of the Evalution Metric Avearage \(R^2\) and PCC values of 12 datasets for DeepCTF and state-of-the-art models

Bar Plot Representation of \(R^2\) and PCC for DeepCTF without DNA shape with DeepCTF with DNA Shape, CRPTS and CRPT models for 12 in-vitro datasets
In the training process of DeepCTF, we minimise the tolerable loss function for each dataset. The loss function used in our proposed model is Mean Squared Error (MSE), which is described below:
where N represents the total number of DNA samples in each training dataset, and \({\bar{y}}_i\) and \(y_i\) denote the ground and the observed value of the i-th sample, respectively. To prevent overfitting of the model, L2 regularisation was employed; \(\lambda \) denotes a regularisation variable, and \(\left\| .\right\| _{2}\) denotes the L2 norm. Mini-batch size is equal to 300, and AdaMax optimises the loss function. In AdaMax, the neural network’s dropout ratio, momentum, and Delta were chosen at random from [0.2,0.5], [0.9,0.99,0.999], and [1e-8,1e-6,1e-4], respectively. We employed five-fold cross-validation to guarantee model accuracy and avoid overfitting. An early stop approach was utilized in addition to choosing 100 training epochs to reduce the model running time. We also utilized a random-search approach to determine the optimal configuration for certain sensitive hyperparameters, such as dropout ratio, Momentum, and Delta, wherein we randomly sampled 30 hyperparameter settings. The training process spanned 100 epochs, during which the accuracy of the validation set was evaluated and monitored after each epoch. The model achieving the highest accuracy on the validation set was saved.
4.2 Evaluation metrics
DeepCTF model performance is evaluated using current competitive techniques to assess the suggested approach. The Pearson correlation coefficient (PCC) and coefficient of determination \((R^2)\) were used to evaluate the proposed model’s predicted binding affinity. Working under the assumption that as these mentioned evaluation metrics approach 1, the model’s efficacy improves. These two metrics were implemented on every dataset to confirm the model’s overall performance. The following defines two performance measures:
\(y_{i}\), \(Y_{i}\), y, and Y stand observed, predicted, average observed, and average predicted binding affinity scores, respectively. Where \(S_{yY}=\sum _{i}(y_{i}-\bar{y})(Y_{i}-\bar{Y})\), \(S_{yy}=(y_{i}-\bar{y})^2\), and \(S_{YY}=(Y_{i}-\bar{Y})^2\). Also Rss=\(\sum _{i}(y_{i}-Y_{i})^2\) is the residuals of sum of squares and Tss=\(\sum _{i}(y_{i}-\bar{y})^2\) is the total sum of squares.
4.3 Performance comparison with competitive models
To assess DeepCTF’s performance, we evaluate it not only with Deepbind, which relied on using the DNA sequences as primary input source processed by CNN but also with four different techniques that combined DNA shapes and sequences, which are two kernel-based approaches, DLBSS and CRPTS. Evaluating DeepCTF’s performance against the state-of-the-art approaches using 12 in vitro datasets is shown by considering the aforementioned PCC and \(R^2\) metrics.
Moreover, Figs. 3 and 4 compare the overall efficacy of DeepCTF with the state-of-the-art methods using 12 in vitro datasets. Concerning PCC and R2, it is clear from Figs. 3 and 4 that DeepCTF performs more well and steadily than the other approaches. As seen from these plots, DeepCTF performance is superior to the two kernel-based techniques due to the use of DNA sequences with the DNA shapes, proving that both significantly influence the identification of TFBSs. DeepCTF attains a statistically significant improvement in average \(R^2\) and PCC, as seen in Fig. 5. In terms of \(R^2\) and PCC, DeepCTF outperforms DLBSS and CRPTS by roughly 7% and 4%, and 3% and 1.4%, respectively. This indicates that our suggested DL model with an attention mechanism outperforms the one that merely uses CNN. In 12 in vitro datasets, DeepCTF’s highest and lowest values outperform the competing approaches. The smaller box of the DeepCTF shows that the two indicators (\(R^2\) and PCC) range is more condensed, demonstrating its strong stability.
The exceptional performance of DeepCTF in comparison to other competitive models (K_spectrum+Shape, Dimismatch+ shape, DeepBind, DLBSS, CRPT, and CPRTS) was attributed to two factors: (1) It utilises the DNA shape information; and (2) By employing an attention mechanism with CNN and RNN, DeepCTF prioritises obtaining global information regarding DNA sequences instead of local information. Thus, the main flaw of the convolution technique is that it only processes local neighbourhoods; as a result, global information is missed. This flaw of convolutional technique performance is overcome with the help of self-attention modules to gather more relational information from the network.
Further, we experimented with the DeepCTF model utilising only DNA sequences (without DNA Shape data) as input to evaluate whether adding DNA shape information impacts the resulting prediction accuracy of TF binding affinities. Figure 6 shows that the DeepCTF model with only DNA sequence data (DeepCTF_without DNAShape) as input has lower values of \(R^2\) and PCC values as compared to DeepCTF with both DNA sequence plus shape data as input, but higher values as compared to CRPT. This shows that the DeepCTF model has good stability. Since DeepCTF consists of an attention layer mechanism that extracts the global representation of the input DNA sequences and combines it with the local features drawn out from the next convolutional layer, the LSTM layer draws out long-term dependence in the DNA sequences and then combines it with DNA shape features generated by a convolutional layer which enhances the model prediction ability.
5 Conclusion
Deep-learning models have effectively reduced the computational cost and time required for exploring the intricate relationships within large-scale biological data, revealing hidden complexities. This paper proposes an attention-based deep learning model (DeepCTF) to use DNA sequences and shape data to predict transcription factor binding specificities. This method uses an attention layer, a CNN layer, and an RNN layer to learn features from DNA sequences, and on the other side, it uses a single convolutional layer to learn features from DNA shape data. The two heterogeneous data sets are suitably integrated and fully utilized by the model. The higher efficiency of our proposed model DeepCTF is due to the usage of the attention layer, which extracts the global representation of DNA sequences and combines it with the local features extracted from the CNN layers and provides it to the RNN layer, which is used to learn the long term dependencies from DNA sequences. Thus, the experimental findings obtained on 12 uPBM datasets demonstrate the high efficiency of our proposed approach, DeepCTF, in TFBS prediction.
Data availability
The data that support the findings of this study is openly available at https://bitbucket.org/wenxiu/sequence-shape/src/master/
References
Nasiri, E., Berahmand, K., Rostami, M., Dabiri, M.: A novel link prediction algorithm for protein-protein interaction networks by attributed graph embedding. Comput. Biol. Med. 137, 104772 (2021)
Stormo, G.D.: DNA binding sites: representation and discovery. Bioinformatics 16(1), 16–23 (2000)
Gerstberger, S., Hafner, M., Tuschl, T.: A census of human rna-binding proteins. Nat. Rev. Genet. 15(12), 829–845 (2014)
Zambelli, F., Pesole, G., Pavesi, G.: Motif discovery and transcription factor binding sites before and after the next-generation sequencing era. Brief. Bioinform. 14(2), 225–237 (2013)
Vamathevan, J., Clark, D., Czodrowski, P., Dunham, I., Ferran, E., Lee, G., Li, B., Madabhushi, A., Shah, P., Spitzer, M., et al.: Applications of machine learning in drug discovery and development. Nat. Rev. Drug Discov. 18(6), 463–477 (2019)
Berger, M.F., Philippakis, A.A., Qureshi, A.M., He, F.S., Estep III, P.W., Bulyk, M.L.: Compact, universal dna microarrays to comprehensively determine transcription-factor binding site specificities. Nat. Biotechnol. 24(11), 1429–1435 (2006)
Newburger, D.E., Bulyk, M.L.: Uniprobe: an online database of protein binding microarray data on protein-dna interactions. Nucleic Acids Res. 37(1), D77–D82 (2009)
Barski, A., Cuddapah, S., Cui, K., Roh, T.-Y., Schones, D.E., Wang, Z., Wei, G., Chepelev, I., Zhao, K.: High-resolution profiling of histone methylations in the human genome. Cell 129(4), 823–837 (2007)
Schmidt, D., Wilson, M.D., Spyrou, C., Brown, G.D., Hadfield, J., Odom, D.T.: Chip-seq: using high-throughput sequencing to discover protein-dna interactions. Methods 48(3), 240–248 (2009)
Stormo, G.D.: Dna binding sites: representation and discovery. Bioinformatics 16(1), 16–23 (2000)
Zhao, X., Huang, H., Speed, T.P.: Finding short dna motifs using permuted markov models. In: Proceedings of the Eighth Annual International Conference on Research in Computational Molecular Biology, pp. 68–75, (2004)
Huang, D.-S., Zheng, C.-H.: Independent component analysis-based penalized discriminant method for tumor classification using gene expression data. Bioinformatics 22(15), 1855–1862 (2006)
Huang, D.-S., Hong-Jie, Yu.: Normalized feature vectors: a novel alignment-free sequence comparison method based on the numbers of adjacent amino acids. IEEE/ACM Trans. Comput. Biol. Bioinf. 10(2), 457–467 (2013)
Deng, S.-P., Huang, D.-S.: Sfaps: An r package for structure/function analysis of protein sequences based on informational spectrum method. Methods 69(3), 207–212 (2014)
Xia, J.-F., Zhao, X.-M., Song, J., Huang, D.-S.: Apis: accurate prediction of hot spots in protein interfaces by combining protrusion index with solvent accessibility. BMC Bioinform. 11, 1–14 (2010)
Zheng, C.-H., Zhang, L., Ng, V.T.-Y., Shiu, C.K., Huang, D.-S.: Molecular pattern discovery based on penalized matrix decomposition. IEEE/ACM Trans. Comput. Biol. Bioinf. 8(6), 1592–1603 (2011)
LeCun, Y., Bengio, Y., Hinton, G.: Deep learning. Nature 521(7553), 436–444 (2015)
Goodfellow, I., Bengio, Y., Courville, A.: Deep Learning. MIT Press (2016)
Yang, S., Zhou, D., Cao, J., Guo, Y.: Rethinking low-light enhancement via transformer-gan. IEEE Signal Process. Lett. 29, 1082–1086 (2022)
Guo, Y., Zhou, D., Ruan, X., Cao, J.: Variational gated autoencoder-based feature extraction model for inferring disease-mirna associations based on multiview features. Neural Netw. 165, 491–505 (2023)
Guo, Y., Zhou, D., Li, P., Li, C., Cao, J.: Context-aware poly (a) signal prediction model via deep spatial–temporal neural networks. IEEE Trans. Neural Netw. Learn. Syst. (2022)
Deng, L., Hinton, G., Kingsbury, B.: New types of deep neural network learning for speech recognition and related applications: an overview. In: 2013 IEEE International Conference on Acoustics, Speech and Signal Processing, pp. 8599–8603. IEEE (2013)
Krizhevsky, A., Sutskever, I., Hinton, G.E.: Imagenet classification with deep convolutional neural networks. Adv. Neural Inf. Process. Syst. 25 (2012)
Li, H.: Deep learning for natural language processing: advantages and challenges. Natl. Sci. Rev. 5(1), 24–26 (2018)
Libbrecht, M.W., Noble, W.S.: Machine learning applications in genetics and genomics. Nat. Rev. Genet. 16(6), 321–332 (2015)
Talukder, A., Barham, C., Li, X., Hu, H.: Interpretation of deep learning in genomics and epigenomics. Briefings Bioinform. 22(3):bbaa177 (2021)
Zou, J., Huss, M., Abid, A., Mohammadi, P., Torkamani, A., Telenti, A.: A primer on deep learning in genomics. Nat. Genet. 51(1), 12–18 (2019)
Li, W., Guo, Y., Wang, B., Yang, B.: Learning spatiotemporal embedding with gated convolutional recurrent networks for translation initiation site prediction. Pattern Recogn. 136, 109234 (2023)
Alipanahi, B., Delong, A., Weirauch, M.T., Frey, B.J.: Predicting the sequence specificities of dna-and rna-binding proteins by deep learning. Nat. Biotechnol. 33(8), 831–838 (2015)
Quang, D., Xie, X.: Danq: a hybrid convolutional and recurrent deep neural network for quantifying the function of dna sequences. Nucleic Acids Res. 44(11), e107–e107 (2016)
Kelley, D.R., Snoek, J., Rinn, J.L.: Basset: learning the regulatory code of the accessible genome with deep convolutional neural networks. Genome Res. 26(7), 990–999 (2016)
Zhang, Q., Zhu, L., Huang, D.-S.: High-order convolutional neural network architecture for predicting dna-protein binding sites. IEEE/ACM Trans. Comput. Biol. Bioinf. 16(4), 1184–1192 (2018)
Trabelsi, A., Chaabane, M., Ben-Hur, A.: Comprehensive evaluation of deep learning architectures for prediction of dna/rna sequence binding specificities. Bioinformatics 35(14), i269–i277 (2019)
Vaswani, A., Shazeer, N., Parmar, N., Uszkoreit, J., Jones, L., Gomez, A.N., Kaiser, Ł., Polosukhin, I.: Attention is all you need. Adv. Neural Inf. Process. Syst. 30 (2017)
Nagoudi, E.M.B., Elmadany, A.R., Abdul-Mageed, M.: Arat5: Text-to-text transformers for arabic language generation. arXiv:2109.12068, (2021)
Ullah, F., Ben-Hur, A.: A self-attention model for inferring cooperativity between regulatory features. Nucleic Acids Res. 49(13), e77–e77 (2021)
Shen, L.-C., Liu, Y., Song, J., Dong-Jun, Y.: Saresnet: self-attention residual network for predicting dna-protein binding. Briefings Bioinform. 22(5), 101 (2021)
Rohs, R., West, S.M., Sosinsky, A., Liu, P., Mann, R.S., Honig, B.: The role of dna shape in protein-dna recognition. Nature 461(7268), 1248–1253 (2009)
Zhou, T., Shen, N., Yang, L., Abe, N., Horton, J., Mann, R.S., Bussemaker, H.J., Gordân, R., Rohs, R.: Quantitative modeling of transcription factor binding specificities using dna shape. Proc. Natl. Acad. Sci. 112(15), 4654–4659 (2015)
Ma, W., Yang, L., Rohs, R., Noble, W.S.: Dna sequence+ shape kernel enables alignment-free modeling of transcription factor binding. Bioinformatics 33(19), 3003–3010 (2017)
Yang, J., Ma, A., Hoppe, A.D., Wang, C., Li, Y., Zhang, C., Wang, Y., Liu, B., Ma, Q.: Prediction of regulatory motifs from human chip-sequencing data using a deep learning framework. Nucleic Acids Res. 47(15), 7809–7824 (2019)
Zhang, Q., Shen, Z., Huang, D.-S.: Predicting in-vitro transcription factor binding sites using dna sequence+ shape. IEEE/ACM Trans. Comput. Biol. Bioinf. 18(2), 667–676 (2019)
Wang, S., Zhang, Q., Shen, Z., He, Y., Chen, Z.-H., Li, J., Huang, D.-S.: Predicting transcription factor binding sites using dna shape features based on shared hybrid deep learning architecture. Molecular Therapy-Nucleic Acids 24, 154–163 (2021)
Zhou, J., Troyanskaya, O.G.: Predicting effects of noncoding variants with deep learning-based sequence model. Nat. Methods 12(10), 931–934 (2015)
Deng, L., Hui, W., Liu, X., Liu, H.: Deepd2v: a novel deep learning-based framework for predicting transcription factor binding sites from combined dna sequence. Int. J. Mol. Sci. 22(11), 5521 (2021)
Hochreiter, S., Schmidhuber, J.: Long short-term memory. Neural Comput. 9(8), 1735–1780 (1997)
Weirauch, M.T., Cote, A., Norel, R., Annala, M., Zhao, Y., Riley, T.R., Saez-Rodriguez, J., Cokelaer, T., Vedenko, A., Talukder, S., et al.: Evaluation of methods for modeling transcription factor sequence specificity. Nat. Biotechnol. 31(2), 126–134 (2013)
Gordân, R., Shen, N., Dror, I., Zhou, T., Horton, J., Rohs, R., Bulyk, M.L.: Genomic regions flanking e-box binding sites influence dna binding specificity of bhlh transcription factors through dna shape. Cell Rep. 3(4), 1093–1104 (2013)
Stella, S., Cascio, D., Johnson, R.C.: The shape of the dna minor groove directs binding by the dna-bending protein fis. Genes Dev. 24(8), 814–826 (2010)
Zhou, T., Yang, L., Yan, L., Dror, I., Machado, A.C.D., Ghane, T., Di Felice, R., Rohs, R.: Dnashape: a method for the high-throughput prediction of dna structural features on a genomic scale. Nucleic Acids Res. 41(W1), W56–W62 (2013)
Author information
Authors and Affiliations
Contributions
S. Tariq and A. Amin did the study’s conception and analysis. S. Tariq gathered the data and information. The manuscript was written by S. Tariq and revised by A. Amin. The final manuscript was read and approved by all writers.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no Conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Tariq, S., Amin, A. DeepCTF: transcription factor binding specificity prediction using DNA sequence plus shape in an attention-based deep learning model. SIViP 18, 5239–5251 (2024). https://doi.org/10.1007/s11760-024-03229-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11760-024-03229-7