
Abstract
Key message
This study enriched the understanding of the mechanism of nitrogen tolerance and starvation of yellowhorn and provided a reference for the breeding of low-nitrogen tolerance germplasm in the future.
Abstract
Yellowhorn is a rare woody oil crop in China, which can survive in barren, drought, cold, and even saline-alkali environments. However, its growth and development can be seriously affected by low-nitrogen stress. A comprehensive understanding of its transcriptional regulation activities under low-nitrogen stress is expected to indicate the key molecular mechanisms of its tolerance to low nitrogen levels. In this study, yellowhorn was divided into control, low-nitrogen, and nitrogen-free groups for treatment. Samples were treated for 15 days before assessing physiological characteristics and transcriptome analysis. Under low-nitrogen and no-nitrogen condition, a total of 10,733 differentially expressed genes were identified, among which 3870 genes were up-regulated and 6,863 genes were down-regulated. Under low-nitrogen stress, the most up-regulated genes were enriched in the phenylpropane synthesis pathway, flavonoid synthesis pathway, and plant hormone signal transduction pathway. Our determination of total flavonoids and proanthocyanidins also verified the upregulation of these three pathways. Brassinosteroid, salicylic, and jasmonic acid (BR, SA and JA, respectively) pathway-related genes were significantly up-regulated in the signal transduction pathway of plant hormones. This study provided a comprehensive review of the transcriptomics changes of yellowhorn under low nitrogen stress and detailed its insights into the relevant mechanism of BR, SA, and JA signaling pathway in resisting low-nitrogen stress, laying a solid foundation to further identify the corresponding molecular mechanism of yellowhorn and other woody oil plants.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Yellowhorn (Xanthocera sorbifolia Bunge) is a unique woody oil tree species in China, belonging to Sapindaceae, and widely distributed in most areas of northern China (Liu et al. 2014). It has high economic value, e.g., the seeds are rich in oil (55–70%), unsaturated fatty acids account for 85–93%, with high antioxidant activity and nutritional value (Zhang et al. 2010, Ruan et al. 2016). In addition, yellowhorn has extremely high ecological value, being different from most energy tree species that can only survive on farmland. Owing to its powerful root system and strong environmental adaptability, yellowhorn can survive in barren, drought, cold, and even saline-alkali environments (Guo et al. 2013; Liu et al. 2013; Jin et al. 2020). Thus, it has attracted much attention in recent years. The Chinese government is also extremely concerned about such excellent tree species, and launched the yellowhorn Breeding Program as a “green environmental solution” to alleviate the energy crisis (Ebert Sustainability 2014), also promoting yellowhorn planting in desert, semi-arid, and arid areas as an effective measure to restore vegetation and prevent soil erosion, such as the demonstration base of one million mu of yellowhorn energy forest in Maigaiti county (Yao et al. 2013). When planted on barren land, yellowhorn susceptibility to various abiotic stresses increases, which affects their growth and development.
Yellowhorn would not only be subjected to environmental stresses such as drought, low temperature, heat, and saline-alkali (Jin et al. 2020; Lang et al. 2020), but also may be stressed by nutrient scarcity such as that of nitrogen. Nitrogen (N) is an important mineral element necessary for plant life, and the main nutrient and component of several macromolecules and signaling compounds (such as nucleic acids, proteins, and hormones) (Lin et al. 2011; Krapp 2015). It affects several aspects of plant growth and development, such as N and C distribution, root branching, leaf growth, and flowering time (Bi et al. 2007; Zhu et al. 2021). In plants, Inorganic N uptake occurs mainly through nitrate and ammonium transporters, which transform nitrate and ammonium N absorbed by roots into amino acids that are subsequently transported to leaves (Wang et al. 2012; Yang et al. 2019; Luo et al. 2015; Black et al. 2002). Although plants absorb N mainly from the soil, soil N is usually limited, especially in barren areas with extremely low available N concentration, such as deserts and other places (Liu et al. 2020). Therefore, lack of nitrogen will seriously limit the growth and development of plants (Chen et al. 2008). N deficiency leads to a decrease in plant growth rate, makes plant leaves yellow, and eventually reduces leaf size and photosynthetic rate (Nunes-Nesi et al. 2010; Babst et al. 2019). In addition, several studies emphasize that the effective N concentration in the soil affects the ability of plants to respond to biological stress (Ballini et al. 2013). Thus, yellowhorn grown in barren areas such as deserts may face a variety of abiotic stresses, making necessary to analyze the molecular mechanism of yellowhorn under low-N stress.
Currently, yellowhorn research mainly focuses on growth (Yao et al. 2009), cultivation management, oil composition and extraction (Shen et al. 2019), and conventional breeding (Liu et al. 2014). Transcriptomic studies have been conducted to shed light on different aspects of yellowhorn biology (Liu et al. 2013; Wang et al. 2018). The recent release of its whole genome sequence (Quanxin et al. 2019, Qiang et al. 2019) should help investigate the underlying molecular mechanisms. So far, several transcriptome studies have been carried out on yellowhorn, e.g., Liu used the buds, leaves, flowers, and seeds of yellowhorn for transcriptome analysis, and then studied and identified the genes related to yellowhorn oil accumulation (Liu et al. 2013). Furthermore, Li sequenced the transcriptome of seeds with different oil content, and found that the seeds with high oil content were mainly consequence of fatty acid biosynthesis and carbon supply increase (Li et al. 2020). However, there are few studies on the molecular mechanisms related to abiotic stress in yellowhorn. At present, only Jin et al. have studied transcriptome sequencing of yellowhorn seedlings treated with NaCl, ABA and low temperature, and the results indicate that yellowhorn may form a specific molecular defense system against a variety of abiotic stresses (Jin et al. 2020). So far, there is no relevant report on the molecular defense mechanism of yellowhorn under low-nitrogen stress.
RNA-Seq is a widely used tool for studying molecular regulation mechanisms and finding stress response genes (Que et al. 2014; Yang et al. 2018a, b), and has been used on several plants to explore the molecular mechanism under low-N stress (Lu et al. 2019; Liu et al. 2020). Quan et al. revealed the molecular mechanism of two kinds of Tibetan wild barley under slow N deficiency, and identified candidate genes related to low-N tolerance (Quan et al. 2019). Sugarcane is one of the most important cash crops worldwide, and its regulation and metabolic pathways under low-N conditions have also been preliminarily revealed (Yang et al. 2019) Yang et al. used transcriptomics to reveal changes in secondary metabolite (flavonoids and caffeine, among others) synthesis pathways in tea under low-nitrogen conditions (Yang et al. 2018a, b). Liang’s results indicated that anthocyanins, the secondary metabolites accumulated in Arabidopsis under low-nitrogen stress, contribute to its resistance to low-N stress (Liang and He 2018). Meanwhile, Wu et al. showed that jasmonic acid (JA) signaling would mediate large-scale systemic changes in nitrogen uptake and distribution in rice (Wu et al. 2018). In conclusion, plants have certain similarities in resistance to low-nitrogen stress, but each plant also has its own unique mechanism of low-nitrogen tolerance, so it is necessary to study the mechanism of low nitrogen tolerance in yellowhorn.
In seed plants, the seedling stage is the most fragile in the life cycle (Jin et al. 2020), and seedling traits phenotyping would be a helpful strategy for pre-selection of plants with strong adaptability to the field stresses (Avramova et al. 2016). Therefore, to fill the knowledge gap regarding yellowhorn molecular defense mechanisms under low-N stress, we used RNA-Seq technology to sequence the transcriptome of yellowhorn seedlings under different N stress conditions.
Methods
Experimental design
Yellowhorn seeds were soaked in water at 50 °C for 3 days. Then, seeds were wrapped with cotton cloth soaked in water to keep them moist until they germinated. Germinated seeds were planted in sand and grown under a 16-h/8-h light cycle, 60% humidity, and 28 °C supplemented with 1/2 Hoagland solution every other day.
The experiment is as follows: yellowhorn which grows for 2 months and is similar in size, is divided into three groups and treated with three different concentrations of nutrient solution. The nutrient solution was 945 mg/l calcium sulfate, 506 mg/l potassium sulfate, 80 mg/l ammonium nitrate, 136 mg/l dipotassium hydrogen phosphate, 493 mg/l magnesium sulfate, 5.56 g/l ferrous sulfate heptahydrate, 7.46 g/l EDTA, 0.83 mg/l potassium iodide, 6.2 mg/l boric acid, 22.3 mg/l manganese sulfate, 8.6 mg/l zinc sulfate, 0.25 mg/l sodium molybdate, 0.025 mg/l copper sulfate, and 0.025 mg/l cobalt chloride. Ammonium nitrate was used as the sole N source, at 0, 16, and 160 mg/l (NN, LN, and CK, respectively) (The general standard of cultivated soil is that the nitrogen concentration is 150–200 mg/Kg.). During 15 days, water plants with 400 ml of nutrient solution every three days.
Physiological index determination and phenotype observation
After 15 days of treatment, stressed (LN, NN) and control (CK) seedlings were collected for the following measurements, which were performed with three independent biological replicates if not otherwise specified:
Relative plant fresh weight determination and phenotypic observation: The average fresh weights and plant height, root length, and lateral root length of seedling in LN, NN and CK groups were measured calculated using three randomly chosen seedlings as independent biological replicates for each group.
Flavonoids and procyanidins: About 0.2 g of fresh yellowhorn leaves were added to 1 ml of 80% methanol, ground thoroughly, and leached overnight. Then, the sample was centrifuged at 8000 × g for 10 min, and the supernatant was collected. Blow the supernatant N to the water phase, 1 ml petroleum ether was added into the water phase, fully shaken, let stand for layering, and the upper petroleum ether phase was discarded. After adjusting the pH to 2.5–3.0 with about 20 μl 2 M HCl solution, it was extracted twice with 1 ml of ethyl acetate, the ethyl acetate phases combined, and the ethyl acetate phase N was blown to dryness. Then, 1.5 ml PBS buffer was added to redissolve, and measurements were made after membrane filtration. Flavonoids and proanthocyanidins were determined using flavonoid and proanthocyanidin kits (Nanjing Jiancheng Institute of Bioengineering).
Determination of plant hormone content: Brassinolide (BIO(Tw)-E07(Pl)-00016, Shanghai Tongwei reagent), jasmonic acid (Ml077234, Shanghai Institute of Enzyme-Linked Biology), abscisic acid (Ml077235, Shanghai Institute of Enzyme-linked Biology) and salicylic acid (Ml077224, Shanghai Institute of Enzyme-linked Biology) were determined by enzyme-linked immunosorbent assay (Sharipova et al. 2016).
Determination of soluble protein content: We used the method described by Bradford (Coomassie brilliant blue staining) to determine the content of soluble protein with bovine serum albumin as the standard (Bradford 1976).
Determination of chlorophyll content: The chlorophyll content is determined by spectrophotometry (Costache M A. 2012).
Sample collection and RNA extraction
RNA samples were prepared from 9 yellowhorn leaf samples (3 treatments × 3 biological replicates) (The leaves of yellowhorn are pinnate compound leaves). The second compound leaves of seedlings were collected at each time point, which were frozen in liquid nitrogen and stored in a refrigerator at − 80 °C for further analysis. The top tender leaves of yellowhorn were selected, frozen in liquid N, and stored at − 80 °C. According to the manufacturer’s instructions, total RNA was extracted using a total RNA extractor (Trizol) kit (B511311, Chinese laborers) and treated with DNase I without RNase to remove genomic DNA contamination. RNA integrity was evaluated with 1.0% agarose gel. Then, RNA quality and quantity were evaluated using spectrophotometer (IMPLEN, CA, USA) and Agilent 2100 bioanalyzer (Agilent Technologies, CA, USA). The transcriptional group was sequenced using the method described by Wang et al. (Wang et al 2016). In short, mRNA was purified from total RNA using poly-T oligo-attached magnetic beads. Fragmentation was carried out using divalent cations under elevated temperature in NEB Next First Strand Synthesis Reaction Buffer (5X). First strand cDNA was synthesized using random hexamer primer and M-MuLV Reverse Transcriptase. Second-strand cDNA synthesis was subsequently performed using DNA Polymerase I and RNase H. Remaining overhangs were converted into blunt ends via exonuclease/polymerase activities. After adenylation of 3’ ends of DNA fragments, NEB Next Adaptor with hairpin loop structure were ligated to prepare for hybridization. To select cDNA fragments of preferentially 240 bp in length, the library fragments were purified with AMPure XP system (Beckman Coulter, Beverly, USA). Then, 3 μl USER Enzyme (NEB, USA) was used with size-selected, adaptor-ligated cDNA at 37 °C for 15 min followed by 5 min at 95 °C before PCR. Then PCR was performed with Phusion High-Fidelity DNA polymerase, Universal PCR primers and Index (X) Primer. At last, the purified PCR product was quantified using the Agilent Bioanalyzer 2100 system. After cluster generation, the final cDNA library was sequenced on Illumina HiSeq platform.
Differential expression analysis
DESeq2 (version 1.12.4) was used to determine the differentially expressed genes (DEGs) between two samples. If q-Value < 0.001 and | FoldChange |> 2, the gene was considered to be significantly differentially expressed. When the normalized expression of the gene was zero between the two samples, its expression value was adjusted to 0.01 because 0 cannot be plotted on the logarithmic graph. If the normalized expression of a certain gene in both libraries was less than 1, it was excluded from further differential expression analysis.
Functional analysis of differentially expressed genes
The unigene sequence assembled and screened was compared with Nr, Nt, Swiss-Prot, GO, KEGG, and other databases by BLAST, and the comparison results with Nr, Nt, Swiss-Prot and the functional annotation of GO, and KEGG pathway were obtained with E-value < 10 as threshold. In addition, Pfam was searched by Hmmscan to obtain unigenes’ functional domain and protein family annotation information. Functional enrichment analyses included Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) to determine which DEG was significantly enriched in GO or metabolic pathways. GO is an international standard classification system for gene function, while KEGG is a public database of metabolic pathway data. KEGG pathway analysis can identify significantly abundant metabolic pathways or signal transduction pathways in DEG. With a P-value ≤ 0.05, GO items and KEGG pathway were considered to have significant changes.
Quantitative reverse transcription PCR verification
To verify the accuracy of RNA-Seq sequencing, eight DEGs were randomly selected for quantitative reverse transcription PCR (qRT-PCR) verification. Real-time quantitative PCR was performed using One Step RT-qPCR Kit (B639277, Chinese GENERATORS) according to the manufacturer's instructions. The EF-1α gene was used as reference gene and gene-specific primers of the selected DEGs were designed using PRIMER5 (Table S7). qRT-PCRs were performed on an ABI Step One plus platform with three reactions for each biological replicate and a total of three biological replicates for each gene.
Results
Effects of nitrogen stress on yellowhorn growth and development
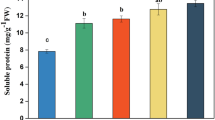
The growth status of yellowhorn under different N conditions was evaluated to assess its growth change under N deficiency. After 15 days of treatment, compared to those in CK, yellowhorn plant height decreased by 5.7 and 14.66% after LN and NN treatment (Table 1), respectively, and fresh weight also decreased by 4.38 and 14.93%, respectively. Contrastingly, yellowhorn root growth and development under N deficiency was stronger than those in CK (Table 1) (Fig. S1). Root length was 21.86% and 38.89% higher than that of CK under LN and NN treatments, respectively, and the number of lateral roots also significantly increased by 52.36 and 50.07%, respectively, compared to that of CK. The root development of yellowhorn may be caused by low-N stress. In this experiment, the soluble protein content of yellowhorn increased after treatment. Compared with CK, the soluble protein of yellowhorn increased by 11.32 and 38.69% after LN and NN treatments, respectively (Table 1), indicating that LN and NN treatments affected the Soluble protein content of plants. Under LN treatment, the contents of chlorophyll a, chlorophyll b and total chlorophyll decreased by 27.83%, 30.5% and 23.38% respectively. Under NN treatment, the contents of chlorophyll a, chlorophyll b and total chlorophyll decreased by 44.77%, 36.61% and 38.33%, respectively. (Table 1). In a word, these results show that low nitrogen and nitrogen deficiency stress have affected the growth and development of yellowhorn.
Illumina sequencing and de novo transcriptome assembly
On the 15th day after treatment, an appropriate amount of yellowhorn leaves were obtained under CK, LN, and NN conditions to reveal the molecular mechanism of yellowhorn response to long-term low-N stress. The RNA integrity number (RIN) of all components is greater than 7.5 (Table 2). Illumina HiSeq™ 2000 was used to sequence yellowhorn seedlings treated with three N concentrations, and nine transcriptome libraries were constructed. After removing low-quality readings, 74.27 Gb clean data were obtained, with clean data reaching 6.86 Gb for each sample and Q30 base percentage above 95.33%. A total of 242,816 unigenes were obtained, with lengths ranging from 200 to over 2000 bp. The average unigene length was 464.84 bp and the single gene splicing length (N50) was 545 bp (Table 3). Clean data for each sample reached 5.90 Gb, and the percentage of Q30 bases was 97.36% or higher (Table S1), which indicated that sequencing data was suitable for gene expression analysis. Additionally, these data can greatly expand the transcriptome resources of yellowhorn in unknown gene mining, molecular genetics, functional genomics and other fields.
Functional annotation and classification
Annotation results: At least 205,824 single genes were annotated in each database, accounting for 84.77% of the total enriched single genes. A total of 187,617 unigenes (77.27%) were annotated in NT database. There were 127,305 (52.43%) and 105,875 (43.6%) unigenes annotated in GO and NR database, respectively. Moreover, 10,088 unigenes were annotated in KEGG database, which is frequently used in plants (Table S2). To study the sequence conservation between yellowhorn and other plant species, we analyzed the species distribution of single gene data set by comparing the sequence with the NR database. The species distribution corresponding to the sequence is shown in Fig. 1. The number of sequences identical to Arabidopsis thaliana was large, reaching 29,531, with 8619 for Citrus sinensis, 5809 for Citrus clementina, 4334 for Phaseolus vulgaris and 2163 for Theobroma cacao.

Hot-species distribution of unigenes in yellowhorn leaf transcription. Different kinds of plants are color coded
The possible functions of unigene in yellowhorn leaves were classified in the GO database. According to the homology comparison results, 127,305 unigenes were annotated in 71 functional groups in the GO database. Among them, a total of 22, 27, and 22 subgroups were annotated in the molecular function, biological process, and cellular component grouping, respectively. In the molecular functional grouping, the subgroups with a large number of unigenes were binding, catalytic activity, and transporter activity (78,134, 62,323, and 8716, respectively). Considering biological processes, there were 86,021, 72,740, and 64,337 unigenes in the cellular, metabolism, and single-organic processes subgroups, respectively, while for cell composition, cells, cell parts, and organelles (Figure S4) were the highest in unigene number (97,843, 97,698, and 73,472, respectively).
All unigenes were matched to 64,849 hits distributed in 25 functional groups in KOG databases (Figure S4). The three groups with the largest number of genes were signal transduction mechanism, general function prediction only, post-translation modification, protein turnover, and molecular modification (protein turn over, chaperones). As shown in Fig. 5, 14,811 unigenes were annotated in 23 channels. Among them, translation, signal transduction, carbohydrate metabolism, and transport and metabolism had a high count of annotated genes (Figure S5).
Analysis of differentially expressed genes under different nitrogen concentrations
Compared with CK, 2475 and 5636 unigenes in the LN group, and 2476 and 3007 unigenes in the NN group were up- and down-regulated, respectively (Fig. 2). To identify active biological pathways enriched with DEGs in yellowhorn, the KEGG pathway database was searched. KEGG enrichment analysis results were shown in Figs. 3A and 4B, with the first 20 top-ranking pathways indicated by the smallest significant Q values. Up-regulated genes were the most enriched in plant hormone signal transduction, flavonoid biosynthesis, and phenylpropanoid biosynthesis. In addition, galactose metabolism, arginine, and proline metabolism, which are involved in regulating osmotic balance, and glutathione metabolism, which can produce and remove reactive oxygen species (ROS), were obviously enriched. In yellowhorn, most genes in these metabolic pathways were up-regulated with lack of N whether in LN or NN treatment group.

Venn diagram of differentially expressed genes regulated by nitrogen concentration, i.e., low-N (LN) and no N (NN) vs control (CK). A up-regulated genes; B down-regulated genes. DESeq2 (version 1.12.4) was used to determine the differentially expressed genes (DEGs) between two samples. If q-Value < 0.001 and | FoldChange |> 2, the gene was considered to be significantly differentially expressed

Bubble chart of up-regulated KEGG genes in different treatments. A low nitrogen vs. control; B no N vs. control. Color represents the qvalue value, and the size of bubbles represents the number of genes

Analysis of plant hormone content under low nitrogen stress. A ABA content in leaves; B BR content in leaves; C SA content in leaves; D JA content in leaves. Perform three biological repetitions. The data were analyzed by one-way ANOVA. The different representations of lowercase letters have statistical significance (P < 0.05)
Yellowhorn genes and pathways related to low-nitrogen response
To explore the low-N response mechanism in yellowhorn, we examined transcriptional changes of potential low-N response genes in LN and NN. We found that genes related to brassinosteroid (BR), JA, and salicylic acid (SA) signal transduction were significantly up-regulated (Tables S3 and S4). The contents of brassinosteroid (BR), jasmonic acid and salicylic acid (SA) in yellowhorn leaves under low-nitrogen stress were also significantly higher than those in the control group (Fig. 5). Additionally, phenylpropanoid biosynthesis was also significantly enriched. As a downstream metabolic pathway of phenylpropanoid biosynthesis, its accumulation was also apparent. Moreover, 33 and 32 DEGs were identified as reactive oxygen scavenging genes (Tables S5 and S6), belonging to glutathione metabolism, and 47 and 33 osmotic potential regulation genes (Tables S5 and S6) were metabolized by arginine, proline, and galactose in LN and NN transcriptase, respectively. The number of related genes identified in the NN transcriptome was significantly lower than that in the LN transcriptome.

Correlation between RNA-Seq expression profile and qRT-PCR results. Their relative expression levels were measured and calculated using EF-1α-F as the internal reference gene. Three biological replicates were performed. The ratios were log2 transformed, > 0 means down-regulated, and < 0 indicates up-regulated
Real-time qPCR validation
To validate the transcriptome data sets, real-time qPCR technology was applied to analyze transcriptional levels of 8 randomly selected genes from transcriptome data sets. Their relative expression levels were measured and calculated using EF-1α-F as the internal reference gene. These eight genes included: leucoanthocyanidin reductase-like, Basic helix–loop–helix DNA-binding family protein, anthocyanidin synthase, chalcone isomerase, hypothetical protein CICLE v10005200mg, NRT1, hypothetical protein CICLE_v10032858mg and abscisic-aldehyde oxidase isoform X1. RT-PCR results confirmed that the transcription changes of these 8 genes were comparable with the fold-changes observed in our transcriptome analysis (Fig. 5).
Determination of brassinosteroid (BR), jasmonic acid and salicylic acid (SA) contents
Transcriptome data showed that the expression of many genes in plant hormone signal transduction pathway was up-regulated in the condition of nitrogen deficiency. The data showed that the contents of brassinosteroid, jasmonic acid and salicylic acid in the treatment group were significantly higher than those in the control group, although there were differences in abscisic acid content, but the differences were not significant. The levels of brassinosteroid in LN and NN groups were 14.48 ng/ml and 23.01 ng/ml, respectively. The jasmonic acid content in LN and NN groups was 115.79 and 193.83 ng/ml, respectively. The contents of salicylic acid in LN and NN groups were 148.83 and 220.1 ng/ml, respectively (Fig. 4).
Determination of total flavonoids and procyanidins
Transcriptome data showed that yellowhorn was active in flavonoid synthesis pathway under N deficiency, and most genes were up-regulated. Data showed that total flavonoid and proanthocyanidin contents in the LN treatment group were significantly higher than those in CK, but those in the NN treatment group were lower than those in CK. The total flavonoid content in the LN group was 4.62 mg/g, while that in NN was only 1.32 mg/g. The content of proanthocyanidins was 0.98 and 0.11 mg/g in the LN and NN group, respectively (Fig. 6).

Contents of total flavonoids and procyanidins under low-N (LN), N-free (NN), and control (CK) treatments. A Total flavonoid content in leaves; B Proanthocyanidins content in leaves. Three biological replicates were performed. Asterisk indicates statistically significant difference (P < 0.05)
Discussion
N is an important essential macronutrients for plants, and its lack in soil will inhibit crop growth (Ologundudu and Adelusi 2013). Nitrates, ammonium, and various amino acids can be used and absorbed from the soil by plant roots (Xuan et al. 2017; Rentsch et al. 2007; Wang et al. 2020a, b). In this study, we controlled the concentrations of nitrate and ammonium in the soil, combined with transcriptome sequencing technology to analyze the mechanism of tolerance to low nitrogen. Compared with CK, single genes in LN and NN groups were down-regulated, suggesting that growth inhibition under nitrogen deficiency was caused by the damage of several genes. Meanwhile, several up-regulated genes may play an important role in yellowhorn resistance to low-N stress.
In plant growth and resistance to abiotic stress, phenylpropane biosynthesis is an important pathway (Wang et al. 2011; Li et al. 2020), with a powerful antioxidant effect, mainly because several antioxidants (simple or complex phenols) come from it (Dixon and Paiva 1995). Depending on the stress type, different phenolic compounds can be induced by up-regulating corresponding biosynthetic enzyme genes (Dixon and Paiva 1995), with the phenylalanine pathway being important to promote the synthesis of plant secondary metabolites, e.g., phenylalanine ammonia lyase (PAL) converts phenylalanine into monohydric phenol and dihydric phenol, which are substrates of polyphenol oxidase (PPO) (Tomás-Barberan et al. 1997). In general, owing to stress caused by various abiotic factors (high temperature and cold, UV-B radiation, physical damage, among others), PAL activity increases and several phenols accumulate (Solecka and Kacperska 2003; Sgarbi et al. 2003), e.g., PAL activity of barley seedlings increased phenolic substance accumulation under NaCl treatment (Wang et al. 2020a, b), and the expression of PAL-related genes was increased and total phenol and flavonoid synthesis was in maize under high temperature stress (Wu et al. 2020). Additionally, PAL is one of the main indicators of environmental pressure in different plant tissues (Sánchez-Ballesta et al. 2000). Numerous studies in wheat, rice, and maize have shown that nitrogen starvation can rapidly induce ROS outbreaks (Lian et al. 2006, Trevisan et al. 2012, Hsieh et al. 2018). In this study, we found that eight genes related to propyl benzene metabolism were up-regulated in the experimental group, including PAL (Trinity _ DN 70437 _ C2 _ G6, Trinity _ DN 73556 _ C3 _ G1, TRINITY_DN74117_c0_g2), caffeoyl-COA O-methyltransferase (TRINITY_DN73937_c1_g1), 8-hydroxygeraniol dehydrogenase-like (TRINITY_DN73343_c1_g1), cytochrome P450 (Trinity _ DN 73127 _ C1 _ G1), caffeoylshikimate esterase (Trinity _ DN 76482 _ C2 _ G1), and aldehyde dehydrogenase family 2 member C4 (Trinity _ DN 66,962 _ C0 _ G1). These results indicate that phenylalanine metabolism may affect the synthesis of phenolic compounds under low-N conditions, participating in yellowhorn resistance to low-N stress under low-N conditions.
Plant growth mainly depends on primary metabolism, while plant defense especially depends on the synthesis of secondary metabolites, such as flavonoids. Flavonoid is one of the main downstream branches of the phenylalanine pathway, which helps plants to resist stress (Li et al. 2020). Flavonoids have several functions in plants, e.g., ultraviolet (UV) damage protection agents and antitoxins of pathogens, reducing oxidative damage (Rajendran et al. 2014), among others, and their concentrations are usually increased owing to such factors, e.g., ultraviolet UV-B radiation promotes flavonoid synthesis in ginkgo biloba leaves (Zhao et al. 2020). In plants, the main biosynthetic pathway of flavonoids is shikimic acid pathway, which provides a common precursor for phenylalanine synthesis, and is not only used for amino acid and protein synthesis, but also for that of secondary metabolites (Taiz and Zeiger 2002, Deng et al. 2019). Moreover, several plants induce flavonoid accumulation in the absence of N, e.g., Potamogeton amplifolius, Nuphar advena, and Arabidopsis (Cronin and Lodge 2003; Lillo et al. 2008). CHS is a key enzyme in flavonoid synthesis and can produce intermediates for the synthesis of all flavonoids, and when CHS reactions are strongly constrained, the production of anthocyanin and nearly all other flavonoids is effectively eliminated (Clark and Verwoerd 2011). In this study, we found that CHS expression was affected by the concentration of mineral elements, with CHS genes (Trinity _ Dn67486 _ C0 _ G3, Trinity _ Dn69687 _ C2 _ G18) being up-regulated in all treatments in the experiment. Meanwhile, the contents of total flavonoids increased significantly in the LN group, while that in the NN group decreased significantly compared to that in CK. Therefore, it can be inferred that flavonoids cannot be synthesized when N concentration is too low.
Anthocyanins, a branch of flavonoids, have several biological functions in higher plants and are one of the most widely studied compounds (Tanaka et al. 2008; Qi et al. 2020). Anthocyanins not only provide colors to plants, but also reduce oxidative damage under biological or abiotic stress (Jaakola 2013). Anthocyanin biosynthesis is also affected by external factors, such as light and low temperature, among others (Zhao et al. 2017, Xie et al. 2018). Anthocyanin synthesis would probably be indicative of the reduction of photosynthetic capacity to balance the lack of N. Recent studies have found that low-N conditions also promote anthocyanin synthesis in plants (Deng et al. 2018; Meng et al. 2020), and our results were consistent with previous findings. And anthocyanin synthesis would probably be indicative of the reduction of photosynthetic capacity to balance the lack of N. We found that anthocyanin content was not consistent with the expression level of structural genes under NN treatment, i.e., the genes related to anthocyanin synthesis were up-regulated but the content of anthocyanin was lower than that of CK. Therefore, we hypothesized that the decrease of yellowhorn anthocyanin content in N-free condition was mainly caused by the deficiency of its precursor, rather than by gene regulation.
In this study, glutathione, arginine, proline, and galactose metabolism were also enriched under hypo-N stress. Glutathione reductases are antioxidant enzymes that can effectively scavenge free radicals and protect plant organisms (Begara Morales et al. 2015). Proline is a protective osmotic-regulatory agent, and at high levels can reduce the water potential and enhance ROS removal by antioxidants as in peas and Stipa purpurea (Sánchez et al. 1998; Yang et al. 2015). In addition, the up-regulated expression of some genes in arginine metabolism was found in this study, and we believe that this situation may be a way to promote proline biosynthesis. Galactose, a soluble sugar, also plays an important role in the plant osmotic regulation, e.g., poplar can enhance the absorption of water by cells through the accumulation of soluble sugar (Jia et al. 2016). Our results showed that glutathione, arginine and proline, and galactose metabolism were the molecular bases allowing yellowhorn to resist low-N stress.
Plant hormones play an important role in plant growth, development, and response to abiotic stress (Bogatek and Gniazdowska 2007; Verma et al. 2016). A large number of studies have shown that plant hormones are important components of nitrogen stress signal transduction (Sultana et al 2020; Kiba et al. 2011). Plants usually respond to external stimuli by activating signaling cascades which modify downstream gene expression patterns and finally realize physiological and metabolic adaptations (Osakabe et al. 2014). Previous studies have found that plants may induce the synthesis of abscisic acid, auxin, gibberellin, ethylene and cytokinin when they are nitrogen deficient (Ma et al. 2020; Kang et al. 2019; Kiba et al. 2011; Merchante et al. 2013). These hormones, such as ethylene, GAs, ABA, and cytokinin, may be involved in the regulation of root growth and development through interactions with auxin (Hu et al. 2020; Kong et al. 2021). In this study, we found that the lower the N concentration, the more developed the root system of yellowhorn. Auxin, gibberellin, ethylene and cytokinin-related genes were not up-regulated in our study, and ABA signal transduction-related genes were only up-regulated in the absence of nitrogen, which was also confirmed by our determination of ABA content in plants. As BR is a major growth-promoting steroid hormone that regulates cell elongation and division and plays an important role in root growth and development (Belkhadir and Jaillais 2015; Vragovic et al. 2015). Therefore, we believed that BR regulated root growth and development of yellowhorn under low-nitrogen stress, and we also confirmed the increase of BR content in plants. Ethylene is not only involved in resisting N hunger, but also plays an important role in anthocyanin synthesis induced by N deficiency (Zhao et al. 2015). In this study, ethylene was not induced under low-N stress, but the cytochrome P450 gene involved in its signal transduction pathway and genes related to proanthocyanidin synthesis were highly expressed (Tiwari et al. 2020). Moreover, we found that low N significantly induced JA signaling pathway and verified that JA content increased in plants. Since cytochrome P450 is also involved in jasmonic acid signal transduction, we speculated that jasmonic acid may be involved in anthocyanin synthesis under low-nitrogen stress. Previous studies have shown that jasmonic acid and salicylic acid are also related to the response to low-nitrogen conditions (Lv et al. 2021, Sun et al. 2020). JA signaling may function by regulating sugar biosynthesis, amino acid biosynthesis, and absorption of NH4+ (Sun et al. 2020). However, the activation of JA or SA-mediated defense responses usually comes at the expense of plant growth (Karasov et al 2017; Campos et al. 2016). Under low-nitrogen stress, compared to those in CK, yellowhorn plant height decreased by 5.7 and 14.66% after LN and NN treatment, respectively, and fresh weight also decreased by 4.38% and 14.93%, respectively. It also seems to confirm that SA and JA signals synergically mediate yellowhorn's resistance to low-nitrogen stress. Therefore, we believe that JA, SA, and BR constitute a highly complex coordinated signal transduction mechanism in yellowhorn.
Conclusion
In summary, we first described yellowhorn physiological response to low-N stress. The transcriptome data sets of different yellowhorn genetic materials were obtained using RNA-Seq technology to explore key genes and metabolic pathways associated with low N. Our results indicated that JA, SA, and BR signaling are activated in yellowhorn under low-N stress. Up-regulated genes involved in the synthesis of phenylpropyl, flavones, and anthocyanins, as well as some genes involved in galactose, arginine and proline, and glutathione metabolism were up-regulated to help yellowhorn fight N hunger. These results expand our understanding of the N-starvation resistance mechanism in yellowhorn and provide reference for the breeding of low-N tolerant germplasm in the future.
Author contribution statement
XL, JZ, HJ, and CJ conceived the idea. XL, CJ, and JZ carried out the laboratory work. XL, and CJ performed the data analysis. XL wrote the manuscript, and HJ completed the final revision. All authors have read and approved the final manuscript.
Data availability
The original data are stored in NCBI database, with SRA registration number: PRJNA726832 and GEO registration number: GSE223332. (https://www.ncbi.nlm.nih.gov/sra/PRJNA726832).
Abbreviations
- DEGs:
-
Differentially expressed genes
- FPKM:
-
Fragments per kb per million fragments
- GO:
-
Gene ontology
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- qRT-PCR:
-
Quantitative real-time PCR
- RNA-seq:
-
RNA sequence
- ABA:
-
Abscisic acid
- SA:
-
Salicylic acid
- JA:
-
Jasmonates
- BR:
-
Gibberellin
- ROS:
-
Scavenging reactive oxygen species
- CK:
-
Control group
References
Babst AB, Gao F, Lucia MAG et al (2019) Three NPF genes in Arabidopsis are necessary for normal nitrogen cycling under low nitrogen stress. Plant Physiol Biochem 143:1–10. https://doi.org/10.1016/j.plaphy.2019.08.014
Ballini E, Nguyen TT, Morel JB (2013) Diversity and genetics of nitrogen-induced susceptibility to the blast fungus in rice and wheat. Rice 6:32. https://doi.org/10.1186/1939-8433-6-32
Begaramorales JC, Sánchezcalvo B, Chaki M et al (2015) Differential molecular response of monodehydroascorbate reductase and glutathione reductase by nitration and S-nitrosylation. J Experim Bot 66(19):5983–5996. https://doi.org/10.1093/jxb/erv306
Belkhadir Y, Jaillais Y (2015) The molecular circuitry of brassinosteroid signaling. New Phytol 206(2):522–540. https://doi.org/10.1111/nph.13269
Bi YM, Wang RL, Zhu T et al (2007) Global transcription profiling reveals differential responses to chronic nitrogen stress and putative nitrogen regulatory components in Arabidopsis. BMC Genom 8:281. https://doi.org/10.1186/1471-2164-8-281
Black BL, Fuchigami LH, Coleman GD (2002) Partitioning of nitrate assimilation among leaves, stems and roots of poplar. Tree Physiol 22(10):717–724. https://doi.org/10.1093/treephys/22.10.717
Bogatek R, Gniazdowska A (2007) ROS and phytohormones in plant–plant allelopathic interaction. Plant Signal Behav 2:317–318. https://doi.org/10.4161/psb.2.4.4116
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein dye binding. Anal Biochem 72(1–2):248–254. https://doi.org/10.1016/0003-2697(76)90527-3
Campos ML, Yoshida Y, Major IT et al (2016) Rewiring of jasmonate and phytochrome B signaling uncouples plant growth-defense tradeoffs. Nat Commun 7:12570. https://doi.org/10.1038/ncomms12570
Chen Y, Ruberson JR, Olson DM (2008) Nitrogen fertilization rate affects feeding, larval performance, and oviposition preference of the beet armyworm, Spodoptera exigua, on cotton. Entomol Exp Appl 126:244–255. https://doi.org/10.1111/j.1570-7458.2007.00662.x
Clark ST, Verwoerd WS (2011) A systems approach to identifying correlated gene targets for the loss of colour pigmentation in plants. BMC Bioinformatics. 12(1):343. http://www.biomedcentral.com/1471-2105/12/343
Costache MA, Campeanu G, Neata G (2012) Studies concerning the extraction of chlorophyll and total carotenoids from vegetables. Romanian Biotechnol Lett 17(5):7702–7708
Cronin G, Lodge DM (2003) Effects of light and nutrient availability on the growth, allocation, carbon/nitrogen balance, phenolic chemistry, and resistance to herbivory of two freshwater macrophytes. Oecologia 137:32–41. https://doi.org/10.1007/s00442-003-1315-3
Deng B, Li YY, Lei G, Liu GH (2018) Effects of nitrogen availability on mineral nutrient balance and flavonoid accumulation in Cyclocarya paliurus. Plant Physiol Biochem 135:111–118. https://doi.org/10.1016/j.plaphy.2018.12.001
Deng B, Li YY, Lei G et al (2019) Effects of nitrogen availability on mineral nutrient balance and flavonoid accumulation in Cyclocarya paliurus. Plant Physiol Biochem 135:111–118. https://doi.org/10.1016/j.plaphy.2018.12.001
Dixon RA, Paiva NL (1995) Stress-induced phenylpropanoid metabolism. Plant Cell 7:1085–1097. https://doi.org/10.1105/tpc.7.7.1085
Ebert S (2014) Potential of underutilized traditional vegetables and legume crops to contribute to food and nutritional security. Income More Sustain Production Syst Sustain 6(1):319–335. https://doi.org/10.3390/su6010319
Guo HH, Wang TT, Li QQ et al (2013) Two novel diacylglycerol acyltransferase genes from Xanthoceras sorbifolia are responsible for its seed oil content. Gene 527(1):266–274. https://doi.org/10.1016/j.gene.2013.05.076
Hsieh PH, Kan CC, Wu HY et al (2018) Early molecular events associated with nitrogen deficiency in rice seedling roots. Sci Rep 8(1):1–23. https://doi.org/10.1038/s41598-018-30632-1
Hu S, Zhang M, Yang Y et al (2020) A novel insight into nitrogen and auxin signaling in lateral root formation in tea plant [Camellia sinensis (L.) O. Kuntze]. BMC Plant Biol 20:232. https://doi.org/10.1186/s12870-020-02448-7
Jaakola L (2013) New insights into the regulation of anthocyanin biosynthesis in fruits. Trends Plant Sci 18(9):477–483. https://doi.org/10.1016/j.tplants.2013.06.003
Jia J, Li S, Cao X et al (2016) Physiological and transcriptional regulation in poplar roots and leaves during acclimation to high temperature and drought. Physiol Plant 157:38–53. https://doi.org/10.1111/ppl.12400
Jin H, Zou J, Li L et al (2020) Physiological responses of yellow-horn seedlings to high temperatures under drought condition. Plant Biotechnol Rep 14:111–120. https://doi.org/10.1007/s11816-019-00590-9
Kang GZ, Wu YF, Li G et al (2019) Proteomics combined with BSMV-VIGS methods identified some N deficiency-responsive protein species and ABA role in wheat seedling. Plant Soil 444(1):177–191. https://doi.org/10.1007/s11104-019-04260-1
Karasov TL, Chae E, Herman JJ, Bergelson J (2017) Mechanisms to mitigate the trade-off between growth and defense. Plant Cell 29:666–680. https://doi.org/10.1105/tpc.16.00931
Kiba T, Kudo T, Kojima M, Sakakibara H (2011) Hormonal control of nitrogen acquisition: roles of auxin, abscisic acid, and cytokinin. J Experim Bot 62:1399–1409. https://doi.org/10.1093/jxb/erq410
Kong L, Zhang Y, Du W et al (2021) Signaling responses to N starvation: focusing on wheat and filling the putative gaps with findings obtained in other plants. A review. Front Plant Sci 12:656696. https://doi.org/10.3389/fpls.2021.656696
Krapp A (2015) Plant nitrogen assimilation and its regulation: a complex puzzle with missing pieces. Curr Opin Plant Biol 25:115–122. https://doi.org/10.1016/j.pbi.2015.05.010
Lang Y, Liu Z, Zheng Z (2020) Investigation of yellow horn (Xanthoceras sorbifolia Bunge) transcriptome in response to different abiotic stresses: a comparative RNA-Seq study. RSC Adv 10:6512–6519. https://doi.org/10.1039/c9ra09535g
Li Z, Xiong F, Guo W et al (2020) The root transcriptome analyses of peanut wild species Arachis correntina (Burkart) Krapov. & W.C. Gregory and cultivated variety Xiaobaisha in response to benzoic acid and p-cumaric acid stress. Genetic Resour Crop Evol 67:9–20. https://doi.org/10.1007/s10722-019-00859-6
Lian X, Wang S, Zhang J et al (2006) Expression profiles of 10,422 genes at early stage of low nitrogen stress in rice assayed using a cDNA microarray. Plant Mol Biol 60:617–631. https://doi.org/10.1007/s11103-005-5441-7
Liang J, He JX (2018) Protective role of anthocyanins in plants under low nitrogen stress. Biochem Biophys Res Commun 498(4):946–953. https://doi.org/10.1016/j.bbrc.2018.03.087
Lillo C, Lea US, Ruoff P (2008) Nutrient depletion as a key factor for manipulating gene expression and product formation in different branches of the flavonoid pathway. Plant Cell Environ 31:587–601. https://doi.org/10.1111/j.1365-3040.2007.01748.x
Lin ZH, Chen RB, Chen CS (2011) Research progress on physiological adaptability of plants to nitrogen deficiency. Hubei Agric Sci 50:4761–4764. https://doi.org/10.14088/j.cnki.issn0439-8114.2011.23.003
Liu Y, Huang Z, Ao Y et al (2013) Transcriptome analysis of yellow horn (Xanthoceras sorbifolia Bunge): a potential oil-rich seed tree for biodiesel in China. PLoS ONE 8(9):e74441. https://doi.org/10.1371/journal.pone.0074441
Liu Y, Huang Z, Ao Y et al (2014) Correction: transcriptome analysis of yellow horn (Xanthoceras sorbifolia Bunge): a potential oil-rich seed tree for biodiesel in China. PLoS ONE. https://doi.org/10.1371/annotation/803f7e8c-0718-41b4-8fc2-cc0b5f776da9
Liu DP, Li MX, Liu Y, Shi LX (2020) Integration of the metabolome and transcriptome reveals the resistance mechanism to low nitrogen in wild soybean seedling roots. Environ Experim Bot. https://doi.org/10.1016/j.envexpbot.2020.104043
Lu M, Chen M, Song J et al (2019) Anatomy and transcriptome analysis in leaves revealed how nitrogen (N) availability influence drought acclimation of Populus. Trees 33:1003–1014. https://doi.org/10.1007/s00468-019-01834-5
Luo J, Zhou J, Li H et al (2015) Global poplar root and leaf transcriptomes reveal links between growth and stress responses under nitrogen starvation and excess. Tree Physiol 35(12):1283–1302. https://doi.org/10.1093/treephys/tpv091
Lv X, Zhang Y, Hu L et al (2021) Low-nitrogen stress stimulates lateral root initiation and nitrogen assimilation in wheat: roles of phytohormone signaling. J Plant Growth Regul 40:436–450. https://doi.org/10.1007/s00344-020-10112-5
Ma N, Dong L, Lü W et al (2020) Transcriptome analysis of maize seedling roots in response to nitrogen-, phosphorus-, and potassium deficiency. Plant Soil 447:637–658. https://doi.org/10.1007/s11104-019-04385-3
Meng JX, Gao Y, Han ML et al (2020) In vitro anthocyanin induction and metabolite analysis in Malus spectabilis leaves under low nitrogen conditions. Hortic Plant J 6:5. https://doi.org/10.1016/j.hpj.2020.06.004
Merchante C, Alonso JM, Stepanova AN (2013) Ethylene signaling: simple ligand, complex regulation. Curr Opin Plant Biol 16:554–560. https://doi.org/10.1016/j.pbi.2013.08.001
Nunes-Nesi A, Alisdair RF, Mark S (2010) Metabolic and signaling aspects underpinning the regulation of plant carbon nitrogen interactions. Mol Plant 3(6):973–996. https://doi.org/10.1093/mp/ssq049
Ologundudu AF, Adelusi A (2013) Effect of nitrogen nutritional stress on some growth parameters of Zea mays L. And Vigna unguiculata (L.) walp. Not Sci Biol 5(1):79–85. https://doi.org/10.15835/nsb518362
Osakabe Y, Osakabe K, Shinozaki K, Tran LS (2014) Response of plants to water stress. Front Plant Sci 5:86. https://doi.org/10.3389/fpls.2014.00086
Qi Y, Wei H, Gu W et al (2020) Transcriptome profiling provides insights into the fruit color development of wild Lycium ruthenicum Murr. from Qinghai-Tibet Plateau. Protoplasma 258:33–34. https://doi.org/10.1007/s00709-020-01542-9
Quan X, Zeng J, Chen G et al (2019) Transcriptomic analysis reveals adaptive strategies to chronic low nitrogen in Tibetan wild barley. BMC Plant Biol 19:68. https://doi.org/10.1186/s12870-019-1668-3
Que YX, Su YC, Guo JL et al (2014) A global view of transcriptome dynamics during Sporisorium scitamineum challenge in sugarcane by RNA-Seq. PLoS ONE 9(8):e106476. https://doi.org/10.1371/journal.pone.0118445
Rajendran P, Rengarajan T, Nandakumar N et al (2014) Kaempferol, a potential cytostatic and cure for inflammatory disorders. Eur J Med Chem 86:103–112. https://doi.org/10.1016/j.ejmech.2014.08.011
Rentsch D, Schmidt S, Tegeder M (2007) Transporters for uptake and allocation of organic nitrogen compounds in plants. FEBS Lett 581:2281–2289. https://doi.org/10.1016/j.febslet.2007.04.013
Ruan CJ, Yan R, Wang BX et al (2016) The importance of yellow horn (Xanthoceras sorbifolia) for restoration of arid habitats and production of bioactive seed oils. Ecol Eng 99:504–512. https://doi.org/10.1016/j.ecoleng.11.073
Sánchez FJ, Manzanares M, de Andres EF et al (1998) Turgor maintenance, osmotic adjustment and soluble sugar and proline accumulation in 49 pea cultivars in response to water stress. Field Crops Res 59:225–235. https://doi.org/10.1016/S0378-4290(98)00125-7
Sanchez-Ballesta MT, Zacarias L, Granell A, Lafuente MT (2000) Accumulation of PAL transcript and PAL activity as affected by heat-conditioning and low-temperature storage and its relation to chilling sensitivity in mandarin fruits. J Agric Food Chem 48:2726–2731. https://doi.org/10.1021/jf991141r
Sgarbi E, Fornasiero RB, Lins AP, Bonatti PM (2003) Phenol metabolism is differentially affected by ozone in two cell lines from grape (Vitis vinifera L.) leaf. Plant Sci 165:951–957. https://doi.org/10.1016/S0168-9452(03)00219-X
Sharipova G, Veselov D, Kudoyarova G et al (2016) Exogenous application of abscisic acid (ABA) increases root and cell hydraulic conductivity and abundance of some aquaporin isoforms in the ABA-deficient barley mutant Az34. Ann Bot 118:777–785. https://doi.org/10.1093/aob/mcw117
Shen Z, Zhang K, Ao Y et al (2019) Evaluation of biodiesel from Xanthoceras sorbifolia Bunge seed kernel oil from 13 areas in China. J for Res 30:869–877. https://doi.org/10.1007/s11676-018-0683-9
Solecka D, Kacperska A (2003) Phenylpropanoid deficiency affects the course of plant acclimation to cold. Physiol Plant 119:253–262. https://doi.org/10.1034/j.1399-3054.2003.00181.x
Sultana N, Islam S, Juhasz A et al (2020) Transcriptomic study for identification of major nitrogen stress responsive genes in Australian bread wheat cultivars. Front Genet 11:583785. https://doi.org/10.3389/fgene.2020.583785
Sun L, Di DW, Li G et al (2020) Transcriptome analysis of rice (Oryza sativa L.) in response to ammonium resupply reveals the involvement of phytohormone signaling and the transcription factor OsJAZ9 in reprogramming of nitrogen uptake and metabolism. J Plant Physiol 246:153137. https://doi.org/10.1016/j.jplph.2020.153137
Taiz L, Zeiger E (2002) Plant physiology, 4th edn. Sinauer Associates Inc, Sunderlan
Tanaka Y, Sasaki N, Ohmiya A (2008) Biosynthesis of plant pigments: anthocyanins, betalains and carotenoids. Plant J 54:733–749. https://doi.org/10.1111/j.1365-313X.2008.03447.x
Tiwari JK, Buckseth T, Zinta R et al (2020) Transcriptome analysis of potato shoots, roots and stolons under nitrogen stress. Sci Rep 10:1152. https://doi.org/10.1038/s41598-020-58167-4
Toma´s-Barberan FA, Gil MI, Castan˜ er M, et al (1997) Effect of Selected Browning Inhibitors on Phenolic Metabolism in Stem Tissue of Harvested Lettuce. J Agric Food Chem 45:583–589. https://doi.org/10.1021/jf960478f
Trevisan S, Begheldo M, Nonis A, Quaggiotti S (2012) The miRNA-mediated post-transcriptional regulation of maize response to nitrate. Plant Signal Behav 7:822–826. https://doi.org/10.4161/psb.20462
Verma V, Ravindran P, Kumar PP (2016) Plant hormone-mediated regulation of stress responses. BMC Plant Biol 16:86. https://doi.org/10.1186/s12870-016-0771-y
Vragovic K, Sela A, Friedlander-Shani L et al (2015) Translatome analyses capture of opposing tissue-specific brassinosteroid signals orchestrating root meristem differentiation. Proc Natl Acad Sci U S A 112(3):923–928. https://doi.org/10.1073/pnas.1417947112
Wang Y, Chen S, Yu O (2011) Metabolic engineering of flavonoids in plants and microorganisms. Appl Microbiol Biotechnol 91:949–956. https://doi.org/10.1007/s00253-011-3449-2
Wang YY, Hsu PK, Tsay YF (2012) Uptake, allocation and signaling of nitrate. Trends Plant Sci 17(8):458–467. https://doi.org/10.1016/j.tplants.2012.04.006
Wang WS, Zhao XQ, Li M et al (2016) Complex molecular mechanisms underlying seedling salt tolerance in rice revealed by comparative transcriptome and metabolomic profiling. J Exp Bot 67:405–419. https://doi.org/10.1093/jxb/erv476
Wang L, Ruan CJ, Liu LG et al (2018) Comparative RNA-seq analysis of high- and low-oil yellow horn during embryonic development. Int J Mol Sci 19(10):3071. https://doi.org/10.3390/ijms19103071
Wang M, Ding YX, Wang Q et al (2020a) NaCl treatment on physio-biochemical metabolism and phenolics accumulation in barley seedlings. Food Chem 331:127282. https://doi.org/10.1016/j.foodchem.2020.127282
Wang R, Qian J, Fang Z et al (2020b) Transcriptomic and physiological analyses of rice seedlings under different nitrogen supplies provide insight into the regulation involved in axillary bud outgrowth. BMC Plant Biol 20:197. https://doi.org/10.1186/s12870-020-02409-0
Wu X, Ding C, Baerson SR et al (2018) The roles of jasmonate signalling in nitrogen uptake and allocation in rice (Oryza sativa L.). Plant Cell Environ 42(2):659–672. https://doi.org/10.1111/pce.13451. (PMID: 30251262)
Wu DG, Zhan QW, Yu HB et al (2020) Genome-wide identification and analysis of maize PAL gene family and its expression profile in response to high-temperature stress. Pak J Bot. https://doi.org/10.30848/PJB2020-5(28)
Xie YP, Chen PX, Yan Y et al (2018) An atypical R2R3 MYB transcription factor increases cold hardiness by CBF-dependent and CBF-independent pathways in apple. New Phytol 218(1):201–218. https://doi.org/10.1111/nph.14952
Xuan W, Beeckman T, Xu G (2017) Plant nitrogen nutrition: sensing and signaling. Curr Opin Plant Biol 39:57–65. https://doi.org/10.1016/j.pbi.2017.05.010
Yang YQ, Li X, Kong XX et al (2015) Transcriptome analysis reveals diversified adaptation of Stipa purpurea along a drought gradient on the Tibetan Plateau. Funct Integr Genom 15(3):295–307. https://doi.org/10.1007/s10142-014-0419-7
Yang Y, Wang F, Wan Q et al (2018a) Transcriptome analysis using RNA-Seq revealed the effects of nitrogen form on major secondary metabolite biosynthesis in tea (Camellia sinensis) plants. Acta Physiol Plant 40:127. https://doi.org/10.1007/s11738-018-2701-0
Yang YT, Yu Q, Yang YY et al (2018b) Identification of cold-related miRNAs in sugarcane by small RNA sequencing and functional analysis of a cold inducible ScmiR393 to cold stress. Environ Exp Bot 155:464–476. https://doi.org/10.1016/j.envexpbot.2018.07.030
Yang YY, Gao SW, Su YC et al (2019) Transcripts and low nitrogen tolerance: regulatory and metabolic pathways in sugarcane under low nitrogen stress. Environ Exp Bot 163:97–111. https://doi.org/10.1016/j.envexpbot.2019.04.010
Yao Z, Wang L, Qi J (2009) Biosorption of methylene blue from aqueous solution using a bioenergy forest waste: Xanthoceras sorbifolia seed coat. Clean 37:642–648. https://doi.org/10.1002/clen.200900093
Yao ZY, Qi JH, Yin LM (2013) Biodiesel production from Xanthoceras sorbifolia in China: opportunities and challenges. Renew Sustain Energy Rev 24:57–65. https://doi.org/10.1016/j.rser.2013.03.047
Zhang S, Zu YG, Fu YJ et al (2010) Supercritical carbon dioxide extraction of seed oil from yellow horn (Xanthoceras sorbifolia Bunge.) and its anti-oxidant activity. Bioresour Technol 101(7):2537–2544. https://doi.org/10.1016/j.biortech.2009.11.082
Zhao WC, Yang XY, Yu HJ et al (2015) RNA-seq-based transcriptome profiling of early nitrogen deficiency response in cucumber seedlings provides new insight into the putative nitrogen regulatory network. Plant Cell Physiol 56(3):455–467. https://doi.org/10.1093/pcp/pcu172
Zhao CL, Yu YQ, Chen ZJ et al (2017) Stability-increasing effects of anthocyanin glycosyl acylation. Food Chem 214:119–128. https://doi.org/10.1016/j.foodchem.2016.07.073
Zhao BB, Wang L, Pang SY et al (2020) UV-B promotes flavonoid synthesis in Ginkgo biloba leaves. Indus Crops Prod 151:112483. https://doi.org/10.1016/j.indcrop.2020.112483
Zhu L, Liu L, Sun H et al (2021) Physiological and comparative transcriptomic analysis provide nsight into cotton (Gossypium hirsutum L.) root senescence in response. Front Plant Sci 12:748715. https://doi.org/10.3389/fpls.2021.748715
Acknowledgements
We thank Sangon Biotech (Shanghai) for helping with transcriptome sequencing.
Funding
This work was financially supported by Science Technology Yuanjiang Project of Xinjiang Uygur Autonomous Region (2016E02045).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
We declare that there is no conflict of interest in this manuscript.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
468_2023_2414_MOESM2_ESM.xlsx
Supplementary file2 (XLSX 11 KB) Table S1. Sample QC data information statistics. Data are presented as the means ± standard error. Asterisk indicates statistically significant difference (p < 0.05).
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Li, X., Zou, J., Jin, C. et al. Transcriptomics analysis reveals Xanthoceras sorbifolia Bunge leaves’ adaptation strategy to low nitrogen. Trees 37, 1153–1166 (2023). https://doi.org/10.1007/s00468-023-02414-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00468-023-02414-4