
Abstract
Background
Although many pediatric nephrologists consider focal segmental glomerulosclerosis (FSGS) and minimal change disease (MCD) as separate clinical entities, whether the initial histology could affect clinical courses in children with steroid-resistant nephrotic syndrome (SRNS) suspected of having an immune-based etiology remains unknown, especially for long-term outcomes.
Methods
We retrospectively reviewed long-term outcomes (> 3 years; median follow-up, 9.1 years) of 21 children with initial SRNS (FSGS, N = 9; MCD, N = 12) who achieved complete remission with immunosuppressive agents, including cyclosporine.
Results
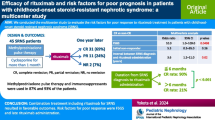
At NS onset, incidence of acute kidney injury (67% vs. 8%, P < 0.05) and proportion of patients with non-selective proteinuria (56% vs. 0%, P < 0.01) were significantly higher in the FSGS group than the MCD group. Furthermore, median days until complete remission after treatment was significantly longer in the FSGS group than the MCD group (116 days vs. 45 days, P < 0.001). Although subsequent biopsy histology of the 12 patients in the MCD group was still identical in all MCD, three of nine patients in the FSGS group were reclassified from FSGS to MCD at second biopsy. At last visit, all patients maintained complete remission, and none developed chronic kidney disease.
Conclusions
Initial presentation in the FSGS group was characterized by more severe clinical manifestations than the MCD group. If complete remission is achieved, FSGS and MCD in children with immune-mediated SRNS may constitute a single disease spectrum because the long-term outcomes are favorable, irrespective of initial histology.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Steroid-resistant nephrotic syndrome (SRNS) is considered a heterogeneous pathological entity, including immune-mediated and monogenic etiologies [1]. Focal segmental glomerulosclerosis (FSGS) and minimal change disease (MCD) are the two key histological diagnoses in children with SRNS as an immune-mediated disease. At present, there are controversies about whether FSGS and MCD in this cohort are distinct glomerular lesions or belong to the same disease spectrum [2,3,4], because the clinical manifestations such as massive proteinuria and generalized edema are indistinguishable, especially in the early stages of the disease.
In a recent review article on children with SRNS who received rituximab [5], Kamei et al. argued that FSGS might simply result from persistent proteinuria because the histological diagnosis was at some point changed from MCD to FSGS in the subsequent kidney biopsy when the patients were not able to achieve remission. The authors concluded that there is a possibility that FSGS and MCD represent a pathologically identical clinical entity. However, whether the long-term outcomes between the two histological groups in children with SRNS after remission are equivalent remains unknown.
In this study, we investigated the relationship between the initial histological diagnosis (such as FSGS and MCD) and the long-term outcomes in children with initial SRNS suspected of having an immune-mediated etiology.
Methods
Patients
Between March 2005 and October 2016, 21 consecutive children with initial SRNS (14 males and 7 females) who achieved complete remission (CR) with immunosuppressive medications, including cyclosporine (CsA) and who had been followed up for > 3 years (median observation period, 9.1 years; interquartile range, 7.2–9.8 years) at Saitama Children’s Medical Center were enrolled in this retrospective study. Patients with SRNS-associated genetic mutations, such as WT1 and NPHS2 (i.e., monogenic, non-immune-mediated disease), were excluded.
In this study, the definitions and criteria adopted for NS, CR, and NS relapse followed the clinical practice guideline for pediatric idiopathic NS in Japan [6]. SRNS was defined as the absence of CR despite prednisolone treatment at a dose of 2 mg/kg/day (maximum 60 mg/day) for 4 weeks. Acute kidney injury (AKI) was defined as an elevated serum creatinine level ≥ 1.5 times the baseline level [7]. Non-selective proteinuria was defined as the selectivity index of ≥ 0.2 (SI, the clearance ratio of IgG/transferrin). This study was approved by the ethics committee of the Saitama Children’s Medical Center (approval number 2019-03-009).
Treatment protocol
Following the diagnosis of initial SRNS, all patients received oral CsA (4–7 mg/kg/day; maximum 150 mg/day) in addition to prednisolone (1 mg/kg/day, maximum 40 mg/day). For patients who had nephrotic-range proteinuria (UP/C of ≥ 2.0, early morning urinary protein-to-creatinine ratio) at the SRNS diagnosis, intravenous pulse methylprednisolone (IVMP; 20 mg/kg/day; maximum 1000 mg/day) was additionally administered for 3 consecutive days over 2–3 weeks. For patients with refractory SRNS who were unresponsive to combined CsA and IVMP, rituximab was administered intravenously at a single dose of 375 mg/m2 (maximum 500 mg/dose) [8]. After achieving a marked decrease in proteinuria (UP/C of < 1.0), prednisolone was gradually tapered off and then discontinued within 12 months. After approximately 2–3 years of CsA treatment, the dosage was gradually tapered off by 20–50 mg every 4 weeks. Relapses were treated with prednisolone 2 mg/kg/day (maximum, 60 mg/day) until proteinuria was undetectable for at least three consecutive days. Prednisolone was administered on alternate days thereafter, and the dose was tapered off within 6 months at a rate of 5–10 mg every 2–4 weeks. Mycophenolate mofetil (MMF; 30–40 mg/kg/day; maximum, 2000 mg/day) was offered as a treatment option if the patients developed frequent-relapsing and/or steroid-dependent NS after the discontinuation of CsA [9]. If NS relapse occurred despite the initiation of MMF, a single infusion of rituximab was administered after CR with prednisolone. For patients who achieved > 12 months of prednisolone-free remission, MMF was gradually tapered off by 250 mg every 4 weeks.
Histological evaluation
Kidney specimens were examined by light, immunofluorescence, and electron microscopies. The first kidney biopsies were performed in all patients at the diagnosis of initial SRNS. The second biopsies were also done in all patients during CR who received CsA for 2–3 years to confirm chronic nephrotoxicity. The histologic variants of FSGS were evaluated using the Columbia classification. The kidney specimens were evaluated by a single pathologist (HM) blinded to the patients’ clinical profiles.
Statistical analysis
Categorical variables were presented as frequencies and percentages and compared using the chi-square or Fisher’s exact test as appropriate. Unless indicated otherwise, continuous variables were expressed as median (interquartile range). The parametric two sample t test or non-parametric Mann–Whitney U test was used as appropriate for comparing continuous variables. All analyses were performed using JMP pro 14 version 14.0 (SAS Institute Inc., Cary, NC, USA). The level of statistical significance was set at P < 0.05.
Results
At the diagnosis of initial SRNS (median age, 3.0 years; interquartile range, 1.8–4.8 years), a kidney biopsy revealed FSGS in 9 and MCD in 12 patients. In the FSGS group, seven patients were classified as “not otherwise specified,” while two patients had “cellular variant.” After immunosuppressive treatment, all patients were able to achieve CR at a median of 73 days (interquartile range, 45–112 days). We compare the baseline characteristics and clinical courses of the patients in the FSGS group with those in the MCD group (Table 1). The incidence of AKI at the onset of NS was significantly higher in the FSGS group than in the MCD group (67% vs. 8%, P < 0.05). Furthermore, the proportion of patients with non-selective proteinuria at NS onset was significantly higher in the FSGS group than in the MCD group (56% vs. 0%, P < 0.01). Through a stratified analysis based on these findings, we found that if the patients had either AKI or non-selective proteinuria at NS onset, then, the sensitivity and specificity of the FSGS diagnosis at the initial biopsy were 89% and 92%, respectively, with a positive predictive value of 89%. The median days taken to achieve CR after commencing treatment was significantly longer in the FSGS group than in the MCD group (116 days vs. 45 days, P < 0.001). In the FSGS group, a delayed response to treatment, defined as requiring more than 8 weeks until CR, developed in all nine patients. However, only two of the 12 patients in the MCD group had a delayed response (P < 0.001). A second kidney biopsy was performed in all patients at a median of 25 months (interquartile range, 25–27 months) after the initial biopsy. Although the subsequent biopsy histology of the 12 patients in the MCD group was still identical to MCD in all cases, three of the nine patients in the FSGS group are reclassified from FSGS to MCD at the second biopsy (Table 2). After discontinuing CsA, NS relapse was observed in 15 patients (71%), leading to the use of MMF and rituximab in ten and six patients, respectively. The proportion of patients with NS relapse, MMF, rituximab, and treatment-free remission after discontinuing CsA did not differ significantly between the FSGS group and the MCD group. At the last visit, although five patients received MMF, treatment-free remission for > 12 months was achieved in 13 patients (62%). Furthermore, all patients maintained CR, and none of the patients developed chronic kidney disease (CKD, estimated glomerular filtration rate of < 90 mL/min/1.73 m2).
Discussion
In this study on children with initial SRNS who achieved CR with immunosuppressive medications, we demonstrated that the initial presentation in the FSGS group was characterized by more severe clinical manifestations than in the MCD group, such as higher incidence of AKI and longer duration until CR. Furthermore, we found that the presence of AKI and non-selective proteinuria at NS onset were significant predictive factors of FSGS diagnosis in the initial kidney biopsy. However, the long-term outcomes after CR in this cohort were favorable, irrespective of the initial histological diagnosis.
Although idiopathic FSGS and MCD have been described as separate entities in most textbooks, Maas et al. recently proposed that they are different phenotypes of the same disease processes: if the initial podocyte injury is severe, FSGS lesions may develop [10]. The authors concluded that the pathogenesis of FSGS and MCD should be investigated together under the definition of idiopathic NS. The results in our study (i.e., severe clinical manifestations at initial presentation in the FSGS group and similarly favorable long-term outcomes after achieving CR in the both groups) are consistent with Maas’s hypothesis.
In a multicenter retrospective study carried out on hospitalized children with idiopathic NS [11], Rheault et al. reported that the risk factors for AKI included nephrotoxic medications, infection, and SRNS. The authors showed that children with SRNS were more likely to develop AKI than children with steroid-sensitive NS (odds ratio, 2.06; 95% confidence interval 1.33–3.19). In a nationwide cohort study on Japanese children with newly diagnosed idiopathic NS [12], Sato et al. showed that the incidence of AKI was relatively high (24%) and AKI was significantly related to the development of mild CKD. However, they were not able to assess the relationship between AKI and SRNS because the number of SRNS cases was very small. In a recent study performed on 101 children at the first onset of idiopathic NS [13], we reported that the rate of SRNS progression was significantly higher in the AKI group than in the non-AKI group (36% vs. 12%, P < 0.05). Therefore, although the etiologies of AKI in idiopathic NS are multifactorial, such as intravascular volume depletion and infectious complications, we speculated that the presence of AKI at NS onset was the risk factor for SRNS progression because of no significant differences in the baseline characteristics, such as serum albumin levels and C-reactive protein levels between the AKI group and the non-AKI group. Furthermore, in this study, we first showed that the presence of AKI at the onset of NS was a significant predictor of FSGS diagnosis in children with initial SRNS.
In the 1960s, the efficacy of SI in predicting the response to prednisolone was established in both children and adults with idiopathic NS [14, 15]. In subsequent decades, the value of SI waned in clinical practice because its assessment did not seem to help in predicting the histological diagnosis and in determining the long-term outcomes. In a retrospective study of adult patients with idiopathic NS (including FSGS (N = 29) and MCD (N = 9)) [16], however, Bazzi et al. revealed that the rate of non-selective proteinuria (SI, > 0.2) was significantly higher in the FSGS group than in the MCD group (62% vs. 11%, P < 0.01), which accorded with the results of our study in children with initial SRNS. Furthermore, we first showed that if the patients had either non-selective proteinuria or AKI at NS onset, the cohort was more likely to have FSGS as initial histological diagnosis (11/12, 89%).
In this study, there were no significant differences in the long-term outcomes between the FSGS group and the MCD group, and none of the patients developed CKD. In previous studies of childhood SRNS [2,3,4], however, FSGS as the initial histological diagnosis was found to be a predictive factor for CKD development, especially in the patients who did not achieve remission. We speculate that the initial aggressive treatment, including CsA, IVMP, and rituximab, improved the kidney outcomes in our study, particularly for patients with FSGS who had severe clinical manifestations at NS onset. Furthermore, although more than 60 genes related to SRNS have been described in recent years and monogenic etiology can account for one-third of the cases of SRNS [1], patients with a monogenic cause were not necessarily excluded from previous studies. In contrast, only patients who achieved CR with immunosuppressive medications suspected of having an immune-mediated SRNS were included, and those with SRNS-associated genes were excluded from our study.
To the best of our knowledge, this is the first study demonstrating the reassignment from FSGS to MCD at the second kidney biopsy in one-third of the children (three of the nine patients) with initial SRNS who achieved CR with immunosuppressive therapy. We speculate that the effective therapy was able to prevent the progression of new scarring in the FSGS group, although the development from segmental sclerotic lesions to obsolescent glomeruli was observed at the second kidney biopsy in two of the three patients. Furthermore, although the small sample size may lead to misclassification of the histological diagnosis, we believe that the number of false-negative FSGS cases may be low because the probability that no glomeruli showing FSGS would be found was very low (12%) if the biopsy contained 20 glomeruli [17]: the number of glomeruli of the three patients reclassified from FSGS to MCD in our study was sufficient (median, 26).
In conclusion, FSGS and MCD in children with SRNS as an immune-mediated disease seem to be variants of a single disease spectrum because the histological diagnosis was changeable on the timing of kidney biopsy and the long-term outcomes after CR were similarly favorable. Therefore, kidney biopsy may not be necessarily required for children with initial SRNS who can achieve CR within 8 weeks after immunosuppressive medications, because no such patients were diagnosed with FSGS in this cohort. However, although the therapeutic protocol for children with initial SRNS was homogeneous at our hospital, this study is limited by its retrospective study design and the fact it was carried out at a single center. Furthermore, we agree that sampling error may occur, and the number of patients in our study was insufficient to draw robust conclusions. Therefore, future prospective studies are required to investigate whether initial histological diagnosis could affect the clinical course in children with initial SRNS.
References
Lee JM, Kronbichler A, Shin JI, Oh J (2020) Current understandings in treating children with steroid-resistant nephrotic syndrome. Pediatr Nephrol. https://doi.org/10.1007/s00467-020-04476-9
Paik KH, Lee BH, Cho HY, Kang HG, Ha IS, Cheong HI, Jin DK, Moon KC, Choi Y (2007) Primary focal segmental glomerular sclerosis in children: clinical course and prognosis. Pediatr Nephrol 22:389–395
Gipson DS, Chin H, Presler TP, Jennette C, Ferris ME, Massengill S, Gibson K, Thomas DB (2006) Differential risk of remission and ESRD in childhood FSGS. Pediatr Nephrol 21:344–349
Abrantes MM, Cardoso LS, Lima EM, Penido Silva JM, Diniz JS, Bambirra EA, Oliveira EA (2006) Predictive factors of chronic kidney disease in primary focal segmental glomerulosclerosis. Pediatr Nephrol 21:1003–1012
Kamei K, Ishikura K, Sako M, Ito S, Nozu K, Iijima K (2020) Rituximab therapy for refractory steroid-resistant nephrotic syndrome in children. Pediatr Nephrol 35:17–24
Ishikura K, Matsumoto S, Sako M, Tsuruga K, Nakanishi K, Kamei K, Saito H, Fujinaga S, Hamasaki Y, Chikamoto H, Ohtsuka Y, Komatsu Y, Ohta T, Nagai T, Kaito H, Kondo S, Ikezumi Y, Tanaka S, Kaku Y, Iijima K, Japanese Society for Pediatric Nephrology; Japanese Society for Pediatric Nephrology (2015) Clinical practice guideline for pediatric idiopathic nephrotic syndrome 2013: medical therapy. Clin Exp Nephrol 19:6–33
Khwaja A (2012) KDIGO clinical practice guidelines for acute kidney injury. Nephron Clin Pract 120:c179–c184
Fujinaga S, Nishino T, Umeda C, Tomii Y, Watanabe Y, Sakuraya K (2019) Long-term outcomes after early treatment with rituximab for Japanese children with cyclosporine- and steroid-resistant nephrotic syndrome. Pediatr Nephrol 34:353–357
Fujinaga S, Ohtomo Y, Umino D, Takemoto M, Shimizu T, Yamashiro Y, Kaneko K (2007) A prospective study on the use of mycophenolate mofetil in children with cyclosporine-dependent nephrotic syndrome. Pediatr Nephrol 22:71–76
Maas RJ, Deegens JK, Smeets B, Moeller MJ, Wetzels JF (2016) Minimal change disease and idiopathic FSGS: manifestations of the same disease. Nat Rev Nephrol 12:768–776
Rheault MN, Zhang L, Selewski DT, Kallash M, Tran CL, Seamon M, Katsoufis C, Ashoor I, Hernandez J, Supe-Markovina K, D’Alessandri-Silva C, DeJesus-Gonzalez N, Vasylyeva TL, Formeck C, Woll C, Gbadegesin R, Geier P, Devarajan P, Carpenter SL, Kerlin BA, Smoyer WE, Midwest Pediatric Nephrology Consortium (2015) AKI in children hospitalized with nephrotic syndrome. Clin J Am Soc Nephrol 10:2110–2118
Sato M, Ishikura K, Ando T, Kikunaga K, Terano C, Hamada R, Ishimori S, Hamasaki Y, Araki Y, Gotoh Y, Nakanishi K, Nakazato H, Matsuyama T, Iijima K, Yoshikawa N, Ito S, Honda M (2019) Prognosis and acute complications at the first onset of idiopathic nephrotic syndrome in children: a nationwide survey in Japan (JP-SHINE study). Nephrol Dial Transplant. https://doi.org/10.1093/ndt/gfz185
Fujinaga S, Kusaba K (2019) Impact of acute kidney injury at the onset of idiopathic nephrotic syndrome in Japanese children. Clin Exp Nephrol 23:1171–1172
Joachim GR, Cameron JS, Schwartz M, Becker EL (1964) Selectivity of protein excretion in patients with the nephrotic syndrome. J Clin Invest 43:2332–2346
Cameron JS, White RH (1965) Selectivity of proteinuria in children with the nephrotic syndrome. Lancet 1:463–465
Bazzi C, Petrini C, Rizza V, Arrigo G, D'Amico G (2000) A modern approach to selectivity of proteinuria and tubulointerstitial damage in nephrotic syndrome. Kidney Int 58:1732–1741
Corwin HL, Schwartz MM, Lewis EJ (1988) The importance of sample size in the interpretation of the renal biopsy. Am J Nephrol 8:85–89
Acknowledgments
The authors wish to thank Dr. Hitohiko Murakami (Division of Pathology, Saitama Children’s Medical Center) for his great contributions to this study.
Funding
SF has received clinical research funding B at Saitama Children’s Medical Center.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the Institutional Research Committee and/or National Research Committee at which the study was conducted (approval number 2019-03-009) with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Watanabe, Y., Fujinaga, S., Endo, A. et al. Baseline characteristics and long-term outcomes of steroid-resistant nephrotic syndrome in children: impact of initial kidney histology. Pediatr Nephrol 35, 2377–2381 (2020). https://doi.org/10.1007/s00467-020-04760-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-020-04760-8