
Abstract
The World Health Organization has recognized that testicular function is temperature dependent. Testicular heat exposure caused by occupational factors, lifestyle, and clinical diseases can lead to different degrees of reproductive problems. The aim of this study was to reveal the transcriptional regulatory network and its potential crucial roles in mediating the effects of testicular heat exposure. Testicular tissue was collected from a group of mice subjected to scrotal heat exposure as well as a control group. RNA was isolated from both groups and used for high-throughput sequencing. Using differential transcriptome expression analysis, 172 circRNAs, 279 miRNAs, 465 lncRNAs, and 2721 mRNAs were identified as significantly differentially expressed in mouse testicular tissue after heat exposure compared with the control group. Through Gene Ontology (GO) term and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses, differentially expressed lncRNAs and mRNAs were found to have potentially important functions in meiotic cell cycle (GO:0051321), cytoplasm (GO:0005737), membrane raft (GO:0045121), MAPK signaling (mmu04010), purine metabolism (mmu00230), and homologous recombination (mmu03440). Some of the most upregulated and downregulated lncRNAs and circRNAs were predicted to be associated with numerous miRNAs and mRNAs through competing endogenous RNA regulatory network analysis, which were validated with molecular biology experiments. This research provides high-throughput sequencing data of a testicular heat exposure model and lays the foundation for further study on circRNAs, miRNAs, and lncRNAs that are involved in male reproductive diseases related to elevated testicular temperature.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Environment and lifestyle are currently considered to be related to the pathogenesis of idiopathic male infertility (Krzysciak et al. 2020; Marcho et al. 2020). Sperm maturation requires adequate nutritional support and the absence of certain substances—such as essential amino acids and trace elements—which can lead to spermatogenesis disorders and reduce the fertilization ability (Lammi and Qu 2018). Sperm production depends on high levels of testosterone; many plastic products contain estrogen-like substances, such as bisphenol A, which can lead to decreased gonadotropin secretion and increase the rate of sperm deformities (Cariati et al. 2019). Long-term exposure to radiation sources, such as computers, mobile phones, and microwave ovens, can affect the amount of sperm a male produces, resulting in decreased sperm motility and even male infertility (Santini et al. 2018; Jaffar et al. 2019). Nicotine in tobacco can affect the development of spermatogenic cells by passing through the blood–testosterone barrier; it can then cause DNA breaks in sperm and worsen sperm deformities (Mohamed and Abdelrahman 2019; Zhou et al. 2019). Low androgen levels induced by alcohol can lead to testicular atrophy and abnormal sperm count, morphology, and function (Horibe et al. 2019). Spermatogenesis is particularly sensitive to temperature; indeed, elevated testicular temperature (heat exposure) can reduce spermatogenesis and sperm motility, increase sperm malformation rates, and damage the testicular morphology (Hamilton et al. 2018; Rao et al. 2019; Shahat et al. 2020). In-depth studies on the etiology and mechanism of idiopathic male sterility have an important guiding role in clinical work.
Spermatogenesis is a complex, orderly process of fine regulation in which spermatogonia develop into mature sperm through continuous mitosis, meiosis, and cell differentiation (Cai et al. 2019; Dunleavy et al. 2019; Cannarella et al. 2020). The World Health Organization has recognized that normal testicular function is temperature dependent. In most mammals, including humans, testes are located in the scrotum, where the average temperature is 33.3 °C. The structural characteristics of the scrotum and inverse heat exchange mechanisms of testicular blood vessels maintain the relatively low temperature in the testis (Molina and Anderson 2018). However, scrotal temperature regulation is limited. When the testis is exposed to a high temperature for longer than the scrotal regulation range, spermatogenesis will inevitably be damaged, resulting in a decline in the concentration and vitality of sperm, an increase in the abnormal sperm rate, and spermatogenesis disorders (Zhang et al. 2018b). The effects and molecular mechanisms of heat stress on reproductive medicine need to be further studied.
Recent studies have demonstrated that noncoding RNA participates in the biological response to heat and cold stress (Yang et al. 2017; Quan et al. 2020; Wang et al. 2020). The long noncoding RNA (lncRNA) MSTRG.80946.2 participates in fatty acid metabolism during exposure to cold stress by modifying the downstream signaling pathway of ACP1, TSPY1, and Tsn (Ji et al. 2020). lncR9A, lncR117, and lncR616 function as competing endogenous RNAs for miRNA-398 to regulate CSD1 expression during cold resistance in winter wheat (Lu et al. 2020). In the summer, high temperatures can lead to markedly decreased concentrations of unsaturated fatty acids in milk from dairy cows by regulating the competing endogenous signals of circular RNAs (circRNAs) (Wang et al. 2020). After being stimulated with heat stress, the chironomid Diamesa tonsa, a cold stenothermal insect, produces lncRNA to affect heat shock protein 70 kDa (HSP70) protein expression by functioning as a ribosomal sponge to maintain a steady state of living (Bernabo et al. 2020). The pathological process caused by heat stress in human umbilical vein endothelial cells can be mediated by several miRNAs and their targets (Liu et al. 2017). The transcriptome and molecular functions of noncoding RNA in heat-induced injury to testicular tissue have not yet been reported in detail.
To investigate the roles of noncoding RNA in mediating the effects of testicular heat exposure, we analyzed the differential transcriptome expression of circRNAs, miRNAs, lncRNAs, and mRNAs, and their molecular functional regulatory pathways through high-throughput sequencing, Gene Ontology (GO) term and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses, competing endogenous RNA regulatory networks, and molecular biology experiments. This research is the first that has explored the signaling pathways for noncoding RNA that mediate the effects of testicular heat exposure in a mouse model at the transcriptome level.
Materials and methods
Mouse testicular heat exposure model and histology
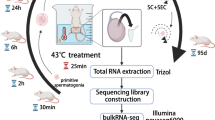
Male ICR mice were purchased from SPF (Beijing) Biotechnology Co., Ltd. (Beijing, China). Mice were first anesthetized with 0.01 ml/g 5% chloral hydrate (Shanghai Macklin Biochemical Co., Ltd., Shanghai, China), and then the scrotum was placed in a water bath for 25 min at 33 °C for the control group or 43 °C for the high-temperature group (Akintayo et al. 2020). Mice were sacrificed 24 h after testicular heat exposure, and the testes and epididymides were separated from the mouse body and used for histology and RNA isolation. This study was approved by the laboratory animal management and ethics committee of Bengbu Medical College (No. 2020024, April 3, 2020).
For histological detection, mouse testes and epididymides were incubated in 4% paraformaldehyde for 24 h, dehydrated, and embedded in paraffin. The paraffin-embedded tissues were cut into 5-μm sections, which were mounted on glass slides, dewaxed, and rehydrated. The sections were then stained with hematoxylin and eosin (H&E) (Service Biological Technology, Wuhan, China). The sections were dehydrated and then sealed with neutral gum. Finally, the slides were observed under a light microscope and photographed. This experiment was performed using three biological replicates.
RNA isolation and high-throughput sequencing
Total RNA was extracted from the testis after heat exposure using the TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. The integrity of the isolated RNA was determined by agarose electrophoresis, and the concentration of total RNA and potential DNA/protein contamination were analyzed using a NanoDrop ND-1000 (Thermo Fisher Scientific, Waltham, MA, USA). Total RNA was obtained after removing ribosomal RNA (rRNA) using the RiboZero Magnetic Gold Kit (Epicentre, an Illumina Company, Madison, WI, USA). The KAPA Stranded RNA-Seq Library Prep Kit (Illumina, San Diego, CA, USA) and the NEB Multiplex Small RNA Library Prep Set (Illumina) were used to generate sequencing libraries for circRNAs/lncRNAs/mRNAs and miRNAs, respectively. These libraries were sequenced using a NovaSeq 6000 (Illumina) to obtain raw sequencing data. Then, the Q30 values and ratios of mapping for transcripts were analyzed to verify the quality of raw sequencing data. This experiment was performed using three biological replicates.
High-throughput sequencing data analysis
To analyze circRNA/lncRNA/mRNA transcripts, the transcriptional abundance and fragments per kilobase of gene/transcript model per million mapped fragments (FPKM) of gene expression levels were calculated by the StringTie and Ballgown software programs, respectively (Mortazavi et al. 2008; Frazee et al. 2015; Pertea et al. 2015). miRNA levels were determined by miRDeep2, and differential expression calculations for miRNA in counts per million reads (CPM) were obtained using the edgeR software (Robinson et al. 2010; Friedlander et al. 2012;). The following cut-offs were used to identify significantly differentially expressed transcripts: fold change ≥ 1.5 and p ≤ 0.05, with q ≤ 1.00 to correct the p value calculation. The obvious differences in the expression profile are displayed with a volcano plot and a heatmap. GO term (http://geneontology.org/) and KEGG pathway analyses (https://www.kegg.jp/) were used to examine the molecular mechanisms involved in testicular heat exposure.
Quantitative real-time PCR and western blotting.
The expression levels of circRNAs, lncRNAs, and mRNAs were analyzed by quantitative real-time PCR. In brief, the PrimeScript™ RT reagent kit (TaKaRa, Otsu, Japan) was used to generate complementary DNA (cDNA) from 500 ng of total RNA. Subsequently, the TB Green™ Premix Ex Taq™ II kit (TaKaRa) was used to determine target gene expression. The target gene expression level was normalized to β-actin using the comparative 2−ΔΔ Ct method (Yao et al. 2014). The sequences of primers used to amplify target genes are shown in Table S1; the β-actin primers were reported in our previous study (Hu et al. 2019b). This experiment was performed using three biological replicates and three technical replicates.
The protein expression level was analyzed by western blotting (Liang et al. 2013). In brief, radioimmunoprecipitation assay (RIPA) buffer (Millipore, Bedford, MA, USA) with a proteinase inhibitor cocktail (Roche, Mannheim, Germany) was used to homogenize the tissue and isolate protein. Proteins were separated with sodium dodecyl sulfate–polyacrylamide gel electrophoresis (Beyotime Biotechnology, Beijing, China). Separated proteins were then transferred to nitrocellulose membranes (Beijing Labgic Technology, Beijing, China). Then, nitrocellulose membranes with proteins were treated with an antibody against RAB7 (ABclonal Technology, Wuhan, China) and β-actin (ABclonal Technology, Danvers, MA, USA). This experiment was performed using three biological replicates.
Competing endogenous RNA network
Any RNA transcripts (circRNAs, lncRNAs, and mRNAs) with miRNA binding sites can compete for and mutually affect the same miRNA; these species are known as competing endogenous RNAs (Salmena et al. 2011). The binding of targets and miRNA was predicted using TargetScan (http://www.targetscan.org/mamm_31/) and miRanda (http://www.microrna.org/microrna/home.do). Some of the top upregulated and downregulated lncRNAs and circRNAs were verified by quantitative real-time PCR. Then, competing endogenous RNA regulatory networks for the confirmed lncRNAs/circRNAs and miRNA–mRNA partners were constructed by the competing endogenous RNA hypothesis.
Statistical analysis
Student’s t-test was used to analyze the quantitative real-time PCR and high-throughput sequencing data. p < 0.05 was considered a statistically significant difference. Bar graphs present data as the mean ± standard error of the mean.
Results
Establishing a mouse testicular heat exposure model and transcriptome profiling
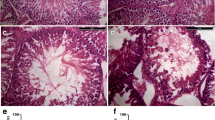
To explore the function of noncoding RNA in mediating the effects of testicular heat exposure, mice were first subjected to scrotal heat exposure for 25 min at 33 °C in the control group and for 25 min at 43 °C in the high temperature group (Fig. 1a–d). After 24 h, some spermatogenic cells in the testis of the high-temperature group were enlarged (Fig. 1c) and vacuolated and spermatozoa in the epididymis were pyknotic (Fig. 1d). Total RNA was isolated from about 0.05 g testicular tissue and assessed for RNA integrity (Fig. S1), quantity, and quality assurance (Table 1). A Q30 value > 80% usually indicates very high sequencing quality; the Q30 values for circRNA/lncRNA/mRNA (Table 2) and miRNA (Table 3) transcripts were > 90%. Furthermore, the ratios of mapping for circRNA, lncRNA, and mRNA transcripts were > 80% (Table 4), and miRNA transcripts were > 60% (Table 5), values that indicate good match rates. The high-throughput sequencing data are available from NCBI with GEO accession numbers GSE165696 for circRNAs, lncRNAs, and mRNAs, and GSE165697 for miRNAs.

Testicular heat exposure damages spermatogenesis. a–d Mouse scrota were exposed for 25 min to either a 33 °C (control group) or a 43 °C (high-temperature group) water bath. After 24 h, the testis (a and c) and epididymis (b and d) were subjected to hematoxylin and eosin staining to examine morphological changes. In the testis of 43 °C treatment (c), arrows indicate enlarged germ cells and arrowheads indicate vacuolated germ cells. In the epididymis of 43 °C treatment (d), arrows indicate pyknotic spermatozoa
Differentially expressed transcriptome exploration
After passing quality control, the high-throughput sequencing data were subjected to differential expression analysis to compare testicular tissue from the control and high-temperature groups. There were obvious differences in the circRNA (Fig. 2a, b), miRNA (Fig. 3a, b), lncRNA (Fig. 4 a,b), and mRNA (Fig. 5a, b) expression profiles, as shown with volcano plots and heatmaps. Overall, 172 circRNAs (91 upregulated and 81 downregulated), 279 miRNAs (138 upregulated and 141 downregulated), 465 lncRNAs (96 upregulated and 369 downregulated), and 2721 mRNAs (2003 upregulated and 718 downregulated) were identified as significantly differentially expressed in the high-temperature compared with the control group using the cut-offs of fold change ≥ 1.5 and p ≤ 0.05, with a q ≤ 1.00 cut-off applied to correct the p value calculation. The frequency distribution of the fold changes was also examined for circRNAs, miRNAs, lncRNAs, and mRNAs. A 2–five fold change was most common for circRNAs and miRNAs, while a 1.5–two fold change was most common for lncRNAs and mRNAs (see Table 6 [upregulated transcripts] and Table 7 [downregulated transcripts]).

Circular RNA (circRNA) expression profiles. a Volcano plot for differentially expressed circRNAs. Red, gray, and green represent upregulated, not differentially expressed, and downregulated circRNAs, respectively. b Heatmap for differentially expressed circRNAs. Red and green represent upregulated and downregulated circRNAs, respectively

MicroRNA (miRNA) expression profile. a Volcano plot for differentially expressed miRNAs. Red, gray, and green represent upregulated, not differentially expressed, and downregulated miRNAs, respectively. b Heatmap for differentially expressed miRNAs. Red and green represent upregulated and downregulated miRNAs, respectively

Long noncoding RNA (lncRNA) expression profile. a Volcano plot for differentially expressed lncRNAs. Red, gray, and green represent upregulated, not differentially expressed, and downregulated lncRNAs, respectively. b Heatmap for differentially expressed lncRNAs. Red and green represent upregulated and downregulated lncRNAs, respectively

Messenger RNA (mRNA) expression profile. a Volcano plot for differentially expressed mRNAs. Red, gray, and green represent upregulated, not differentially expressed, and downregulated mRNAs, respectively. b Heatmap for differentially expressed mRNAs. Red and green represent upregulated and downregulated mRNAs, respectively
GO term and KEGG pathway analyses for mRNA
The molecular mechanisms involved in mediating the effects of testicular heat exposure were analyzed by GO term and KEGG pathway analyses. The GO terms of biological process (BP), cellular component (CC), and molecular function (MF) for upregulated and downregulated mRNAs are shown in Tables S2–S7, and the top 10 terms are shown in Figs. 6a and 7a. The number of upregulated and downregulated mRNAs for the KEGG pathway analysis were 152 and 29, respectively (Tables S8 and S9), and the top 10 pathways are shown in Fig. 6b and 7b. Regarding upregulated mRNAs, the most enriched GO terms for BP/CC/MF and KEGG pathways were negative regulation of cellular process (GO:0048523), cytoplasm (GO:0005737), amide binding (GO:0033218), and MAPK signaling (mmu04010) (Fig. 6a and b). Regarding downregulated mRNAs, the most enriched GO terms for BP/CC/MF and KEGG pathways were cell cycle (GO:0007049), microtubule cytoskeleton (GO:0015630), motor activity (GO:0003774), and purine metabolism (mmu00230) (Fig. 7a and b).

Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses of upregulated messenger RNAs (mRNAs). a Top 10 enriched GO terms for upregulated mRNAs. BP indicates biological process, CC indicates cellular component, and MF indicates molecular function. b Top 10 enriched KEGG pathway for upregulated mRNAs

Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses of downregulated messenger RNAs (mRNAs). a Top 10 enriched GO terms for downregulated mRNAs. BP indicates biological process, CC indicates cellular component, and MF indicates molecular function. b Top 10 enriched KEGG pathway for downregulated mRNAs
Prediction of lncRNA targets
GseaPrerank of gene set enrichment analysis was performed to elucidate the potential function of the target genes of the top 10 upregulated and downregulated lncRNAs after testicular heat exposure (Mootha et al. 2003; Subramanian et al. 2005). For GO term analysis, the top 10 differentially expressed lncRNAs have a potential function in the meiotic cell cycle (GO:0051321), DNA repair (GO:0006281), condensed chromosome kinetochore (GO:0000777), membrane raft (GO:0045121), DNA dependent ATPase activity (GO:0008094), and extracellular matrix binding (GO:0050840) (Figs. 8, S2,S3). For KEGG pathway analysis, the top 10 differentially expressed lncRNAs were classified into spliceosome (mmu03040), homologous recombination (mmu03440), RNA transport (mmu03013), nod-like receptor signaling pathway (mmu04621), and lysosome (mmu04142) (Fig. 9). These results suggest that the differentially expressed lncRNAs mediate the effects of testicular heat exposure through changes in DNA metabolism.

Biological process of Gene Ontology (GO) analysis for target genes of the top 10 upregulated (pink) and downregulated (green) lncRNAs was performed by GseaPrerank

Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis for target genes of the top 10 upregulated (pink) and downregulated (green) lncRNAs was performed by GseaPrerank
Verification of select differentially expressed lncRNA and circRNA
The expression of the most upregulated and downregulated lncRNAs and circRNAs based on deep-sequencing data were verified by quantitative real-time PCR. Of the 12 selected differentially expressed lncRNAs (upregulated: AC167229.2, Gm19705, BC021767, C430049B03Rik, Gm13067, and Gm14221; downregulated: 4930405N21.RIK, AC154404.1, Gm10619, Gm16976, Speer9-ps1, and Zfp89), only Gm14221 showed an opposite expression trend, namely upregulated based on high-throughput sequencing and downregulated based on quantitative real-time PCR (Fig. 10a). The other lncRNAs showed consistent expression trends (Fig. 10a). The change in expression of the 12 selected differentially expressed circRNAs (upregulated: Rras2, Scfd2, Lyst, Mad1l1, Bbx, Cnot6l, and Fam160b1; downregulated: 4932438A13Rik, Lrp1b, Phkb, Rbm33, and Rint1), except for Lrp1b and Rbm33, was consistent between high-throughput sequencing and quantitative real-time PCR (Fig. 10b).

Verification of the selected top differentially expressed long noncoding RNAs (lncRNAs) and circular RNAs (circRNAs) in deep-sequencing data by quantitative real-time PCR. Log2 fold change of 43 °C/33 °C was calculated to compare the expression trend of lncRNAs (a) and circRNAs (b) in deep-sequencing data and real-time quantitative PCR. Bar graphs present the mean ± standard error of the mean
Construction of a competing endogenous RNA regulatory network
The top three upregulated and downregulated lncRNAs (upregulated: Gm19705; downregulated: 4930405N21Rik and Speer9-ps1) and circRNAs (upregulated: Scfd2; downregulated: Phkb and Rint1) were used to construct the competing endogenous RNA regulatory network by integrating the miRNA and mRNA transcriptomes using TargetScan and miRanda (Fig. S4). Many reproduction-related genes were involved in the competing endogenous RNA regulatory network, such as Rab7, Tardbp, Gpx5, and Mgat1 (Fig. S4 and 11a). Furthermore, RAB7 protein expression was inhibited by overexpression of miRNA-1187 (miR-1187) and rescued by overexpression of Gm19705, which further indicated that Gm19705 may serve as a competing endogenous RNA for miR-1187 to regulate RAB7 expression in testicular heat exposure (Fig. 11b). The molecular mechanisms by which noncoding RNA mediate the effects of testicular heat exposure need to be further explored in future studies.

Construction of competing endogenous RNA network. a The competing endogenous RNA regulatory network of upregulated lncRNA Gm19705 was constructed by integrating microRNA (miRNA) and messenger RNA (mRNA) using TargetScan and miRanda. Red, blue, and green represent lncRNA, miRNAs, and mRNAs, respectively. b The lncRNA Gm19705 alleviates the repressive effects of miR-1187 on RAB7 protein expression. The spermatocyte cell line GC-2 was transfected with the indicated oligonucleotides and plasmids, and isolated protein was then detected by western blotting
Discussion
In humans, the common causes of testicular temperature increase are occupational factors, lifestyle factors, and clinical diseases (Pastuszak and Wang 2015; Baert et al. 2018; Shen et al. 2019). Workers in certain occupations, such as welders and boiler workers, are exposed to induced heat radiation, which causes the scrotal temperature to rise and results in spermatogenic problems due to long-term work in a high-temperature environment (Shen et al. 2019). Lifestyle factors, such as frequent steam baths, tight pants, and prolonged sitting, can increase local scrotal temperature, which can affect testicular spermatogenesis and lead to decreased fertility (Baert et al. 2018). Clinical diseases, such as varicocele, cryptorchidism, and persistent fever, can lead to an increase in testicular temperature, germ cell apoptosis, and semen quality decline (Pastuszak and Wang 2015). Thus, various heat sources directly or indirectly lead to testicular temperature increase, which can negatively affect spermatogenesis and even induce male infertility. This research was dedicated to explore the transcriptional regulatory network and its potential role in a mouse testicular heat exposure model on the transcriptome level. The findings have laid the foundation for further study of noncoding RNA and mRNA in male reproductive diseases related to elevated testicular temperature. Mouse scrota exposed to 43 °C for 25 min had enlarged, vacuolated spermatogenic cells in the testis and pyknotic spermatozoa in the epididymis. Total RNA was isolated from testicular tissues and prepared for high-throughput sequencing.
Heat exposure has a cumulative effect on testicular injury, which is attenuated by the cooling interval after heat exposure (Durairajanayagam et al. 2015). Short-term heat-stimulation-induced sperm damage can be reversible, but long-term heat stimulation leads to irreversible spermatogenic damage (Zhang et al. 2018a). Studies have shown that the mechanism of spermatogenic cell apoptosis induced by high temperatures may be related to reactive oxygen species, nitric oxide synthase, tumor suppressor proteins, transfer of pro-apoptotic factor Bax from the cytoplasm to the nucleus, and the release of cytochrome c from mitochondria (Badr et al. 2018). After passing quality control, the high-throughput sequencing data were evaluated with differentially expressed transcriptome exploration, GO term and KEGG pathway analyses, and network regulation. Overall, 172 circRNAs, 279 miRNAs, 465 lncRNAs, and 2721 mRNAs were significantly differentially expressed in the mouse testicular tissue after heat exposure. Furthermore, most circRNAs and miRNAs showed a 2–fivefold change in expression, while most lncRNAs and mRNAs showed a 1.5–twofold change in expression. The GO terms cellular process (GO:0048523), cytoplasm (GO:0005737), amide binding (GO:0033218), and MAPK signaling (mmu04010) were enriched for the upregulated mRNAs, while cell cycle (GO:0007049), microtubule cytoskeleton (GO:0015630), motor activity (GO:0003774), and purine metabolism (mmu00230) were enriched for the downregulated mRNAs.
circRNAs and lncRNAs have been reported to participate in the development of many diseases, such as immune disorders, nerve problems, cancer, and angiocardiopathy (Hu et al. 2019a; Li et al. 2020a, b; Yu et al. 2020). The lncRNA GAS5 suppresses T helper 17 cells and reduces immune thrombocytopenia by facilitating TRAF6-induced STAT3 ubiquitination (Li et al. 2020b). The expression level of the circRNA hsa_circ_0067582 is significantly decreased in human gastric cancer; it may serve as a potential biomarker for gastric cancer diagnosis (Yu et al. 2020). In the present study, we found that many circRNAs and lncRNAs were significantly differentially expressed in mouse testicular tissue after heat exposure. GseaPrerank analysis revealed that the top 10 upregulated and downregulated lncRNAs due to testicular heat exposure were part of various classical reproduction-related activities, such as the meiotic cell cycle (GO:0051321), DNA repair (GO:0006281), condensed chromosome kinetochore (GO:0000777), membrane raft (GO:0045121) homologous recombination (mmu03440), RNA transport (mmu03013), spliceosome (mmu03040), and nod-like receptor signaling pathway (mmu04621). Many classical signaling pathways, especially DNA metabolism, are mediated by these lncRNAs and circRNAs to regulate the effects of testicular heat exposure.
Competing endogenous RNA regulatory network analysis has been widely used in high-throughput sequencing research to identify circRNA/lncRNA–miRNA–mRNA signaling pathways that play important roles in physiological and pathological processes (Bahari et al. 2018; Zhao et al. 2019; Yang et al. 2020). Our results showed that 465 lncRNAs and 172 circRNAs were significantly differentially expression in the heat-exposed testicular tissue compared with the control group. When verifying the high-throughput sequencing results with real-time PCR, 1 of the 12 lncRNAs and 10 of 12 circRNAs with the most significant differences between the groups showed consistent expression trends with quantitative real-time PCR analysis. To further explore the function of noncoding RNA in testicular heat exposure, some top upregulated and downregulated lncRNAs (Gm19705, 4930405N21Rik, and Speer9-ps1) and circRNAs (Scfd2, Phkb, and Rint1) were chosen to construct a regulatory network. It revealed that reproduction-related genes may be affected by the Gm19705/Scfd2-miRNA regulatory network. For example, Gm19705 reversed the suppressive effect of miR-1187 on RAB7 protein expression level. The molecular mechanisms that describe interactions between circRNAs/lncRNAs and targets that regulate the testicular tissue after heat exposure need to be further studied.
In summary, high-throughput sequencing revealed differentially expressed circRNAs, lncRNAs, miRNAs, and mRNAs after testicular heat exposure. Various analyses were performed, namely, differentially expressed transcriptome exploration, GO term and KEGG pathway analyses, and regulatory network regulation. These findings have laid the foundation for further study of noncoding RNA in male reproductive diseases related to elevated testicular temperature.
Change history
02 July 2021
A Correction to this paper has been published: https://doi.org/10.1007/s00441-021-03480-1
References
Akintayo A, Liang M, Bartholdy B, Batista F, Aguilan J, Prendergast J, Sabrin A, Sundaram S, Stanley P (2020) The golgi glycoprotein MGAT4D is an intrinsic protector of testicular germ cells from mild heat stress. Sci Rep 10:2135
Badr G, Abdel-Tawab HS, Ramadan NK, Ahmed SF, Mahmoud MH (2018) Protective effects of camel whey protein against scrotal heat-mediated damage and infertility in the mouse testis through YAP/Nrf2 and PPAR-gamma signaling pathways. Mol Reprod Dev 85:505–518
Baert Y, Onofre J, Van Saen D, Goossens E (2018) Cryopreservation of human testicular tissue by isopropyl-controlled slow freezing. Methods Mol Biol 1748:287–294
Bahari MNA, Sakeh NM, Abdullah SNA, Ramli RR, Kadkhodaei S (2018) Transciptome profiling at early infection of Elaeis guineensis by Ganoderma boninense provides novel insights on fungal transition from biotrophic to necrotrophic phase. BMC Plant Biol 18:377
Bernabo P, Viero G, Lencioni V (2020) A long noncoding RNA acts as a post-transcriptional regulator of heat shock protein (HSP70) synthesis in the cold hardy Diamesa tonsa under heat shock. PLoS ONE 15:e0227172
Cai B, Sun DL, Deng WM, Jin BF (2019) [Roles of cyclins in the progression of spermatogenesis]. Zhonghua nan ke xue = Ntl J Androl 25:168–171
Cannarella R, Condorelli RA, Mongioi LM, La Vignera S, Calogero AE (2020) Molecular biology of spermatogenesis: novel targets of apparently idiopathic male infertility. Int J Mol Sci 21
Cariati F, D’Uonno N, Borrillo F, Iervolino S, Galdiero G, Tomaiuolo R (2019) Bisphenol a: an emerging threat to male fertility. Reproductive Biology and Endocrinology : RB&E 17:6
Dunleavy JEM, O’Bryan MK, Stanton PG, O’Donnell L (2019) The cytoskeleton in spermatogenesis. Reproduction 157:R53–R72
Durairajanayagam D, Agarwal A, Ong C (2015) Causes, effects and molecular mechanisms of testicular heat stress. Reprod Biomed Online 30:14–27
Frazee AC, Pertea G, Jaffe AE, Langmead B, Salzberg SL, Leek JT (2015) Ballgown bridges the gap between transcriptome assembly and expression analysis. Nat Biotechnol 33:243–246
Friedlander MR, Mackowiak SD, Li N, Chen W, Rajewsky N (2012) miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res 40:37–52
Hamilton T, Siqueira AFP, de Castro LS, Mendes CM, Delgado JC, de Assis PM, Mesquita LP, Maiorka PC, Nichi M, Goissis MD, Visintin JA, Assumpcao M (2018) Effect of heat stress on sperm DNA: protamine assessment in ram spermatozoa and testicle. Oxid Med Cell Longev 2018:5413056
Horibe A, Eid N, Ito Y, Otsuki Y, Kondo Y (2019) Ethanol-induced autophagy in sertoli cells is specifically marked at androgen-dependent stages of the spermatogenic cycle: potential mechanisms and implications. Int J Mol Sci 20
Hu JZ, Rong ZJ, Li M, Li P, Jiang LY, Luo ZX, Duan CY, Cao Y, Lu HB (2019a) Silencing of lncRNA PKIA-AS1 attenuates spinal nerve ligation-induced neuropathic pain through epigenetic downregulation of CDK6 expression. Front Cell Neurosci 13:50
Hu K, He C, Ren H, Wang H, Liu K, Li L, Liao Y, Liang M (2019b) LncRNA Gm2044 promotes 17beta-estradiol synthesis in mpGCs by acting as miR-138-5p sponge. Mol Reprod Dev 86:1023–1032
Jaffar FHF, Osman K, Ismail NH, Chin KY, Ibrahim SF (2019) Adverse effects of Wi-Fi radiation on male reproductive system: a systematic review. Tohoku J Exp Med 248:169–179
Ji H, Niu C, Zhan X, Xu J, Lian S, Xu B, Guo J, Zhen L, Yang H, Li S, Ma L (2020) Identification, functional prediction, and key lncRNA verification of cold stress-related lncRNAs in rats liver. Sci Rep 10:521
Krzysciak W, Papiez M, Bak E, Morava E, Krzysciak P, Ligezka A, Gniadek A, Vyhouskaya P, Janeczko J (2020) Sperm antioxidant biomarkers and their correlation with clinical condition and lifestyle with regard to male reproductive potential. J Clin Med 9
Lammi MJ, Qu C (2018) Selenium-related transcriptional regulation of gene expression. Int J Mol Sci 19
Li H, Xu JD, Fang XH, Zhu JN, Yang J, Pan R, Yuan SJ, Zeng N, Yang ZZ, Yang H, Wang XP, Duan JZ, Wang S, Luo JF, Wu SL, Shan ZX (2020a) Circular RNA circRNA_000203 aggravates cardiac hypertrophy via suppressing miR-26b-5p and miR-140-3p binding to Gata4. Cardiovasc Res 116:1323–1334
Li J, Tian J, Lu J, Wang Z, Ling J, Wu X, Yang F, Xia Y (2020b) LncRNA GAS5 inhibits Th17 differentiation and alleviates immune thrombocytopenia via promoting the ubiquitination of STAT3. Int Immunopharmacol 80:106127
Liang M, Yao G, Yin M, Lu M, Tian H, Liu L, Lian J, Huang X, Sun F (2013) Transcriptional cooperation between p53 and NF-kappaB p65 regulates microRNA-224 transcription in mouse ovarian granulosa cells. Mol Cell Endocrinol 370:119–129
Liu J, Zhu G, Xu S, Liu S, Lu Q, Tang Z (2017) Analysis of miRNA expression profiling in human umbilical vein endothelial cells affected by heat stress. Int J Mol Med 40:1719–1730
Lu Q, Guo F, Xu Q, Cang J (2020) LncRNA improves cold resistance of winter wheat by interacting with miR398. Functional Plant Biology : FPB 47:544–557
Marcho C, Oluwayiose OA, Pilsner JR (2020) The preconception environment and sperm epigenetics. Andrology 8:924–942
Mohamed DA, Abdelrahman SA (2019) The possible protective role of zinc oxide nanoparticles (ZnONPs) on testicular and epididymal structure and sperm parameters in nicotine-treated adult rats (a histological and biochemical study). Cell Tissue Res 375:543–558
Molina SL, Anderson KL (2018) Adult male with scrotal swelling and pain. Ann Emerg Med 71:e113–e114
Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, Groop LC (2003) PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34:267–273
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5:621–628
Pastuszak AW, Wang R (2015) Varicocele and testicular function. Asian J Androl 17:659–667
Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL (2015) StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 33:290–295
Quan J, Kang Y, Luo Z, Zhao G, Ma F, Li L, Liu Z (2020) Identification and characterization of long noncoding RNAs provide insight into the regulation of gene expression in response to heat stress in rainbow trout (Oncorhynchus mykiss). Comp Biochem Physiol d: Genomics Proteomics 36:100707
Rao M, Ke D, Cheng G, Hu S, Wu Y, Wang Y, Zhou F, Liu H, Zhu C, Xia W (2019) The regulation of CIRBP by transforming growth factor beta during heat shock-induced testicular injury. Andrology 7:244–250
Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140
Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP (2011) A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell 146:353–358
Santini SJ, Cordone V, Falone S, Mijit M, Tatone C, Amicarelli F, Di Emidio G (2018) Role of mitochondria in the oxidative stress induced by electromagnetic fields: focus on reproductive systems. Oxid Med Cell Longev 2018:5076271
Shahat AM, Rizzoto G, Kastelic JP (2020) Amelioration of heat stress-induced damage to testes and sperm quality. Theriogenology 158:84–96
Shen H, Fan X, Zhang Z, Xi H, Ji R, Liu Y, Yue M, Li Q, He J (2019) Effects of elevated ambient temperature and local testicular heating on the expressions of heat shock protein 70 and androgen receptor in boar testes. Acta Histochem 121:297–302
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 102:15545–15550
Wang D, Chen Z, Zhuang X, Luo J, Chen T, Xi Q, Zhang Y, Sun J (2020) Identification of circRNA-associated-ceRNA networks involved in milk fat metabolism under heat stress. Int J Mol Sci 21
Yang F, Chen Y, Xue Z, Lv Y, Shen L, Li K, Zheng P, Pan P, Feng T, Jin L, Yao Y (2020) High-throughput sequencing and exploration of the lncRNA-circRNA-miRNA-mRNA network in type 2 diabetes mellitus. Biomed Res Int 2020:8162524
Yang Y, Zhang X, Su Y, Zou J, Wang Z, Xu L, Que Y (2017) miRNA alteration is an important mechanism in sugarcane response to low-temperature environment. BMC Genomics 18:833
Yao G, Liang M, Liang N, Yin M, Lu M, Lian J, Wang Y, Sun F (2014) MicroRNA-224 is involved in the regulation of mouse cumulus expansion by targeting Ptx3. Mol Cell Endocrinol 382:244–253
Yu X, Ding H, Yang L, Yu Y, Zhou J, Yan Z, Guo J (2020) Reduced expression of circRNA hsa_circ_0067582 in human gastric cancer and its potential diagnostic values. J Clin Lab Anal 34:e23080
Zhang MH, Zhai LP, Fang ZY, Li AN, Qiu Y, Liu YX (2018a) Impact of a mild scrotal heating on sperm chromosomal abnormality, acrosin activity and seminal alpha-glucosidase in human fertile males. Andrologia
Zhang MH, Zhai LP, Fang ZY, Li AN, Xiao W, Qiu Y (2018) Effect of scrotal heating on sperm quality, seminal biochemical substances, and reproductive hormones in human fertile men. J Cell Biochem 119:10228–10238
Zhao R, Li FQ, Tian LL, Shang DS, Guo Y, Zhang JR, Liu M (2019) Comprehensive analysis of the whole coding and non-coding RNA transcriptome expression profiles and construction of the circRNA-lncRNA co-regulated ceRNA network in laryngeal squamous cell carcinoma. Funct Integr Genomics 19:109–121
Zhou J, Zhu C, Luo H, Shen L, Gong J, Wu Y, Magdalou J, Chen L, Guo Y, Wang H (2019) Two intrauterine programming mechanisms of adult hypercholesterolemia induced by prenatal nicotine exposure in male offspring rats. FASEB Journal : Official Publication of the Federation of American Societies for Experimental Biology 33:1110–1123
Acknowledgements
We thank Professor Fei Sun (Nantong University, Nantong, China) for experimental discussion and guidance.
Funding
This study was supported by the National Natural Science Foundation of China (81801516), the Outstanding Young Talents Support Program in Colleges and Universities of Anhui Province (China) (gxyq2020021), the 512 Talent Cultivation Plan of Middle-aged Backbone Teachers of Bengbu Medical College (China) (by51201207), the Open Project of NHC Key Laboratory of Male Reproduction and Genetics (China) (KF201909), the Key Project of Translational Medicine of Bengbu Medical College (China) (BYTM2019007), and the Undergraduate Training Program for Innovation and Entrepreneurship of Anhui Province (China) (S202010367043).
Author information
Authors and Affiliations
Contributions
Meng Liang and Ke Hu conceived this study and wrote the paper. Ke Hu, Chaofan He, Xunying Sun, and Longhui Li performed the experiments and analyzed the data. Yifan Xu, Kejia Zhang, and Xiaohua Liu reviewed the paper.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original version of this article was revised. Affiliation 1 and 2 are now corrected.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Hu, K., He, C., Sun, X. et al. Integrated study of circRNA, lncRNA, miRNA, and mRNA networks in mediating the effects of testicular heat exposure. Cell Tissue Res 386, 127–143 (2021). https://doi.org/10.1007/s00441-021-03474-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-021-03474-z