
Abstract
The testis serves as the primary site for spermatogenesis in mammals and is a crucial organ for the secretion of male hormones. Heat stress (HS) can have adverse effects on the seminiferous tubules, sperm quality, and sperm fertilization capability within the testis. Despite numerous previous studies describing various time points after heat stress in mice, a systematic and comprehensive dataset on heat stress and recovery in mice has been lacking. This study aimed to explore the gene expression changes in the recovery of multiple seminiferous epithelial cycles and spermatogenic cycles in mouse testicles after heat stress. We obtained high-throughput bulk RNA-seq data from testicular tissue of 4 NC mice and 32 HS mice (divided into 9 groups: NC, 30 min, 2 h, 6 h, 24 h, 3d, 8d, 24d, 47d, and 95d) and illustrated the dynamic changes in differential genes. This data set provides valuable insights into the detailed dynamic changes of one or more spermatogenic cycles after heat stress in mouse testicles, as well as the molecular mechanisms involved.
Similar content being viewed by others

Background & Summary
In modern society, one in seven couples of childbearing age suffers from infertility, with male infertility patients taking up half of the cases1, and it has now become a health issue of great concern2. As the core organ of the male reproductive system, the normal development of the testes is crucial to the maintenance of the function of spermatogenesis and the expression of male sexual characteristics3. The testes are the primary site of production. Within the tubular system of the testes (seminiferous tubules), spermatogonial stem cells differentiate into various types of spermatogonia. Among them, B-type spermatogonia undergo meiotic division to form spermatocytes, and as meiosis progresses, eventually develop into mature spermatozoa4,5. These spermatozoa carry genetic information and participate in the reproductive process.
Previous studies have shown that the testes are temperature-sensitive organs and that the testes control dissociation distance and thus temperature by regulating the scrotum of the organism6,7,8. Environmental changes such as global warming, sedentary lifestyles and unusual work environments can have a huge impact on the spermatogenic function of the male testes9,10. However heat stress(HS) has been recognised as an important factor influencing the decline in semen quality in men worldwide11,12. Lin et al.13 showed that after heat treatment of the posterior third of the body of male mice given a 43 °C water bath for 15 min, the testicular index of the mice was significantly reduced, with severe damage to the seminiferous tubules and abnormal testicular morphology. Wang et al.14 found that 30 min and 6 h after undergoing acute heat stress treatment at 43 °C degrees can gene transcriptional regulation of porcine testis effects. Gan et al.15 found that the testicular weight of mice dropped to 68.45% on the 7th day after heat treatment at 43 °C for 25 minutes, and there were a large number of small RNAs involved in epigenetic modification that were regulating gene functions. At the same time, mammals will continue to repair damaged testicular function during growth and development. Garcia-Oliveros et al.16 observed in Nellore bulls post-scrotal Heat Stress that, from days 14–42, there was a notable decreased sperm motility, membrane integrity, and mitochondrial membrane potential (MMP), accompanied by increase in abnormal sperm, and this symptom recovered by 70–77 days. These studies show that heat treatment not only has adverse effects on the phenotype, sperm quality, and quantity of mammalian testicles, but also affects gene expression and epigenetic regulation in testicular tissue. These adverse effects are attenuated over multiple spermatogenic cycles. In mice, the cycle of the seminiferous epithelium cycle(SEC) is approximately 8.6 days, and the complete spermatogenic cycle(SC) is around 35 days17. However, there are few reports on the process of restoring one or more complete spermatogenic cycles and seminiferous epithelial cycles after testicular heat stress.
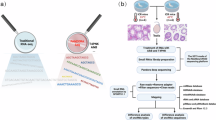
This study aims to describe the dynamic expression of genes in intact testicular tissue after heat stress. According to the seminiferous epithelium cycle and spermatogenic cycle time of mice, we divided into 9 approximately close time points, namely 30 min, 2 h, 6 h, 24 h, 3d, 24d, 47d, 95d after heat stress. we performed bulk RNA-seq at these time points and obtained high-quality sequencing results(Fig. 1). In summary, we constructed a comprehensive testicular heat stress and recovery model and displayed transcriptome data to provide a reference for the prevention and treatment of testicular heat stress injury.

The flow chart of the composition, design concept, and analysis process of the study sample, among which we selected 9 time points after heat stress that are similar to the spermatogenic cycle and the seminiferous epithelial cycle. The inner circle of arrows represents the dynamic processes of the mouse seminiferous epithelial cycle and complete spermatogenic cycle. Within this cycle, spermatogonia differentiate into A/B types during development. B-type spermatogonia, undergoing two rounds of meiotic division, eventually differentiate into spermatids, completing the spermatogenic cycle. SEC: seminiferous epithelial cycle, SC: spermatogenic cycle.
Methods
Animals and sample collection
This study used a total of 36 male ICR mice, 10 weeks old, obtained from CHENGDU DOSSY EXPERIMENTAL ANIMALS CO., LTD., Chengdu, China. Each mouse was housed individually in a cage, and all mice were maintained at a temperature of 22 ± 3 °C. Mice were provided with free access to water and food. Approval for this research was obtained from the Ethics Committee of Sichuan Agricultural University (Sichuan, China, No. 20210156).
These 36 mice with similar body weights were randomly assigned to two groups: the control group (NC, 4 mice) and the heat stress group (HS). Within the heat stress group, mice were further divided into 9 time intervals after heat treatment (HS30min: 3 mice, HS2h: 4 mice, HS6h: 3 mice, HS24h: 3 mice, HS3d: 3 mice, HS8d: 4 mice, HS24d: 4 mice, HS47d: 4 mice, HS95d: 4 mice). Among them, 8d is to recover approximately 1 SEC, 24d is to recover approximately 3 SECs, and 47d is about 1 SEC + 1 SC. 95 days is twice the time to recover. Building upon methodologies from prior studies to establish a testicular heat stress model18,19. Briefly, mice were first anesthetized with 0.01 mL/g 5% chloral hydrate, and then the mouse scrotum was placed in a water bath at 33 °C (NC group) or 43 °C (HS group) for 25 minutes. The mice were then dried and fed with a standard-fat diet (11.2% fat) in a normal environment until the end of the experiment. Anesthetized mice (0.01 mL/g 5% chloral hydrate) were placed flat on the test platform, and complete testicular samples were peeled off under conditions that complied with animal welfare. Tissue samples were collected and stored at −80 °C for subsequent experiments.
RNA extraction, library construction and sequencing
First, total RNA was extracted from the RNA samples. Following the manufacturer’s guidelines, the NEBNext Ultra RNA Library Prep Kit for Illumina (NEB, USA, Catalog #: E7530L) was used to construct sequencing libraries. Simultaneously, index code sequences were incorporated into the libraries to serve as unique identifiers for sample recognition.
In detail, poly-T oligo-attached magnetic beads were employed to isolate mRNA from total RNA. Subsequently, fragmentation was conducted using divalent cations in the NEB Next First Strand Synthesis Reaction Buffer (5X) at elevated temperature. The first step involves synthesizing the first strand cDNA using a random hexamer primer and M-MuLV Reverse Transcriptase (RNase H). Subsequently, the second strand cDNA is synthesized using DNA Polymerase I and RNase H. Any remaining overhangs were converted into blunt ends through the activity of exonuclease/polymerase. Before hybridization, the DNA fragments with adenylated 3′ ends are ligated with NEB Next Adaptor containing hairpin loop structures. The library fragments are then purified using the AMPure XP system (Beverly, USA) to select for cDNA fragments with lengths preferentially ranging from 370–420 bp. After incubating 3 µL of USER enzyme (NEB, USA) with size-selected, adaptor-ligated cDNA at 37 °C for 15 minutes, followed by 5 minutes at 95 °C, PCR was conducted. Subsequently, PCR products were purified using the AMPure XP system, and library quality was assessed on the Agilent 5400 system (Agilent, USA) before quantification by QPCR (1.5 nM).
Based on the predetermined effective library concentration and data amount, qualified library samples will undergo PE150 sequencing at Novogene Bioinformatics Technology Co., Ltd (Beijing, China).
RNA-seq data analysis process
Raw sequence data in FASTQ format generated by the Illumina novaseq 6000 sequencing platform was used for further analysis. Quality control of rawdata(Fig. 2A) was performed using TrimGalore (Version 0.6.8, https://github.com/FelixKrueger/TrimGalore) according to the following regulations (1) reads with a quality score lower than Q25 were removed (2) reads with an output reads length lower than 35 bp were removed (3) reads with an error rate of 10% or higher were removed (4) bases overlapping the front and back adapters were higher than 4 bp will be removed. And the cleandata (Fig. 2B) obtained after QC was compared to the mouse reference genome (GRCm39) using STAR (v2.7.10a)20. All samples with high mapping ratio can be used for subsequent operations. (Fig. 2C). Quantification was performed using Kallisto (v0.44.0)21 and normalized by TPM (transformed transcripts per kilobase million). At the same time, TPM was used to perform principal component analysis (PCA) clustering on the samples to verify the repeatability and availability of the data (Fig. 3A–J).

Sequencing Quality Profile Analysis. (A,B) Number of raw reads and clean reads for sequencing data. (C) Boxplot of mapping ratio. (D) Pearson’s correlation coefficient between sample replicates. (E,F) The proportion of Q20 and Q30 quality scores for sequenced bases. (G–I) The 3 types of splicing.

Principal component analysis of TPM values for all samples. (A) PCA analysis for 9 groups. (B–J) PCA analysis for NC-HS30min, NC-HS2h, NC-HS6h, NC-HS24h, NC-HS3d, NC-HS8d, NC-HS24d, NC-HS47d, NC-HS95d.
Identifcation of diferentially expressed genes
We analyzed the quantitative data for Differential expression of genes(DEG) using Deseq2(v 1.38.3)22 with |log2(fold change) | > 1.5 and pvalue < 0.05 (Fig. 4). HS3d, HS8d, HS24d showed a large number of differential genes(Fig. 4A). According to the pre-specified number of clusters, multiple groups of clusters (genes) with different dynamic patterns are finally obtained through time series analysis. Among them, for the genes in each cluster in cluster 4, they have similar temporal expression characteristics as the previous results (Fig. 4B). In addition, the number of differential genes is shown in the Fig. 4C, where red represents up-regulation and blue represents down-regulation. Through the volcano plot of differential analysis (Fig. D-L), we found a large number of differential genes in 3d, 8d,and 24d. These results indicate that this data set can more completely and realistically describe the gene expression trends of mouse testicular recovery after heat stress.
Functional enrichment analysis
In order to facilitate researchers to deeply explore the functional characterization of mouse testicular tissue recovery after heat stress and prove the analyzability of this data set, we used differential genes to conduct simple enrichment analysis of this data set. The DEG were subjected to gene ontology (GO) enrichment analyses by using Clusterprofiler package(v4.10.0)23, and then the 9 groups of up-regulated (left) and down-regulated (right) differential genes were mapped for subsequent analyses using the R language and an online analysis platform (https://www.bioinformatics.com.cn/, www.genescloud.cn/home/, accessed January 23, 2024) for mapping (Fig. 5).
Data Records
The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive (Genomics, Proteomics & Bioinformatics 2021) in National Genomics Data Center (Nucleic Acids Res 2022), China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (GSA: CRA014981) that are publicly accessible at https://ngdc.cncb.ac.cn/gsa24. The Gene Expression data reported in this paper have been uploaded to figshare25 (https://doi.org/10.6084/m9.figshare.26411167.v2).
Technical Validation
Sequencing quality statistics
We obtained a total of 460 Gb rawdata by RNA-seq (the data volume was calculated as reads *PE150 *2 paired). Subsequently, we performed a basic analysis of the data obtained by sequencing, and the number of reads of the samples was around 4 × 107, as can be seen from the graphs of our data profiles, The mapping rate to the reference genome was above 91% in all cases (Fig. 2A–C). The Pearson correlation coefficient among the samples exceeded 0.85, confirming the robustness of sample reproducibility (Fig. 2D), Q20 and Q30 confirm the reliability of data quality (Fig. 2E,F). Similarly We identified 3 splice variants in the samples by STAR software (Fig. 2G–I). These results are sufficient to prove that we have obtained a complete set of RNA-seq data sets with good quality.
Data repeatability
Principal component analysis revealed distinct clusters of HS-3d, HS-8d, and HS-24d, suggesting that gene expression in the seminiferous epithelial cycle is more distinct from NC. whereas gene expression from 30 min to 24 h was very similar to NC. Similar patterns were observed at 47 and 95 days. This shows that the effects of heat stress are repaired at the beginning and end of the second spermatogenic cycle after the complete spermatogenic cycle, and the gene expression trend gradually converges with NC (Fig. 3A). The distances between sample points represent the similarity between samples, with samples from the same tissue type exhibiting closer proximity. Clearly, samples from the same tissue type exhibit closer distances between them (Fig. 3B–J).

Profile analysis of differential genes. (A) Upset plots for 9 groups. (B) Clustering by time series analysis. (C) Number of genes for up- and down-regulated genes. (D–L) volcano diagram for NC-HS30min, NC-HS2h, NC-HS6h, NC-HS24h, NC-HS3d, NC-HS8d, NC-HS24d, NC-HS47d, NC-HS95d.

Gene ontology (GO) enrichment analyses of up-regulated(left) and down-regulated(right).
Usage Notes
This study constructed a heat stress and recovery model of mouse testicular tissue at 9 time points, which covered the recovery of one or more approximate complete seminiferous epithelial cycle and complete spermatogenic cycle (30 min, 2 h, 6 h, 24 h, 3d, 8d, 24d 47d, 95d) after heat stress. This dataset can provide a data base for research projects investigating the dynamics of gene expression in testicular heat stress. The comprehensive time points provide a reference for male testicular heat stress studies.
An important factor to be considered by the researcher is the key role of mouse strain and age in this study, as different strains of mice have different heat resistance in testicular tissue and may have different results on RNA-seq data. Due to the relatively larger size of ICR mice and their testicular tissues compared to other strains, which facilitates experimental procedures, the data obtained in this study are based on ICR mice, and the subsequent analysis is conducted on this strain of mice. Moreover, 10-week-old mice were mainly selected for the heat stress test in this study, and any interpretation of the data should be based on that age.
Also outlined in our manuscript are the methods used for heat-stressed animal handling, RNA extraction, library preparation, sequencing, and data processing, which may be helpful to other researchers seeking replication of this experiment.
Code availability
Raw sequencing data were analyzed using publicly available bioinformatics sofwares. We used common data analysis sofware packages and no custom code was created. Sofware tools used are as follows:
TrimGalore (v0.6.8, https://github.com/FelixKrueger/TrimGalore)
STAR (v2.7.10a, https://github.com/alexdobin/STAR)
Kallisto (v0.44.0, https://github.com/pachterlab/kallisto)
R sofware (v4.2.0, https://www.r-project.org/)
Clusterprofiler (v4.10.0, https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html)
Deseq2 (v1.38.3, https://bioconductor.org/packages/release/bioc/html/DESeq2.html)
Bioinformatics analysis platform (https://www.bioinformatics.com.cn/)
Genescloud tools (https://www.genescloud.cn/home)
GraphPad Prism 8 (GraphPad Sofware Inc., USA) was used for statistical analyses and data visualization.
References
Agarwal, A. et al. Male infertility. The Lancet 397, 319–333, https://doi.org/10.1016/S0140-6736(20)32667-2 (2021).
Focusing on male infertility. Nature Reviews Urology 21, https://doi.org/10.1038/s41585-024-00856-0 (2024).
Nordkap, L., Joensen, U. N., Jensen, M. B. & Jørgensen, N. Regional differences and temporal trends in male reproductive health disorders: semen quality may be a sensitive marker of environmental exposures. Molecular cellular endocrinology 355, 221–230, https://doi.org/10.1016/j.mce.2011.05.048 (2012).
Di Persio, S. et al. Spermatogonial kinetics in humans. Development 144, 3430–3439, https://doi.org/10.1242/dev.150284 (2017).
Wang, F. et al. Deficient spermiogenesis in mice lacking Rlim. Elife 10, e63556, https://doi.org/10.7554/eLife.63556 (2021).
Kim, B., Park, K. & Rhee, K. Heat stress response of male germ cells. Cellular Molecular Life Sciences 70, 2623–2636, https://doi.org/10.1007/s00018-012-1165-4 (2013).
Hjollund, N. H. I., Bonde, J. P. E., Jensen, T. K., Olsen, J. & Team, D. F. P. P. S. Diurnal scrotal skin temperature and semen quality. International journal of andrology 23, 309–318, https://doi.org/10.1046/j.1365-2605.2000.00245.x (2000).
Pérez-Crespo, M., Pintado, B. & Gutiérrez-Adán, A. Scrotal heat stress effects on sperm viability, sperm DNA integrity, and the offspring sex ratio in mice. Molecular Reproduction Development: Incorporating Gamete Research 75, 40–47, https://doi.org/10.1002/mrd.20759 (2008).
De Toni, L., Finocchi, F., Jawich, K. & Ferlin, A. Global warming and testis function: A challenging crosstalk in an equally challenging environmental scenario. Frontiers in Cell Developmental Biology 10, 1104326, https://doi.org/10.3389/fcell.2022.1104326 (2023).
Hoang-Thi, A.-P. et al. The Impact of High Ambient Temperature on Human Sperm Parameters: A Meta-Analysis. Iranian Journal of Public Health 51, 710, https://doi.org/10.18502/ijph.v51i4.9232 (2022).
Aldahhan, R. A. & Stanton, P. G. Heat stress response of somatic cells in the testis. Molecular cellular endocrinology 527, 111216, https://doi.org/10.1016/j.mce.2021.111216 (2021).
Boni, R. Heat stress, a serious threat to reproductive function in animals and humans. 86, 1307–1323, https://doi.org/10.1002/mrd.23123 (2019).
Lin, C. et al. Enhanced protective effects of combined treatment with β-carotene and curcumin against hyperthermic spermatogenic disorders in mice. BioMed Research International 2016, https://doi.org/10.1155/2016/2572073 (2016).
Wang, Y. et al. Identification of internal reference genes for porcine immature Sertoli cells under heat stress. Reproduction in Domestic Animals 57, 1344–1352, https://doi.org/10.1111/rda.14211 (2022).
Gan, M. et al. Potential Function of Testicular MicroRNAs in Heat-Stress-Induced Spermatogenesis Disorders. International Journal of Molecular Sciences 24, 8809, https://doi.org/10.3390/ijms24108809 (2023).
Garcia-Oliveros, L. N. et al. Chronological characterization of sperm morpho-functional damage and recovery after testicular heat stress in Nellore bulls. Journal of Thermal Biology 106, 103237 (2022).
Mishra, R. K. & Singh, S. K. Safety assessment of Syzygium aromaticum flower bud (clove) extract with respect to testicular function in mice. Food Chemical Toxicology 46, 3333–3338, https://doi.org/10.1016/j.fct.2008.08.006 (2008).
Hu, K. et al. Integrated study of circRNA, lncRNA, miRNA, and mRNA networks in mediating the effects of testicular heat exposure. Cell Tissue Research 386, 127–143, https://doi.org/10.1007/s00441-021-03474-z (2021).
Akintayo, A. et al. The Golgi glycoprotein MGAT4D is an intrinsic protector of testicular germ cells from mild heat stress. Scientific reports 10, 2135, https://doi.org/10.1038/s41598-020-58923-6 (2020).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21, https://doi.org/10.1093/bioinformatics/bts635 (2013).
Bray, N. L., Pimentel, H., Melsted, P. & Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nature biotechnology 34, 525–527, https://doi.org/10.1038/nbt.3519 (2016).
Anders, S. & Huber, W. Differential expression analysis for sequence count data. Nature Precedings, 1-1, https://doi.org/10.1038/npre.2010.4282.1 (2010).
Wu, T. et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. The innovation 2, https://doi.org/10.1016/j.xinn.2021.100141 (2021).
NGDC/CNCB Genome Sequence Archive https://ngdc.cncb.ac.cn/gsa/browse/CRA014981 (2024).
Mailin, G. et al. The RNA-seq mapping of Testicular Development after Heat Stress in Sexually Mature Mice. figshare https://doi.org/10.6084/m9.figshare.26411167.v2 (2024).
Acknowledgements
This work was supported by National Natural Science Foundation of China (32302689); Sichuan Science and Technology Program (2021YFYZ0030, 2021YFYZ0007, 2024NSFSC1176); the Program for Pig Industry Technology System Innovation Team of Sichuan Province (sccxtd-2024-08-09); China Agriculture Research System (CARS-35). China National Postdoctoral Program For Innovative Talents (BX20230250); China Postdoctoral Science Foundation (2023M732509).
Author information
Authors and Affiliations
Contributions
M.G. and L.Z. designed the experiment. Y.J., Y.W., J.M. and T.L. collected samples and constructed a sequencing library. Y.Y. and C.L. analyzed the data and wrote the paper. L.S. and L.Z. revised the paper. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Mailin, G., Yang, Y., Liu, C. et al. The RNA-seq mapping of Testicular Development after Heat Stress in Sexually Mature Mice. Sci Data 11, 913 (2024). https://doi.org/10.1038/s41597-024-03782-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03782-3
- Springer Nature Limited