
Abstract
Toxoplasma (T.) gondii is able to infect various cell types in different hosts. The replication of this parasite within different peripheral mononuclear blood cell populations in chicken has not yet been fully understood. Aim of the present study was to investigate the impact of chicken erythrocytes and thrombocytes as potential host cells for T. gondii. Cultures of primary avian erythrocytes and thrombocytes were inoculated with tachyzoites of T. gondii type II strain ME49. Parasite replication was detected by a quantitative real-time PCR at different times postinoculation until 24 or 48 h, respectively, displaying long-term investigations for the chosen cultures. The parasite replication curve showed a continuous decrease of parasite stages in erythrocytes and thrombocytes. Observations by light microscopy showed massive destruction for both cell populations. Few macrophages in between the infected thrombocytes were viable during the investigation period and showed internalised tachyzoites by confocal laser scanning microscopy. These findings show that T. gondii is not capable of replication in chicken erythrocytes and thrombocytes; therefore, both cannot be considered as potential host cells. In further consequence, monocyte-derived macrophages seem to be the key to the dissemination mechanisms for T. gondii in chicken.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Toxoplasma gondii (T. gondii) belongs to the class coccidia and is known to be an important parasite in mammals but in other vertebrates as well. Moreover, the number of potential host cells is as vast as the nucleated cell populations of an organism. Regarding the investigations into infection and invasion, respectively, the focus of most experiments was on leukocytes and their pathogen host interaction with the parasite.
Previously, different authors have shown the capability of T. gondii to replicate in chicken macrophages (Malkwitz et al. 2013; Quéré et al. 2013) inferring the important role of these professional phagocytes of the innate immune defence as a possible harbours for tachyzoites during the infection. However, other cell populations of tissue or blood might be of potential importance for the pathogenicity of tachyzoites.
Thus, erythrocytes of different host species may be invaded by tachyzoites of T. gondii (Schupp et al. 1978; Tanabe et al. 1980). Since these red blood cells show a variable metabolism between different species and are even nucleated in birds, the question of their susceptibility to the T. gondii infection and the capacity of T. gondii to survive develop and replicate within these cells arises. The same applies to chicken thrombocytes (DaMatta et al. 1998; St Paul et al. 2012). Erythrocytes and thrombocytes together represent about two-thirds of the blood cells in circulation in chickens.
Aim of the present study was to gain further knowledge about the susceptibility of chicken erythrocytes and thrombocytes for T. gondii infection. Both cell populations were infected in vitro with tachyzoites of T. gondii. The activity of parasite replication was determined by calculating the number of parasitic stages (tachyzoites) in erythrocytes and thrombocytes, respectively. The observations were made using a long-term infection and collated to similar experiments with the according population of macrophages to compare the host cell skills of thrombocytes and erythrocytes, respectively.
Materials und methods
Experimental design
Host cells
Erythrocytes (culture 1)
Erythrocytes were isolated as previously described by St Paul et al. (2013). Briefly, erythrocytes were gained ex vivo from chicken (Brown Leghorn chickens). Repeated blood sampling of the chickens was approved by local authorities in accordance with current German and European animal welfare legislations (animal trial notification no. V13/12, at Landesdirektion Sachsen, Leipzig, Germany). Blood was taken from the wing vein (Vena cutanea ulnaris) into citrate coated specimen cups (WDT, Garbsen, Germany), diluted with phosphate buffered saline (PBS) and centrifuged in Biocoll® separating solution (density 1.077 g/ml; Biochrom AG, Berlin, Germany) to separate the leukocyte part of the peripheral blood mononuclear cell fraction (PBMC). During the whole isolation and cultivation, blood samples were handled separately to prevent individual cross-reaction. For each isolate, the sedimented erythrocytes were washed with PBS and counted by use of a Neubauer chamber to generate a constant cell number per well. Erythrocytes were suspended in RPMI-1640 medium (PAA, Coelbe, Germany), supplemented with 10 % chicken serum, penicillin (100 U/ml, PAA), streptomycin (0.1 mg/ml, PAA) and amphotericin B (0.0025 mg/ml, PAA), seeded in 24-well tissue culture dishes (about 1 × 106 per ml, TPP, Trasadingen, Switzerland) and incubated at 40 °C with 5 % CO2.
Thrombocytes (culture 2)
The leukocyte part of the peripheral blood mononuclear cells (PBMC) was generated ex vivo from chicken blood (Brown Leghorn chickens) following the same procedure as previously described (Malkwitz et al. 2013). After the gradient centrifugation, the separated PBMC fraction was washed with PBS and cultivated from pooled blood using 6-well tissue culture dishes (TPP, Trasadingen, Switzerland) instead. After 24 h, the supernatant was removed and the remaining adherent cell cultures were washed with PBS and incubated with fresh medium for further usage.
Monocyte-derived macrophages (culture 3)
For isolation of monocyte-derived macrophages (MM), parts of the primary cell cultures generated using the isolation described above (2.1.1.2 Thrombocytes) were cultured without any manipulation for 48 h to let derived macrophages adhere, as previously described (Malkwitz et al. 2013). Then, gained primary MM were washed with PBS and replaced with fresh medium.
T. gondii tachyzoites
Tachyzoites of the type II ME49 strain (Lunde and Jacobs 1983) were maintained at 37 °C in VERO cell cultures (ECACC catalogue no. 84113001) using IMDM medium with 5 % foetal calf serum and penicillin (100 U/ml, PAA), streptomycin (0.1 mg/ml, PAA) and amphotericin B (0.0025 mg/ml, PAA). Parasites were acquired directly before use by harvesting tachyzoites from destroyed VERO cultures.
For visual demonstration of tachyzoite replication in MM only, isolated adherent cultures were infected with type I strain RH-GFP (strain RH, genetically modified to express green fluorescent protein (GFP), kind gift of Prof. Dominique Soldati-Favre, University of Geneva Medical School, Switzerland).
Infection
Infections were performed at a multiplicity of infection (MOI) of 1 tachyzoite per blood cell for all investigated cell types, which is within the usual dose range (Guillermo and DaMatta 2004; Ong et al. 2011; Seabra et al. 2002). The absolute number of blood cells per well varied slightly between the cultures for adherent PBMC and MM at the time point of infection. Thus, cell numbers in representative wells were counted directly before infection to calculate the applicable MOI.
For erythrocytes, infection was performed directly after ex vivo generation. Samples were analysed at different time points after infection until 24 h postinfection (p.i.). Thrombocytes were infected 12 h after seeding and samples of infected cultures were collected for analysis until 48 h p.i. Infection of MM was performed at 72 h after seeding and cells were collected for analysis at different time points until 48 h p.i.
Flow cytometry
To characterise the adherent primary cell populations used for infection, representative wells were harvested after 24 h of cultivation as described before (Malkwitz et al. 2013) and tested for cellular composition. Cellular characteristics were determined by flow cytometry using a FACS Canto II (BD Biosciences, Heidelberg, Germany) equipped with a 488- and 633-nm laser. Cell populations were detached from the plate by incubation with accutase (PAA, Piscataway, USA) for 30 min, washed with PBS and scraped. Gained primary cells were immediately stained with fluorescein isothiocyanate (FITC)—or phycoerythrin (PE)-conjugated mouse anti-chicken monoclonal antibodies (Southern Biotechnology Associates, Eching, Germany) directed against the following immune cell subpopulations: monocytes/macrophages (clone KUL01), alpha/beta T cells (TCRαβ/Vβ1; clone TCR-2), B cells (Bu-1; clone AV20) and thrombocytes (clone K1, kind gift from Prof. Bernd Kaspers, LMU Munich). In addition, size scatter properties were regarded.
The proportion of dead cells was calculated by propidium iodide (Sigma, Taufkirchen, Germany) staining. Data acquisition and analysis were performed as previously described (Pieper et al. 2011).
Light microscopy
Growth, morphology and the course of infection were monitored by light microscopy (DM IRB, Leica, Bensheim, Germany) before and after infection for all cultures over the whole study period.
CLSM
The adherent primary cells of the thrombocyte isolation (culture 2) were additionally examined by means of two confocal laser scanning microscopes (TCS-SP2 and TCS SP8 Leica, Bensheim, Germany).
To demonstrate cell morphology as well as infection of the cells with the parasites, thrombocyte cultures and MM cultures were grown on micro culture slides (BD Biosciences, San Jose, California, USA), infected or not with the tachyzoites (described below) and fixed with methanol for 10 min. Cell morphology was analysed by staining of cell nuclei by 4′,6-diamidino-2-phenylindole (DAPI, Sigma) and cytoplasm using Evans blue (Sigma). DAPI was additionally used for visualising intracellular and extracellular tachyzoites within the cultures. Staining procedures, adjustment of the vonfocal laser scanning microscopy (CLSM) and CLSM analysis was performed as described (Berndt et al. 2007; Malkwitz et al. 2013).
Quantitative assessment of parasite replication
The parasite burden was assessed in duplicates for every time point including uninfected negative control wells at the end (n = 6 wells per time point for every cell culture type).
For erythrocytes (culture 1), the duplicates were performed at 1 h p.i., 4 h p.i., 8 h p.i., 12 h p.i., 18 h p.i., and 24 h p.i. due to the limited viability of infected cells, which did not survive longer.
For thrombocytes (culture 2) and MM (culture 3), samples were collected at 1 h p.i., 8 h p.i., 24 h p.i., 36 h p.i., and 48 h p.i.
The number of tachyzoites at each sampling time point was determined by quantitative T. gondii deoxyribonucleic acid (DNA) detection employing a real-time quantitative PCR (qPCR) technique based on the 529-bp fragment specific for T. gondii as described formerly (Edvinsson et al. 2006).
For data analysis, the absolute replicate numbers were normalised (as percentages of the 1 h p.i. value) because of the variance of the actual number of tachyzoites applied on the respective culture wells (erythrocytes, thrombocytes, MM).
Statistical evaluation of the parasite replication data was performed using IBM SPSS® Statistics 20 package (IBM Corporation, New York, USA). Data was analysed for normal distribution by Kolmogorov-Smirnov test. Because of non-normal distribution, group differences were tested by Kruskal-Wallis and subsequent Mann-Whitney U test.
Results
Flow-cytometric characterisation of host cell cultures 2 and 3
Thrombocyte culture (culture 2)
A mean total of 80.9 % of the living cells were positively stained for the avian thrombocyte-marker K1. Additionally, about 13.8 % of all viable cells were stained positively for the avian monocyte/macrophage marker KUL01. Besides, T- and B-lymphocytes were present to a low extent. The average viability in the thrombocyte culture was 89.5 %.
Primary MM culture (culture 3)
The MM culture showed a purity similar to prior investigations with approx. 79.3 % macrophages and 16.2 % T and 3.1 % B-lymphocyte contamination. The culture viability was 91.9 % in average.
Parasite replication in erythrocytes (culture 1)
In infected erythrocyte cultures, the number of parasites detected by real-time PCR was significantly decreased (P < 0,002) to 25 % of the initial infection dose at 8 h p.i. and 10 % at 12 h p.i., respectively (Fig. 1). However, during further 12 h of cultivation, there were no additional changes in the measurable replication number.

Median parasite replication in chicken erythrocyte cultures (normalised based on 1 h p.i. values)
The observations at light microscopy showed cellular damage at 4 h p.i. already with increasing amount of cellular detritus during the course of the infection. Neither morphologically intact erythrocytes nor tachyzoites of T. gondii were seen microscopically at 12 h p.i. besides the remainders of damaged cells (Fig. 2a–c). In the uninfected control, erythrocytes remained intact until the end of the observation period (Fig. 2d).

Erythrocyte cultures. a Erythrocytes 1 h p.i. with cellular detritus (arrow) and free tachyzoites (arrowhead) in between; b 8 h p.i. few erythrocytes left, considerable amounts of cellular detritus (arrow) and free tachyzoites (arrowheads) are observed; c 24 h p.i. minor changes compared with 8 h p.i., some erythrocytes are present as well as single free tachyzoites (arrowhead) and cellular detritus (arrow); and d uninfected control (time point according to 24 h p.i.). (a–d light microscopy, ×400 optical magnification)
Parasite replication in adherent cell cultures 2 and 3
Quantification of infection
In general, the replicate number measured by qPCR showed an initial decrease (P < 0. 01) of the number of parasites in the adherent thrombocyte culture as well as in the MM culture during the first 12 h p.i. (Fig. 3a, b).

a Median parasite replication in chicken thrombocyte cultures (culture 2); b median parasite replication in derived macrophage cultures (culture 3); and c extrapolated curve of parasite replication in pure thrombocyte cultures (normalised basing on 1 h p.i. values)
In thrombocytes, the level of parasite stages at 12 h p.i. is about 3 % of the initial infection dose. This was followed by a moderate increase (P < 0.01) of nearly 50 % of the initial infection dose until 48 h p.i.
The replication rate for T. gondii-tachyzoites in infected MM cultures showed a significant increase (P = 0.019) from 12 h p.i. until 48 h p.i. reaching a proliferation rate of nearly 300 % compared to the initial infection dose (Fig. 3b).
Since the thrombocyte cultures still contained approx. 13 % macrophages which are suitable for parasite replication as shown above, we extrapolated the potential parasite replication curve (Fig. 3c) for a hypothetical pure thrombocyte culture by the following formula:
hv = median parasite copy no. in culture 21 − (median parasite copy no. in culture 31 × 0,132)
where hv is the hypothetical value of parasite copy no. by replication in a pure thrombocyte culture, 1 is the copy no. for each time point normalised based on 1 h p.i. values, and 2 complies with the percentage of contained macrophages in thrombocyte culture.
The hypothetic, estimated replication curve for T. gondii in infected pure thrombocyte cultures is given in Fig. 3c and indicates a steady decrease in parasitic stages.
Light microscopy
Observations of T. gondii-infected cultures by light microscopy showed similar results (Fig. 4) for both adherent cultures. At 3 h p.i., the adherent PBMC included a heterologous cell population of mainly thrombocytes and additionally few macrophages. There seemed to be no morphological changes in the thrombocytes, whereas the macrophages appeared more granulated. The number of viable cells was reduced obviously at 24 h p.i. Nearly no viable thrombocytes were left, but mostly macrophages were found as surviving cell population in the culture. In contrast, the control group of primary cultured thrombocytes showed no constitutional changes at the corresponding time points.

Thrombocyte cultures (culture 2) a 3 h p.i. thrombocytes with some macrophages (arrow) b 24 h p.i. decreased number of thrombocytes, while macrophages (arrow) show proliferation, thrombocytes (star) are granulated in between cellular detritus; c 48 h p.i. single attached thrombocytes (star) in-between enhanced cellular detritus and some macrophages (arrow); and d uninfected control (according to time point 48 h p.i.). (a–d light microscopy, ×400 and ×100 optical magnification)
At 3 h p.i., the MM (culture 3), which had been sub-cultivated out of the thrombocyte isolation, showed some vacuolisation. No free infectious stages of T. gondii were found until 24 h p.i. and no morphological changes could be detected in the infected cell cultures. Then, macrophages started to proliferate, to balloon and to detach in cell clusters, while single tachyzoites were rarely observed. At 48 h p.i., the number of proliferating macrophages as well as of free tachyzoites was highly increased. There were high amounts of detached cell clusters and hardly any morphologically unaltered macrophages.
The uninfected controls, in contrast, remained viable until the end of the observation period without peculiar morphological changes (Fig. 4d).
Confocal Laser Scanning Microscopy
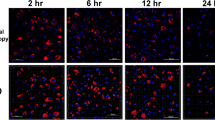
Using DAPI stain for infected thrombocyte cultures, nuclei of tachyzoites were detected intracellularly in the macrophages close to the host cell nucleus (Fig. 5) but not in the thrombocytes. Uninfected cultures showed no evident alterations. In culture 3, it was obvious that parasite replication took place in MM (Fig. 6) which was additionally confirmed by CLSM on RH-GFP-infected cultures (Fig. 7).

CLSM picture of thrombocyte culture (24 h p.i.) stained with DAPI (blue nuclei). a Infected culture showing intracellular localisation of ME49 tachyzoite nuclei (arrow) next to host cell nuclei (arrowhead) in macrophages only. b Uninfected control (according to time point 24 h p.i.)

CLSM picture of MM infected with ME49 showing a Evans blue stain of macrophage cytoplasm, b DAPI stain of nuclei, and c merge demonstrating intracellular localisation of tachyzoites

CLSM picture of MM culture (culture 3) stained with DAPI and using GFP-labelled T. gondii illustrating T. gondii invasion and replication in MM. a Merge of light and confocal microscopy (arrow: T. gondii tachyzoites). b Confocal microscopy picture showing macrophage nuclei (M; blue) and replicating tachyzoites (arrow; green)
Discussion
Erythrocytes are known to be invaded by tachyzoites of T. gondii in mammals (Schupp et al. 1978) and in chickens (Tanabe et al. 1980), respectively. Here, we demonstrate that chicken erythrocytes are not suitable host cells for T. gondii tachyzoite replication undergoing cellular death after infection which might be part of the primary immune response after in vivo infection. There are already detailed results on effects of red blood cells having enhancing effects on the immune system in mammals (Nelson 1953; Hess and Schifferli 2003) like modulating T cells (Porto et al. 2001). For vertebrates with nucleated erythrocytes, even more physiological and immunological activities regarding intracellular signalling, transcription, metabolism activity and secretion of immunoactive substances (Simons 1983) have to be considered. Chicken erythrocytes are known to be an intrinsic part of the innate immune system being capable to react to pathogen-associated molecular patterns (PAMPs) expressing transcripts for Toll-like receptors (Morera et al. 2011; St Paul et al. 2013). They are also able to express transcripts of cytokines and chemokines inducing for instance the activation of macrophages or heterophils (Kogut et al. 2005; Passantino et al. 2007). As yet the complex biological functions of avian erythrocytes are not clarified, as they might interact also as antigen-presenting cells (Salomonsen et al. 1991).
Regarding the difficulties of experiments with primary cells, especially thrombocytes with a low viability in culture systems (DaMatta et al. 1999), we were able to generate primary cultures of high purity and stability. As observations from light microscopy indicate, the viability of thrombocytes was factual until the end of the experiments. Thrombocytes are known to contribute to aspects of host immunity as well (Assinger 2014). Additionally to their hemostatic effects, platelets display a phagocytic and oxidative activity towards bacterial and viral pathogens (Lam 1997; Wigley et al. 1999). They can also secrete several different cytokines, which are stored in granules (Lindemann et al. 2001) and stimulate different types of immune cell-like monocytes, macrophages, lymphocytes and heterophils (Clark et al. 2007; Feng et al. 2014). Furthermore, thrombocytes express different Toll-like receptors after stimulation by different pathogens (St Paul et al. 2012) and play an important role in the inflammatory response (Ferdous and Scott 2015).
There is also information on platelet activation and immune activity opposing different parasites. Control of parasites by thrombocytes seems to be different from other pathogens since aggregation of the pathogen is the first important action (Da’dara and Skelly 2014; Goddard et al. 2015). Additionally, infection of thrombocytes by tachyzoites of the parasite T. gondii was found to be an active penetration process rather than phagocytosis (DaMatta et al. 1998). During our long-term investigations on parasite replication of T. gondii, we have demonstrated that thrombocytes do not hold a host cell function for this parasite. The initial and massive decrease of parasite stages of T. gondii during the first 12 h after infection is proving the lacking suitability of thrombocytes to host that parasite. Taking into account that the thrombocyte culture still contained macrophages at a level of roughly 10 %, it can be assumed that replication takes place in these according to the results from the macrophage cultures and confocal laser-scanning microscopy. The monocyte-derived macrophages contained in the thrombocyte culture are known to represent a replication reservoir for T. gondii (Malkwitz et al. 2013; Quéré et al. 2013). Even low numbers may exert a crucial impact on the replication of parasitic stages in mixed adherent PBMC cultures since high replication rates are to be seen in macrophages as we have shown by further cultivation of adherent macrophages from thrombocyte cultures and infection of these (culture 3).
Therefore, it seems reasonable to extrapolate a potentially constant decrease in pure thrombocyte cultures. However, the increase of parasite stages in the thrombocytes at 12 h after the infection needs to be investigated more detailed to clarify this aspect. In conclusion, we demonstrate in stable cultures that erythrocytes as well as thrombocytes, although nucleated, are not suitable host cells for T. gondii tachyzoites. However, monocyte-derived macrophages could be confirmed as important host cells. Moreover, thrombocytes were not able to prevent the infection of co-cultured monocyte-derived macrophages in spite of their innate immune activity. Further studies are required aiming at the details of immune reaction of thrombocytes during T. gondii infection and their interaction with monocyte-derived macrophages as well as other immunocompetent cell-like lymphocytes or granulocytes.
References
Assinger A (2014) Platelets and infection - an emerging role of platelets in viral infection. Front Immunol 5:649
Berndt A, Wilhelm A, Jugert C, Pieper J, Sachse K, Methner U (2007) Chicken cecum immune response to Salmonella enterica serovars of different levels of invasiveness. Infect Immun 75(12):5993–6007
Clark SR, Ma AC, Tavener SA (2007) Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Net Med 13:463–469
Da’dara AA, Skelly PJ (2014) Schistosomes versus platelets. Thromb Res 134:1176–1181
Damatta RA, Seabra SH, de Souza W (1998) Further studies on the phagocytic capacity of chicken thrombocytes. J Submicrosc Cytol Pathol 30:271–277
Damatta RA, Manhães L, Lassounskaia E, de Souza W (1999) Chicken thrombocytes in culture: lymphocyte-conditioned medium delays apoptosis. Tissue Cell 31:255–263
Edvinsson B, Lappalainen M, Evengård B (2006) ESCMID Study Group for Toxoplasmosis. Real-time PCR targeting a 529-bp repeat element for diagnosis of toxoplasmosis. Clin Microbiol Infect 12:131–136
Feng Y, Dorhoi A, Mollenkopf HJ, Yin H, Dong Z, Mao L, Zhou J, Bi A, Weber S, Maertzdorf J, Chen G, Chen Y, Kaufmann SH (2014) Platelets direct monocyte differentiation into epithelioid-like multinucleated giant foam cells with suppressive capacity upon mycobacterial stimulation. J Infect Dis 210:1700–1710
Ferdous F, Scott T (2015) Bacterial and viral induction of chicken thrombocyte inflammatory responses. Dev Comp Immunol 49:225–230
Goddard A, Leisewitz AL, Kristensen AT, Schoeman JP (2015) Platelet activation and platelet-leukocyte interaction in dogs naturally infected with Babesia rossi. Vet J 205:387–392
Guillermo LV, Damatta RA (2004) Nitric oxide inhibition after Toxoplasma gondii infection of chicken macrophage cell lines. Poult Sci 83:776–782
Hess C, Schifferli JA (2003) Immune adherence revisited: novel players in an old game. News Physiol Sci 18:104–108
Kogut MH, Iqbal M, He H, Philbin V, Kaiser P, Smith A (2005) Expression and function of Toll-like receptors in chicken heterophils. Dev Comp Immunol 29:791–807
Lam KM (1997) Activation, adhesion, migration and death of chicken thrombocytes. Comp Haem Int 1:81–87
Lindemann S, Tolley ND, Dixon DA, McIntyre TM, Prescott SM (2001) Activated platelets mediate inflammatory signaling by regulated interleukin 1beta synthesis. J Cell Biol 154:485–490
Lunde MN, Jacobs L (1983) Antigenic differences between endozoites and cystozoites of Toxoplasma gondii. J Parasitol 69:806–808
Malkwitz I, Berndt A, Daugschies A, Bangoura B (2013) Long-term investigations on Toxoplasma gondii-infected primary chicken macrophages. Parasitol Res 112:3115–3122
Morera D, Roher N, Ribas L, Balasch JC, Doñate C, Callol A, Boltaña S, Roberts S, Goetz G, Goetz FW, Mackenzie SA (2011) RNA-Seq reveals an integrated immune response in nucleated erythrocytes. PLoS One 6(10):e26998
Nelson RA Jr (1953) The immune-adherence phenomenon; an immunologically specific reaction between microorganisms and erythrocytes leading to enhanced phagocytosis. Science 118(3077):733–737
Ong YC, Boyle JP, Boothroyd JC (2011) Strain-dependent host transcriptional responses to Toxoplasma infection are largely conserved in mammalian and avian hosts. PLoS One 6(10):e26369
Passantino L, Massaro MA, Jirillo F, Di Modugno D, Ribaud MR, Modugno GD, Passantino GF, Jirillo E (2007) Antigenically activated avian erythrocytes release cytokine-like factors: a conserved phylogenetic function discovered in fish. Immunopharmacol Immunotoxicol 29:141–152
Porto B, Fonseca AM, Godinho I, Arosa FA, Porto G (2001) Human red blood cells have an enhancing effect on the relative expansion of CD8+ T lymphocytes in vitro. Cell Prolif 34:359–367
Pieper J, Methner U, Berndt A (2011) Characterization of avian gamma/delta T-cell subsets after Salmonella Typhimurium infection of chicks. Infect Immun 79:822–829
Quéré P, Pierre J, Hoang MD, Esnault E, Domenech J, Sibille P, Dimier-Poisson I (2013) Presence of dendritic cells in chicken spleen cell preparations and their functional interaction with the parasite Toxoplasma gondii. Vet Immunol Immunopathol 153:57–69
Salomonsen J, Dunon D, Skjødt K, Thorpe D, Vainio O, Kaufman J (1991) Chicken major histocompatibility complex-encoded B-G antigens are found on many cell types that are important for the immune system. Proc Natl Acad Sci U S A 88:1359–1363
Schupp E, Michel R, Raether W, Niemeitz H, Uphoff M (1978) Invasion of erythrocytes by Toxoplasma gondii. Z Parasitenkd 55:189–193
Seabra SH, de Souza W, Damatta RA (2002) Toxoplasma gondii partially inhibits nitric oxide production of activated murine macrophages. Exp Parasitol 100:62–70
Simons TJ (1983) Characterization of sugar transport in the pigeon red blood cell. J Physiol 338:477–499
St Paul M, Paolucci S, Barjesteh N, Wood RD, Schat KA, Sharif S (2012) Characterization of chicken thrombocyte responses to Toll-like receptor ligands. PLoS One 7(8):e43381
St Paul M, Paolucci S, Barjesteh N, Wood RD, Sharif S (2013) Chicken erythrocytes respond to Toll-like receptor ligands by up-regulating cytokine transcripts. Res Vet Sci 95:87–91
Tanabe K, Kimata I, Takada S (1980) Penetration of chick embryo erythrocytes by Toxoplasma gondii tachyzoites in simplified incubation media. J Parasitol 66:240–244
Wigley P, Hulme SD, Barrow PA (1999) Phagocytic and oxidative burst activity of chicken thrombocytes to Salmonella. Escherichia coli and other bacteria. Avian Pathol 28:567–572
Acknowledgments
The study was funded by a grant of the Karl-Enigk-Stiftung which is gratefully acknowledged. The authors thank the Centre for Infectious Diseases of the Faculty of Veterinary Medicine, University of Leipzig, for the continuous technical support. In addition, we deeply thank Prof. Soldati-Favre for providing the RH-GFP strain.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
All animal trials were performed in accordance with current German and European animal welfare legislations.
Furthermore, we wish to confirm that there are no known conflicts of interest associated with this publication and there has been no significant financial support for this work apart from the funding by Karl-Enigk-Stiftung named above. The funding did not influence the outcome of the study.
Rights and permissions
About this article

Cite this article
Malkwitz, I., Berndt, A., Zhang, R. et al. Replication of Toxoplasma gondii in chicken erythrocytes and thrombocytes compared to macrophages. Parasitol Res 116, 123–131 (2017). https://doi.org/10.1007/s00436-016-5268-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-016-5268-y