
Abstract
Toxoplasma (T.) gondii is known to infect various cell types including macrophages. In the present study, we generated monocyte-derived macrophage cultures from chicken blood. By flow cytometrical analysis, 84.5 % of the cultivated cells showed typical macrophage properties. Macrophage cultures were cultivated at either 37 °C or 40 °C, respectively, and were infected 72 to 96 h post isolationem with tachyzoites of the T. gondii type II strain ME49 at a rate of 7.5 tachyzoites per host cell. Light microscopical investigations revealed incorporation of tachyzoites into the macrophages and gradual destruction of the infected macrophage culture. Parasite multiplication was observed by a quantitative real time PCR (qPCR) based on the 529-bp fragment specific for T. gondii. Samples were drawn 1 h post infectionem (p.i.), as well as 12, 24, 36, 48, and 72 h p.i. The parasite replication curve showed a transient decrease of parasite stages 12 h p.i. followed by a tachyzoite multiplication. The comparison of different culture conditions showed a significantly higher replication rate of T. gondii at 37 °C (median value 48 h p.i., 289.2 % of the initial tachyzoite number) compared to cultures incubated at 40 °C (median value 48 h p.i., 73.1 % of the initial tachyzoite number) throughout the observation period (P < 0.05). In general, replication rates were significantly lower than in a standard VERO cell cultures at 37 °C (P < 0.05). The observed differences were attributed to the physiological chicken macrophage reaction at 40 °C probably approximating the situation in vivo.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Toxoplasma (T.) gondii is a protozoon of the class Coccidia which is distributed globally (Dubey et al. 2011a, 2011b, 2011c). Besides Felines which represent the definite hosts, many warm blooded animals serve as intermediate hosts. To these belong different avian species including chicken (de Camargo et al. 1995; Kaneto et al. 1997; Devada et al. 1998; Dubey et al. 2002; da Silva et al. 2003; Zia-Ali et al. 2007; Dubey 2010). It can be hypothesised that poultry functions as reservoir host because of high seroprevalences in the field (Dubey et al. 2008) in association with the detection of viable tissue stages, e.g. in chickens (Dubey et al. 2005). Except for epidemiologic studies and few infection experiments, little is known up to date about the properties of T. gondii in poultry. Especially the parasite–host interaction including parasite distribution over the organism has been barely investigated. Some previous in vitro investigations gave rise to the assumption that T. gondii is able to infect and replicate within avian macrophages (Onaga et al. 1983; Meirelles and de Souza 1985; Channon et al. 2000; Unno et al. 2008) which could be proven recently by Quéré et al. (2013). Nonetheless, available data is limited to a short-term-after infection.
In mammals, monocyte-derived macrophages are initially the most important population in the course of protozoal infections (Jones et al. 1972; Murray 1986). Macrophages are also the main border of the immune system when it comes to defeat protozoal infection in different organs and tissues. Therefore we established an infection model based on ex vivo isolated chicken primary monocyte-derived macrophages (in the following referred to as macrophages).
The primary cultures were infected with ME49 strain (type II) tachyzoites of T. gondii. The infection experiments were run comparatively at 37 °C or 40 °C, respectively. These temperatures were chosen because 37 °C are known to be optimal for the multiplication of T. gondii tachyzoites (Pfefferkorn and Pfefferkorn 1976; Diab and El-Bahy 2008) and used frequently in in vitro studies while 40 °C mimic the physiologic conditions in poultry. The multiplication of T. gondii tachyzoites of the different strains under different conditions was monitored by quantitative polymerase chain reaction (qPCR) over a prolonged period of 72 h.
Materials and methods
Host cells
The peripheral blood mononuclear cells (PBMC) were generated ex vivo from chicken blood (Brown Leghorn chickens). Whole blood was taken from the wing vein (Vena cutanea ulnaris) into citrate coated specimen cups (WDT, Garbsen, Germany). It was diluted with phosphate buffered saline (PBS) and centrifuged in Biocoll® separating solution (density 1,077 g/ml; Biochrom AG, Berlin, Germany) to separate the peripheral blood mononuclear cell fraction (PBMC). The separated PBMCs suspended in RPMI-1640 medium (PAA, Coelbe, Germany) supplemented with 10 % chicken serum, penicilline (100 U/ml, PAA), streptomycine (0.1 mg/ml, PAA), and amphotericine B (0.0025 mg/ml, PAA) were seeded in 6-well tissue culture dishes (TPP, Trasadingen, Switzerland) and incubated at 37 °C or 40 °C, respectively, with 5 % CO2. After 24 h, the supernatant was removed and the remaining adherent cells (monocyte-derived macrophages) were washed with PBS and incubated for another 48 h to let derived macrophages adhere and remove thrombocytes (Davies and Lloyd 1989; de Almeida et al. 2000; Wigley et al. 2002). At this, cell culture supernatant was removed after the first 24-h period and replaced by fresh medium. The gained supernatant was seeded into other dishes to obtain a second culture of derived adherent macrophages which did not adhere in the first dishes because of crowding. Thereby, the number of harvested macrophages was enhanced.
Flow cytometry
After 4 days of cultivation, the adherent cells from both cultivation temperatures and “first” and “second adherence” cultures were harvested and tested for purity and macrophage characteristics by flow cytometry using a FACS Canto II (BD Biosciences, Heidelberg, Germany) equipped with a 488 and 633-nm laser. Cell populations cultured at 37 °C and 40 °C, respectively, were detached from the plate by incubation with accutase (PAA, Piscataway, USA) for 30 min, washed with PBS and immediately stained with fluorescein isothiocyanate (FITC)- or phycoerythrin (PE)-conjugated mouse anti-chicken monoclonal antibodies (Southern Biotechnology Associates, Eching, Germany) directed against the following chicken surface antigens: KUL1 (macrophage-specific), MHC class I, MHC class II, TCR2 (alpha/beta T-cell-specific), BU1 (B-cell-specific), and CD44. The monoclonal antibodies against the avian CD14 and CD86 antigens (both AbD Serotec, Düsseldorf, Germany) were detected by means of an anti-IgG1 FITC-labelled secondary antibody (BD Biosciences). To detect the chicken CD80 antigen (AbD Serotec), an anti-IgG2a-PerCP-conjugate (BD Biosciences) was used.
The proportion of dead cells was calculated by propidium iodide (Sigma, Taufkirchen, Germany) staining. Data acquisition and analysis were performed as previously described (Pieper et al. 2011).
Light microscopy
To control growth and morphology of the cultured cells, light microscopy (DM IRB, Leica, Bensheim, Germany) was used. Development and morphology of the cells as well as the expected invasion of tachyzoites into the macrophages were monitored at 37 °C and 40 °C. Infected cell cultures were observed for 72 h after infection by light microscopy.
Confocal laser scanning microscopy (CLSM)
The adherent monocyte-derived macrophages were additionally examined by means of a confocal laser scanning microscope (TCS-SP2, Leica, Bensheim, Germany). To demonstrate cell morphology as well as infection of the cells with the parasites, the cells were cultivated on micro culture slides (BD Biosciences, San Jose, California ,USA), inoculated or not with the tachyzoites (described below) and fixed with methanol for 10 min. Cell morphology was analysed by staining of cell nuclei with 4’,6-diamidino-2-phenylindole (DAPI, Sigma) and cytoplasm using Evans blue (Sigma). DAPI was additionally used for visualising of intra- and extracellular tachyzoites within the cultures. Staining procedures, adjustment of the CLSM and CLSM analysis were performed as described (Berndt et al. 2007). All examinations were performed on cell populations cultured at either temperature (37 °C and 40 °C).
In addition, CLSM was performed on infected cultures 24 h after infection to visualise the nuclei of parasites and host cells (DAPI) and the host cell cytoplasm (Evans blue).
T. gondii tachyzoites
The tachyzoites of ME49 (type II) strain was maintained at 37 °C in VERO cell cultures (ECACC catalogue no. 84113001) using IMDM medium with 5 % foetal calf serum and penicilline (100 U/ml, PAA), streptomycine (0.1 mg/ml, PAA) and amphotericine B (0.0025 mg/ml, PAA). Parasites were acquired directly before use by harvesting tachyzoites from destroyed VERO cultures.
Infection
The infection was performed 72 to 96 h after ex vivo generation of the primary cells at a multiplicity of infection (MOI) of 7.5 tachyzoites of the ME49 strain, respectively, per host cell which is within a usual dose range per host cell (Bouchot et al. 2001; Guillermo and DaMatta 2004; Ong et al. 2011; Seabra et al. 2002). The absolute number of host cells per well varied slightly between the cultures (37 °C, 40 °C, and VERO cultures). Thus, cell numbers in representative wells were counted directly before infection to calculate the applicable MOI.
Quantitative assessment of parasite replication
Twice, parasite burden was assessed in triplicates at 1 h post infectionem (p.i.), 12 h p.i., 24 h p.i., 36 h p.i., 72 h p.i. (if intact host cells were left after 48 h p.i.), and uninfected negative control wells 72 h p.i., respectively (n = 6 wells per time point). The number of tachyzoites at each sampling time point was determined by quantitative T. gondii desoxyribonucleic acid (DNA) detection employing a real time quantitative PCR (qPCR) technique as described by Edvinsson et al. (2006) based on the 529 bp fragment specific for T. gondii.
In order to evaluate the replication potential of T. gondii inside the primary macrophages we performed a standard replication trial in the common laboratory T. gondii tachyzoite maintenance VERO cell culture under the culturing conditions described above (one triplicate). VERO cells were cultivated at same number (density) of cells per well as macrophages and infected with same MOI of 7.5 tachyzoites per host cell. Samples were taken at the same specific points in time p.i. as in macrophage cultures (at 1, 12, 24, 36, 48, and 72 h p.i.). They were tested by the same qPCR to determine the replicate numbers of tachyzoite genomes.
For data analysis, the absolute replicate numbers were normalised (as percentages of the 1-h p.i. value) because of the variance of the actual number of tachyzoites applied on the respective culture wells. Statistical evaluation of the parasite replication data was performed using IBM SPSS® Statistics 20 package (IBM Corporation, New York, USA). Data was analysed for normal distribution by Kolmogorov-Smirnov test. Because of non-normal distribution, group differences were tested by Mann–Whitney U test.
Results
Characterisation of the host cell cultures
Microscopy
By light microscopy and CLSM, the cultured cells showed typical morphological features of macrophages (Figs. 1a and 2). The monocyte-derived macrophages were round to irregular shaped with a diameter that varied from approximately 14 to 58 μm. The large and bulky nuclei were mostly centrally located and surrounded by unequally stained wide cytoplasm containing indifferent vacuoles. Cell walls appeared fluently. Comparison of the cell populations kept at 37 °C and 40 °C, respectively, revealed no difference in consistence or purity, though, macrophages seem to have a shorter lifespan at 40 °C than at 37 °C. The isolated cells stayed vital throughout until 10 days post isolationem at either temperature which is the interval needed for the conducted experiments. During the first days of culturing, cell numbers increased while in populations cultured for 20 days viability was reduced markedly down to a low percentage of the initially seeded cells.

Macrophage cultures. a Uninfected control (according to time point 72 h p.i.); b intact macrophages and some free excess tachyzoites (arrow) at 1 h p.i.; c 24 h p.i. cells show proliferation, enhanced vacuolization, and ballooning (arrow); d 48 h p.i. cell detachment, clustering (arrowhead), and free floating tachyzoites (arrow) are observed; e 72 h p.i. single attached macrophages (arrowhead) and many free tachyzoites (arrow) are present. (a–e light microscopy, ×400 optical magnification). f Infected cultures 24 h p.i. with DAPI stained cell nuclei of macrophages (arrowhead) and intracellular T. gondii tachyzoites (arrow). (CLSM, ×100 optical magnification)

Cell culture morphology 96 h post isolationem. a Evans blue stain of cytoplasm (red), b DAPI stain of cell nuclei (blue), and c merge
Flow cytometry
Primary “first” and “second adherence” cultures of both cultivation temperatures delivered similar results. By flow cytometry, 84.5 % (with a range from 73.1 % to 92.7 %) of the cultured cells exhibited typical macrophage scatter properties in the FSC vs. SSC dot plot (Fig. 3a). Within this population, the average rate of dead cells was 15.5 % (Fig. 3b); 98.5 % (with a range from 97.1 % to 99.1 %) of the living cells with macrophage morphology was positively stained for the avian monocyte/macrophage marker KUL01 and most of them also for the MHC class I, MHC class II and CD44 antigen additionally confirming the macrophage identity of the gated cells (Fig. 3c). Hardly any CD80- or CD86-positive cell was detected.

Flow cytometry of cultured cells after 4 days of seeding (n = 4; mean values of one representative culture of 37 °C first adherence, 37 °C second adherence, 40 °C first adherence, and 40 °C second adherence each). a Percentages of cells with typical macrophage scatter characteristic (macrophages) and cells showing lymphocyte/monocyte scatter properties (lymphocytes/monocytes) in the FSC vs. SSC plot. b Percentages of dead cells within the population of macrophages and lymphocytes/monocytes. c Percentages of positively stained cells for a range of avian immune cell markers within the population of macrophages and lymphocytes/monocytes
About 24.85 % of all cultured cells revealed lymphocyte/monocyte scatter characteristics in the FSC vs. SSCdotplot (with a percentage of about 6 % dead cells). Within the gated population, proportions of T cells, B cells as well as 27.9 % (with a range from 24.9 % to 31.0 %) KUL01-positive monocytes were found (Fig. 3c).
Infection model
Microscopy
Light microscopical observations
Light microscopical observations with similar results were performed at several time points after infection in both cultures kept at 37 °C or 40 °C, respectively (Fig. 1).
At 1 h p.i., T. gondii tachyzoites were lying next to the primary cells floating free in the culture medium. Until 24 h p.i., no free infectious stages of T. gondii could be found and the infected cells looked inconspicuous. At 24 h p.i., macrophages started to proliferate and to balloon. Later on, these cells formed clusters and burst while single tachyzoites were rarely observed. At 48 h p.i., the numbers of proliferated macrophages as well as of free tachyzoites was highly increased. There were many detached cell clusters and only single morphologically unchanged macrophages. The total number of macrophages decreased. At 72 h p.i., nearly all cells within the clusters were destroyed. Numerous tachyzoites lay free in the medium.
CLSM
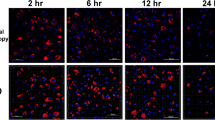
Using DAPI stain for infected cell cultures (37 °C and 40 °C culturing), nuclei of tachyzoites were visualised intracellularly in the macrophages close to the host cell nucleus (Fig. 4).

CLSM of infected macrophage culture (24 h p.i., incubated at 40 °C). a Unstained, b DAPI stain of cell nuclei (blue), and c merge showing intracellular localisation of tachyzoite nuclei (arrow) next to host cell nuclei (arrowhead)
Quantification of parasite replication by qPCR
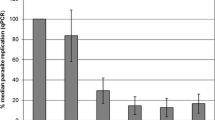
In general, the replicate number measured by qPCR shows an initial decrease of the number of parasites during the first 12 h. Subsequently, the replication rate increases from 12 to 24 h until 48 h p.i. The last detection point at 72 h p.i. signals a final decrease of the number of parasite stages in the dying cultures.
Cultivation temperature
The cultivation temperature had a significant impact on the parasite replication (P < 0.05) at any time point from 12 h p.i. onwards (Table 1). The median relative number of T. gondii stages (percentage of initial stage number measured at 1 h p.i.) was higher at 37 °C throughout. Though, the general course of infection was similar under both culturing conditions, the level of parasite genome copies varied greatly (Fig. 5). At 37 °C, parasite multiplication leads to a steady increase in replicates from 12 h p.i. peaking at 48 h p.i. (median 289.2 %). The final concentration measured 72 h p.i. (220.1 %) showed a significant increase of parasite numbers at 37 °C compared to the initially present stages at 1 h p.i.

Median parasite replication in chicken macrophage cultures compared to standard VERO culture (normalised based on 1 h p.i. values; NC negative control)
In contrast, cultures kept at 40 °C displayed a parasite multiplication on a lower level which was not predominating in the degradation of tachyzoites. The initial number of DNA copies was not reached at any later time point p.i. Maximum parasite stage number was also achieved 48 h p.i. (median percentage 73.1 %) but following decrease led to a final median of 38.2 %.
Comparison with common T. gondii-infected VERO culture
The dynamics of the standard replication curve of T. gondii which was detected in VERO cells showed an initial decrease 12 h p.i. similar to macrophage cultures (Fig. 5). Afterwards, a nearly linear progression was observed. The final normalised replicate number (as percentage of the 1 h p.i. value) measured 72 h p.i. accounted for 1267.4 % (median value) which was significantly more than in both macrophage culture settings (P < 0.05).
Discussion
The isolated macrophages showed a purity of approximately 84.5 % which is comparable to similar mammalian model cultures, e.g. from man or mice (Vissers et al. 1988; Davies and Lloyd 1989, Bennett 1966). The sum of cells not identified as viable macrophages was acceptably low with 15.5 % and represented a mixed population of lymphocytes and monocytes as well as macrophages altered by processing (e.g. accutase treatment) or aging which were not marked by monocyte-macrophage specific KUL01. The light-microscopical observations confirmed the flow cytometrical results, and macrophage-typic morphology of the isolated cells was evident, i.e. large nuclei surrounded by a wide cytoplasm with indifferent vacuoles (Enbergs and Kriesten 1969). The reproducibility of the isolation results and the homogeneity of the population purity between the „first“ and „second adherence“ cultures demonstrated the suitability of the utilised blood isolation protocol for primary macrophage culture generation.
Previous trials with T. gondii in avian macrophages were partly performed in immortalised cells, e.g. in the cell lines HD11 or NCSU (Guillermo and DaMatta 2004). Since T. gondii has been described to be effective in inhibiting apoptosis pathways (Lüder and Gross 2005, Hwang et al. 2010) and other important host cell pathways, immortalised cells are less suited for the investigation of parasite-host cell interactions. Therefore, primary cells are preferable in regard to this aspect, though, propagation, standardisation and purity are obvious advantages of cell lines.
Experiments with T. gondii in primary macrophage cultures have also been described before (Onaga et al. 1983; Meirelles and De Souza 1985; Quéré et al. 2013). At this, a principal ability of the parasite to survive and replicate in the host cell was confirmed (Onaga et al. 1983; Meirelles and De Souza 1985; Quéré et al. 2013) while the observation period of 24 h after infection in general was too short to obtain data on the dynamics of the parasite-host interaction and the parasite replication curve. In addition, the body material from which the cells are isolated is to be considered in respect of the experimental use. The presented in vitro infection model aims at the further investigation of the potential function of monocytes or macrophages, respectively, as transport cells initially after the infection of the avian organism. Such investigations are well possible with naive blood monocyte-derived macrophages which are short-term cultivated under stimuli-poor conditions, and results may be more realistic than after use of prestimulated macrophages e.g. from the spleen or peritoneal fluid as described previously (Onaga et al. 1983; Quéré et al. 2013).
Nonetheless, also significance of the results from ex vivo generated macrophage cultures as single populations is limited to a certain extent because important interactions with other cell populations of the innate and adoptive immune response are missing (Nguyen and Stadtsbaeder 1976; Murray 1986; Kasper et al. 1992; Channon et al. 2000; Fouts and Boothroyd 2007; Davison et al. 2008).
The primary cultures were infected with tachyzoites of the strain ME49 at a MOI of 7.5 which is consistent with infection doses common for T. gondii in cell cultures (Guillermo and DaMatta 2004; Bouchot et al. 2001; Seabra et al. 2002; Ong et al. 2011). Parasite incorporation and multiplication in macrophages was observed by light microscopy and CLSM performed in 12-h intervals. Thereby, intracellular vacuoles containing multiple tachyzoites were mainly visible from 24 h p.i. onwards.
Quéré et al. (2013) showed recently that tachyzoites are able to invade macrophage-like cells as well as dendritic cells actively. Mammalian macrophages have been demonstrated to act as transport host cells during parasite dissemination within the organism (Lambert and Barragan 2010). T. gondii-infected macrophages have been shown to be hypermotile compared to non-infected populations which confirms this assumption (Lambert et al. 2011). In general, different blood cell populations have been described to be differentially susceptible to T. gondii infection (Channon et al. 2000; Unno et al. 2008). However, in vivo infected macrophages not only carry the parasite but also contribute to its control by direct killing of intracellular parasitic stages (Anderson and Remington 1974, Jones et al. 1975) as well as T-cell activation (Canessa et al. 1988, Mordue and Sibley 2003).
According to earlier short-term studies (Onaga et al. 1983; Meirelles and De Souza 1985; DaMatta et al. 2000; Quéré et al. 2013), this study over a 72-h period proved the long-term functionality of at least a subpopulation of primary chicken macrophages as host cells for the acute phase tachyzoite stage of T. gondii.
During the first 24 h after infection, a decrease of the parasitic stage number was observed by qPCR. Simultaneously, free extracellular tachyzoites were visible during the first 12 h p.i. indicating a competitive inhibition of host cell invasion by the infection material excess compared to the number of macrophages. Because no dead tachyzoites were seen in the wells afterwards, an active phagocytosis of dead parasites seems plausible as described by Quéré et al. (2013). The initial decrease of parasitic DNA in parallel to microscopically visible parasite replication in some of the macrophages is assumed to base on differential host cell reaction. In mice, a macrophage subpopulation activating the cellular immune response has been identified after in vivo infections (Mordue and Sibley 2003). In chicken, similar differentiation of macrophages may be possible and macrophages may act differently, i.e. some cells may serve as host cells for parasite replication while others kill the parasite. This hypothesis is supported by observations of Onaga et al. (1983) who detected active T. gondii replication only in approximately 30 % to 35 % of the total macrophage population infected. Interestingly, there has to be a tight interaction between parasites and macrophages since from 24 h p.i. onwards, parasite replication clearly predominates (Fig. 5). Investigations into gene expression to study this interaction are in progress.
In the present experiments, two temperature regimens (37 °C versus 40 °C) were compared directly. In compliance with previous studies (Pfefferkorn and Pfefferkorn 1976) it could be shown that in principle T. gondii well tolerated the elevated cultivation temperature and parasite multiplication occurred in both settings with a minor increase in replicates at 40 °C followed by a plateau phase from 36 to 48 h p.i. ended by the death of all potential host cells. The replication was significantly more efficient and endured over a more prolonged phase at 37 °C. This is probably owed to the host cell metabolism which is altered by the chicken unphysiological temperature of 37 °C. Therefore, we conclude that in further investigations in the avian model, the normal avian body temperature of approximately 40 °C should be considered because of the physiological host cell reactivity and metabolism. Differences in T. gondii population dynamics are attributed to host cell activation and reaction under physiological temperature conditions rather than generalised temperature-based death of the parasite over time. Dead parasites would have been phagocytised and degraded, or lysed and the total replicate number would have decreased instead of displaying a plateau phase. Cell morphology and lysis as observed microscopically also were comparable between the different culture conditions.
In direct comparison to VERO cell cultures replication of tachyzoites was less effective in chicken macrophages. However, it has to be considered that primary macrophages are differentiated cells which do not multiply or only to a very limited extent (Marim et al. 2010). Thus the potential of a culture to supply host cells for the parasite is restricted. VERO cells, in contrast, multiply consistently under the given culture conditions (Jensen et al. 1974) also during the course of infection and provide a sufficient host cell population over 72 h of parasite cultivation. Additionally, VERO cells do not exhibit any immune reaction so parasite replication is not inhibited in this host cell population by defence mechanisms contrary to macrophage cultures.
Conclusion
The presented model of primary chicken macrophages displayed a high purity and suitability for in vitro infection trials. Long-term infections with T. gondii revealed a general non-linear multiplication of the parasite in the macrophage culture until complete culture lysis. The dynamics of parasite replication shown in such a model for avian toxoplasmosis for the first time, to our knowledge, indicated a distinct parasite–host cell interaction putatively based on the macrophage character as cells of the innate immune system. This will be further scrutinised in following studies.
References
Anderson SE Jr, Remington JS (1974) Effect of normal and activated human macrophages on Toxoplasma gondii. J Exp Med 139:1154–1174
Bennett B (1966) Isolation and cultivation in vitro of macrophages from various sources in the mouse. Am J Pathol 48(1):165–181
Berndt A, Wilhelm A, Jugert C, Pieper J, Sachse K, Methner U (2007) Chicken cecum immune response to Salmonella enterica serovars of different levels of invasiveness. Infect Immun 75:5993–6007
Bouchot A, Millot JM, Charpentier S, Bonhomme A, Villena I, Aubert D, Pinon JM (2001) Membrane potential changes after infection of monocytes by Toxoplasma gondii. Int J Parasitol 31:1114–1120
Canessa A, Pistoia V, Roncella S, Merli A, Melioli G, Terragna A, Ferrarini M (1988) An in vitro model for Toxoplasma infection in man. Interaction between CD4+ monoclonal T cells and macrophages results in killing of trophozoites. J Immunol 140:3580–3588
Channon JY, Seguin RM, Kasper LH (2000) Differential infectivity and division of Toxoplasma gondii in human peripheral blood leukocytes. Infect Immun 68:4822–4826
DaMatta RA, Seabra SH, Manhães L, de Souza W (2000) Nitric oxide is not involved in the killing of Trypanosoma cruzi by chicken macrophages. Parasitol Res 86:239–243
da Silva DS, Bahia-Oliveira LM, Shen SK, Kwok OC, Lehman T, Dubey JP (2003) Prevalence of Toxoplasma gondii in chickens from an area in southern Brazil highly endemic to humans. J Parasitol 89:394–396
Davies DE, Lloyd JB (1989) Monocyte-to-macrophage transition in vitro. A systematic study using human cells isolated by fractionation on Percoll. J Immunol Methods 118:9–16
Davison F, Kaspers B, Schat KA (2008) Avian Immunology. Academic Press by Elsevier Ltd., London
de Camargo MC, Antunes CM, Chiari Cde A (1995) Epidemiology of Toxoplasma gondii infection in the municipality of Ribeirâo das Neves, MG. I. Importance of domestic animals as sources of T. gondii infection in humans. Rev Soc Bras Med Trop 28:211–214
de Almeida MC, Silva AC, Barral A, Barral Netto M (2000) A Simple Method for Human Peripheral Blood Monocyte Isolation. Mem Inst Oswaldo Cruz 95:221–223
Devada K, Anandan R, Dubey JP (1998) Serologic prevalence of Toxoplasma gondii in chickens in Madras, India. J Parasitol 84:621–622
Diab MR, El-Bahy MM (2008) Toxoplasma gondii: virulence of tachyzoites in serum free media at different temperatures. Exp Parasitol 118:75–79
Dubey JP, Graham DH, Blackston CR, Lehmann T, Gennari SM, Ragozo AM, Nishi SM, Shen SK, Kwok OC, Hill DE, Thulliez P (2002) Biological and genetic characterisation of Toxoplasma gondii isolates from chickens (Gallus domesticus) from São Paulo, Brazil: unexpected findings. Int J Parasitol 32:99–105
Dubey JP, Edelhofer R, Marcet P, Vianna MC, Kwok OC, Lehmann T (2005) Genetic and biologic characteristics of Toxoplasma gondii infections in free-range chickens from Austria. Vet Parasitol 133:299–306
Dubey JP, Velmurugan GV, Chockalingam A, Pena HF, de Oliveira LN, Leifer CA, Gennari SM, Bahia Oliveira LM, Su C (2008) Genetic diversity of Toxoplasma gondii isolates from chickens from Brazil. Vet Parasitol 157:299–305
Dubey JP (2010) Toxoplasma gondii infections in chickens (Gallus domesticus): prevalence, clinical disease, diagnosis and public health significance. Zoonoses Public Health 57:60–73
Dubey JP, Velmurugan GV, Rajendran C, Yabsley MJ, Thomas NJ, Beckmen KB, Sinnett D, Ruid D, Hart J, Fair PA, McFee WE, Shearn-Bochsler V, Kwok OC, Ferreira LR, Choudhary S, Faria EB, Zhou H, Felix TA, Su C (2011a) Genetic characterisation of Toxoplasma gondii in wildlife from North America revealed widespread and high prevalence of the fourth clonal type. Int J Parasitol 41:1139–1147
Dubey JP, Rajendran C, Ferreira LR, Martins J, Kwok OC, Hill DE, Villena I, Zhou H, Su C, Jones JL (2011b) High prevalence and genotypes of Toxoplasma gondii isolated from goats, from a retail meat store, destined for human consumption in the USA. Int J Parasitol 41:827–833
Dubey JP, Passos LM, Rajendran C, Ferreira LR, Gennari SM, Su C (2011c) Isolation of viable Toxoplasma gondii from feral guinea fowl (Numida meleagris) and domestic rabbits (Oryctolagus cuniculus) from Brazil. J Parasitol 97:842–845
Edvinsson B, Lappalainen M, Evengård B (2006) ESCMID Study Group for Toxoplasmosis. Real-time PCR targeting a 529-bp repeat element for diagnosis of toxoplasmosis. Clin Microbiol Infect 12:131–136
Enbergs H, Kriesten K (1969) Fine structure of the hen's monocytes. Z Zellforsch Mikrosk Anat 97:377–382
Fouts AE, Boothroyd JC (2007) Cellular Response to Infection. Ajioka J., Soldati D. Toxoplasma Molecular and cellular biology, Horizont Bioscience, Norfolk, UK, pp 171–190
Guillermo LV, DaMatta RA (2004) Nitric oxide inhibition after Toxoplasma gondii infection of chicken macrophage cell lines. Poult Sci 83:776–782
Hwang IY, Quan JH, Ahn MH, Ahmed HA, Cha GH, Shin DW, Lee YH (2010) Toxoplasma gondii infection inhibits the mitochondrial apoptosis through induction of Bcl-2 and HSP70. Parasitol Res 107:1313–1321
Jensen MD, Wallach DF, Lin PS (1974) Comparative growth characteristics of VERO cells on gas-permeable and conventional supports. Exp Cell Res 84:271–281
Jones TC, Yeh S, Hirsch JG (1972) The interaction between Toxoplasma gondii and mammalian cells. I. Mechanism of entry and intracellular fate of the parasite. J Exp Med 136:1157–1172
Jones TC, Len L, Hirsch JG (1975) Assessment in vitro of immunity against Toxoplasma gondii. J Exp Med 141:466–482
Kaneto CN, Costa AJ, Paulillo AC, Moraes FR, Murakami TO, Meireles MV (1997) Experimental toxoplasmosis in broiler chicks. Vet Parasitol 69:203–210
Kasper LH, Khan IA, Ely KH, Buelow R, Boothroyd JC (1992) Antigen-specific (p30) mouse CD8+ T cells are cytotoxic against Toxoplasma gondii-infected peritoneal macrophages. J Immunol 148:1493–1498
Lambert H, Barragan A (2010) Modelling parasite dissemination: Host cell subversion and immune evasion by Toxoplasma gondii. Cell Microbiol 12:292–300
Lambert H, Dellacasa-Lindberg I, Barragan A (2011) Migratory responses of leukocytes infected with Toxoplasma gondii. Microbes Infect 13:96–102
Lüder CG, Gross U (2005) Apoptosis and its modulation during infection with Toxoplasma gondii: molecular mechanisms and role in pathogenesis. Curr Top Microbiol Immunol 289:219–237
Marim FM, Silveira TN, Lima DS Jr, Zamboni DS (2010) A method for generation of bone marrow-derived macrophages from cryopreserved mouse bone marrow cells. PLoS One 5:e15263
Meirelles MN, De Souza W (1985) Killing of Trypanosoma cruzi and Leishmania mexicana, and survival of Toxoplasma gondii, in chicken macrophages in vitro. J Submicrosc Cytol 17:327–334
Mordue DG, Sibley LD (2003) A novel population of Gr-1+−activated macrophages induced during acute toxoplasmosis. J Leukoc Biol 74:1015–1025
Murray HW (1986) Cellular resistance to protozoal infection. Annu Rev Med 37:61–69
Nguyen BT, Stadtsbaeder S (1976) Spontaneous interaction in vitro between lymphocytes and syngeneic peritoneal macrophages of mice. Infect Immun 13:884–889
Onaga H, Tajima M, Ishii T (1983) Activation of macrophages by culture fluid of antigen-stimulated spleen cells collected from chickens immunized with Eimeria tenella. Vet Parasitol 13:1–11
Ong YC, Boyle JP, Boothroyd JC (2011) Strain-dependent host transcriptional responses to Toxoplasma infection are largely conserved in mammalian and avian hosts. PLoS One 6(10):e26369. doi:10.1371/journal.pone.0026369
Pfefferkorn ER, Pfefferkorn LC (1976) Toxoplasma gondii: Isolation and preliminary characterization of temperature- sensitive mutants. Exp Parasitol 39:365–376
Pieper J, Methner U, Berndt A (2011) Characterization of avian gamma/delta T-cell subsets after Salmonella Typhimurium infection of chicks. Infect Immun 79:822–829
Quéré P, Pierre J, Hoang MD, Esnault E, Domenech J, Sibille P, Dimier-Poisson I (2013) Presence of dendritic cells in chicken spleen cell preparations and their functional interaction with the parasite Toxoplasma gondii. Vet Immunol Immunopathol 153:57–69
Seabra SH, de Souza W, DaMatta RA (2002) Toxoplasma gondii partially inhibits nitric oxide production of activated murine macrophages. Exp Parasitol 100:62–70
Unno A, Suzuki K, Xuan X, Nishikawa Y, Kitoh K, Takashima Y (2008) Dissemination of extracellular and intracellular Toxoplasma gondii tachyzoites in the blood flow. Parasitol Int 57:515–518
Vissers MC, Jester SA, Fantone JC (1988) Rapid purification of human peripheral blood monocytes by centrifugation through Ficoll-Hypaque and Sepracell-MN. J Immunol Methods 110:203–207
Wigley P, Hulme SD, Bumstead N, Barrow PA (2002) In vivo and in vitro studies of genetic resistance to systemic salmonellosis in the chicken encoded by the SAL1 locus. Microbes Infect 4:1111–1120
Zia-Ali N, Fazaeli A, Khoramizadeh M, Ajzenberg D, Dardé M, Keshavarz-Valian H (2007) Isolation and molecular characterization of Toxoplasma gondii strains from different hosts in Iran. Parasitol Res 101:111–115
Acknowledgements
The study was funded by a grant of the Karl-Enigk-Stiftung. The authors wish to thank the cooperating institutes of the Centre for Infectious Diseases of the Faculty of Veterinary Medicine, University of Leipzig, for the consistent technical support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Malkwitz, I., Berndt, A., Daugschies, A. et al. Long-term investigations on Toxoplasma gondii-infected primary chicken macrophages. Parasitol Res 112, 3115–3122 (2013). https://doi.org/10.1007/s00436-013-3486-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-013-3486-0