
Abstract
Toxoplasma gondii has the ability to infect various nucleated cell types in different hosts. The aim of the present study was to investigate which chicken blood cells were targeted by T. gondii in a mixed blood cell culture similar to in vivo conditions and to evaluate parasite–host cell interactions. The study consisted of two subsequent experiments. In experiment 1, we applied T. gondii tachyzoites (ME49) at a multiplicity of infection of 1 tachyzoite per blood cell and examined parasite replication, cytokine, and inducible nitric oxide synthase (iNOS) mRNA expression between 1 h and 48 h post-infection (p.i.) by quantitative PCR. By using T. gondii RH-GFP tachyzoites expressing the green fluorescent protein (GFP) in experiment 2, we aimed for visualizing infected cells by confocal laser scanning microscopy (CLSM) and flow cytometric analysis at 24 h p.i. The parasite replication curve showed a massive decrease of parasite stages until 24 h p.i. followed by an approximately plateau phase. We observed mainly significantly increased iNOS mRNA expression levels in T. gondii-infected culture compared to uninfected cells. Flow cytometry and CLSM data confirmed monocytes/macrophages as main target cells for T. gondii. Moreover, different lymphocytes like B cells and cytotoxic T cells seem to be targeted to a low extent. Our findings indicate that monocytes/macrophages play a key role during T. gondii infection in chicken as host cells and triggering of immune response. To the best of our knowledge, this is the first report of a mixed chicken blood cell culture experimentally infected with T. gondii.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Toxoplasma gondii is a widely spread apicomplexan parasite (Dubey 2010) and known to be an important zoonotic pathogen. Felids represent definitive hosts and many warm-blooded animals as well as different avian species, including chickens, serve as intermediate hosts (Dubey 2008). The seroprevalence of T. gondii in chickens of organic and backyard farms reaches up to 100% (Dubey 2010). Free-range chickens can easily become infected by feeding from oocyst-contaminated soil and represent a possible source for infection of cats (Dubey 2010).
After oral ingestion of cysts or oocysts, T. gondii disseminates to various organs as tachyzoites (fast-growing stages) and differentiate in peripheral tissues to bradyzoites (slow-growing stages) by forming persisting tissue cysts (Munoz et al. 2011).
As an obligate intracellular parasite, T. gondii has a wide host cell spectrum and can invade and replicate in almost any nucleated cell in mammals (Sibley 2004; Munoz et al. 2011).
Channon et al. (2000) reported a preference of T. gondii tachyzoites to infect and multiply in human monocytic cells compared to neutrophils and lymphocytes in vitro. Several studies in mice indicate a parasite transport function of dendritic cells and monocytes/macrophages during T. gondii infection (Courret et al. 2006; Lambert et al. 2006, 2009; Lambert and Barragan 2010). In addition, natural killer (NK) cells and T cells seem to be involved in parasite dissemination as well (Persson et al. 2007, 2009; Lambert and Barragan 2010).
In chickens, it is already proven that T. gondii is able to replicate in monocytes and dendritic cells, using them for its dissemination in the host (Malkwitz et al. 2013; Quéré et al. 2013).
Although erythrocytes and their precursor cells (Schupp et al. 1978; Tanabe et al. 1980a) of different host species can be invaded by T. gondii, parasite multiplication in chicken erythrocytes and thrombocytes could not be confirmed in vitro (Tanabe et al. 1980b; Malkwitz et al. 2017). Malkwitz et al. (2017) observed a continuous decrease of parasite stages in primary cultures of chicken erythrocytes and thrombocytes. Therefore, it can be concluded that both cell types cannot be considered potential host cells for T. gondii in chicken.
In order to study the immune response to T. gondii infection, murine models are used most frequently (Munoz et al. 2011; Dupont et al. 2012). Regarding the innate immune response, T. gondii infection leads to recruitment and activation of macrophages, NK cells, neutrophils, and dendritic cells (DCs) (Munoz et al. 2011; Dupont et al. 2012). Macrophages play a major role by producing reactive oxygen and nitrogen intermediates (Gazzinelli et al. 1993) as well as cytokines like interleukin (IL)-12 and IL-10 (Munoz et al. 2011; Dupont et al. 2012). NK cells are able to produce high amounts of interferon γ (IFN-γ) with a high activity at an early stage of infection (Sher et al. 1993; Hunter et al. 1994). For protective immunity against T. gondii, IFN-γ plays a crucial role (Suzuki et al. 1988; Filisetti and Candolfi 2004). Neutrophils and DCs are essential sources of IL-12 for promotion of NK responses, T-cell proliferation, and IL-10 and IFN-γ production (Munoz et al. 2011; Dupont et al. 2012). Adaptive immune response to T. gondii is characterized mainly by synergetic action between CD4+ and CD8+ T lymphocytes (Gazzinelli et al. 1991).
Little is known about the immune response to T. gondii infection in chickens. Malkwitz et al. (2018) described distinct primary chicken macrophage activation during T. gondii infection with upregulation of messenger RNA (mRNA) levels for the inducible nitric oxide synthase (iNOS), lipopolysaccharide-induced TNF-alpha factor (LITAF), IL-1, and IL-12 at different time points after infection. However, targeted investigations into concerted actions of chicken blood immune cells have not been published to the best of our knowledge, even though it is probable that the interaction between different blood cell populations is crucial for parasite defense mechanisms under natural conditions. Since prior studies of our working group (Hiob et al. 2017) indicate a complex local immune response, the present study was designed to investigate interactions between a representative mixed chicken blood cell population and T. gondii.
Materials and methods
Experimental design
Two subsequent experiments were conducted. The objective of experiment 1 was to gain further knowledge about the replication of T. gondii in a chicken mixed blood cell culture and immune response of these cells. Therefore, we infected a chicken mixed blood cell culture with T. gondii tachyzoites (ME49) and examined total parasite replication and mRNA levels of a pro-inflammatory response mediator and several cytokines at different time points post-infection (p.i.) by using quantitative PCR (qPCR). T. gondii ME49 was chosen as a representative of type II strain infections, which are most common in chicken and other animal species in Europe (Dubey et al. 2005; Schlüter et al. 2014).
In order to examine the cell type-specific susceptibility of chicken lymphocytes and other leukocytes for T. gondii in a mixed blood cell culture, we performed a subsequent second experiment. In experiment 2, we used T. gondii RH-GFP strain tachyzoites for infection of the obtained blood cell culture. This strain expresses the green fluorescent protein (GFP). We investigated the composition of the obtained blood cell culture by using flow cytometry and visualized infected cells by confocal laser scanning microscopy (CLSM). As our chicken mixed blood cell culture was obtained by using Biocoll® separating solution (density 1.077 g/mL; Biochrom AG, Berlin, Germany), it consisted of peripheral blood mononuclear cells (PBMCs) including nucleated chicken thrombocytes.
Host cells
Individual blood from eight (experiment 1) or six (experiment 2) Brown Leghorn chickens, respectively, was used for generation of PBMC. Whole blood was drawn from the wing vein (Vena cutanea ulnaris) into 2.5-mL EDTA-coated tubes (WDT, Garbsen, Germany). Each blood sample was processed separately in order to avoid immunological clotting reactions of lymphocytes. Samples were diluted 1:1 with Dulbecco’s modified Eagle medium (DMEM; Gibco®, Paisley, Scotland) and carefully layered over Biocoll® separating solution (density 1.077 g/mL; Biochrom AG, Berlin, Germany) to separate the PBMC fraction. After centrifugation (600×g, 20 min, room temperature, without acceleration and brake), the buffy coats containing PBMC were collected and washed twice with DMEM (centrifugation 500×g, 10 min, 4 °C, without acceleration and brake). Cells were counted using a Neubauer chamber, seeded in 24-well tissue culture dishes (Greiner Bio-One GmbH, Frickenhausen, Germany) and cultivated in RPMI-1640 medium (Gibco®, Paisley, Scotland) supplemented with 10% chicken serum (Gibco®, Paisley, Scotland), penicillin (100 U/mL; Gibco®, Grand Island NY, USA), streptomycin (0.1 mg/mL; Gibco®, Grand Island NY, USA), and amphotericin B (0.0025 mg/mL; Biochrom GmbH, Berlin, Germany). Cells were incubated at 41 °C with 5% CO2. In experiment 1, three replicates for iNOS and cytokine expression analyses and one replicate for parasite replication assessment were seeded per chicken for each infection group (negative control (NC) and T. gondii infected) and each time point (1 h, 12 h, 24 h, 48 h p.i.). In experiment 2, the amount of replicates for flow cytometry and CLSM per chicken and group varied between four and eight replicates, depending on the total amount of generated blood cells per chicken.
T. gondii tachyzoites
In experiment 1, tachyzoites of the ME49 (type II) strain were used and grown in HFF cells cultivated with Iscove’s modified Dulbecco’s (IMDM; Gibco®, Paisley, Scotland), 5% fetal calf serum (Gibco®, Paisley, Scotland), penicillin (100 U/mL), streptomycin (0.1 mg/mL), and amphotericin B (0.0025 mg/mL) at 37 °C, 5% CO2 as described before (Zintl et al. 2009). In contrast, for experiment 2, tachyzoites of the RH-GFP (type I) strain (kindly provided by Prof. D. Soldati-Favre) were used. RH-GFP tachyzoites were grown in HFF cells as indicated above but cultivated with penicillin (250 U/mL) and streptomycin (0.25 mg/mL).
To harvest the parasites, the cell culture supernatant was centrifuged at 2000×g for 5 min. The pellet was resuspended in 1 mL IMDM and tachyzoites were counted using a Neubauer chamber to adjust the infection dose. Viability was determined microscopically by assessing morphology and motility of the tachyzoites.
Infection
Infection with T. gondii ME49 type II strain tachyzoites (experiment 1) or RH-GFP type I strain tachyzoites (experiment 2) was performed immediately after cultivation of primary blood cells in the infection wells. The infection dose was established as a multiplicity of infection (MOI) of 1 tachyzoite per blood cell, which is within a usual dose range per host cell (Bouchot et al. 2001; Seabra et al. 2002; Guillermo and DaMatta 2004; Ong et al. 2011; Malkwitz et al. 2017). Cell numbers were counted directly before seeding (1.25 × 106 cells per well) to adjust the applicable MOI. Control wells containing the same number of mixed blood cells remained uninfected.
Cell cultures were monitored twice daily until 48 h p.i. by light microscopy (DMI LED; Leica, Bensheim, Germany) to observe morphology and growth of the cultured cells and tachyzoites (data not shown).
Quantitative assessment of parasite replication (experiment 1)
DNA extraction of cell culture samples (infected and uninfected NC wells) was performed for individual samples on four different sampling time points (1 h p.i., 12 h p.i., 24 h p.i., and 48 h p.i.). Extraction was done using the NucleoSpin® Tissue Kit (Macherey-Nagel, Düren, Germany) following the manufacturer’s instructions for cultured cells.
The number of T. gondii tachyzoites at each sampling time point was determined as DNA copy number by using qPCR based on the T. gondii-specific 529-bp fragment as described by Edvinsson et al. (2006). All qPCR measurements were performed in technical triplicates. Briefly, the 25 μL master mix consisted of 5 μL DNA eluate, 12.5 μL 2× Maxima Probe/ROX qPCR Master Mix (Thermo Fisher Scientific, Schwerte, Germany), 0.9 μL of each primer (25 μM), 5 μL TaqMan probe (2 μM), and nuclease-free water. In every qPCR batch, a positive control containing a known amount of T. gondii DNA from cell culture-derived ME49 tachyzoites and three negative controls (two aliquots of water and one aliquot of DNA elution buffer) were carried along.
Cytokine and iNOS expression profile (experiment 1)
Cell culture samples of uninfected and infected wells were collected at the same time points when T. gondii replication was assessed, i.e., 1 h p.i., 12 h p.i., 24 h p.i., and 48 h p.i., and stored at − 80 °C until RNA extraction. Expression of iNOS and the following cytokines was examined: IFN-γ, tumor necrosis factor α (TNF-α), and IL-10. Therefore, target-specific reverse transcriptase quantitative polymerase chain reactions (RT-qPCR) were employed.
Total RNA extraction and cDNA synthesis
For RNA extraction, the RNeasy® Mini kit (Qiagen, Hilden, Germany) was used following the manufacturer’s instructions for purification of total RNA from animal cells using spin technology. The RNA concentration was measured by a nanophotometer (NanoDrop Thermo Scientific 2000c, Schwerte, Germany). For complementary DNA (cDNA) synthesis, approximately 10 ng/μL RNA was used per sample. Reverse transcription into cDNA was done using the Revert-Aid® first-strand cDNA synthesis kit (Thermo Fisher Scientific, Schwerte, Germany) as described by Hong et al. (2006). The mixture was incubated for 5 min at 25 °C, followed by 60 min at 42 °C and a final heating reaction at 70 °C for 5 min.
qPCR
The mRNA expression of iNOS and cytokines were quantified by a qPCR assay (Park et al. 2008) which was slightly modified as described below. Transcript levels of genes were normalized to those of the two reference genes glyceraldehyde 3-phosphate dehydrogenase (GAPDH), as described by Park et al. (2008), and glucose-6-phosphate dehydrogenase (G6PDH; De Boever et al. 2008). Sequences of the primers used for RT-qPCR are listed in Table 1.
The qPCR assay was performed according to Hong et al. (2006). Ten microliters of 2× SYBR Green master mix (Thermo Fisher Scientific, Schwerte, Germany), 6.6 μL nuclease-free water, 0.4 μL ROX solution (0.5 μM), 2 μL cDNA template, and 0.5 μL of the corresponding primers (25 μM) were used per reaction.
The following thermal profile setup was applied: initial denaturation at 95 °C for 10 min, followed by 40 cycles at 95 °C for 30 s, 60 °C for 30 s, and 72 °C for 1 min, and termination at 95 °C for 1 min, 55 °C for 30 s, and 95 °C for 30 s. Reference target stability and calibrated normalized relative quantities (CNRQ) were calculated using qBase+ v.2.3 (Biogazelle NV, Gent, Belgium).
Flow cytometry (experiment 2)
After 24 h of cultivation, the supernatants (containing the non-adherent cells) of all wells were collected in glass tubes (four to eight individual samples per chicken and group), and the corresponding adherent blood cells were detached from the plate by incubation with accutase (PAA, Piscataway, USA) for 45 min at 37 °C. Cells were washed twice with PBS, centrifuged at 300×g for 20 min, and resuspended in 1000–1500 μL PBS, depending on cell density. A portion of the obtained primary cells were immediately stained with phycoerythrin (PE)-, allophycocyanin (APC)-, or biotin (BIOT)-conjugated mouse anti-chicken monoclonal antibodies (Southern Biotechnology Associates, Eching, Germany) and incubated for 30 min at room temperature in the dark. The biotin-conjugated monoclonal antibody was detected by Streptavidin-DyLight 405 (Dianova, Hamburg). An overview of all used monoclonal antibodies is shown in Table 2. The proportion of dead cells was calculated by 4′,6-diamino-2-phenylindole (DAPI; Sigma, Taufkirchen, Germany) staining. Analysis of dead cells was performed within the CD45+ leukocyte population of the PBMCs. Thus, all material smaller than lymphocytes (apoptotic cells, debris) has been excluded from the analysis.
Cellular characteristics were examined by flow cytometry using a FACS Canto II (BD Biosciences, Heidelberg, Germany) equipped with a 405-, 488-, and 633-nm laser. After gating of CD45-positive leukocytes, the percentage of T. gondii-positive cells in each single cell population (CD4+ T cells, CD8α+ T cells, γδ T cells, B cells, and monocytes/macrophages) was determined.
Confocal laser-scanning microscopy (CLSM; experiment 2)
Remaining cells of every chicken and group (24 h p.i.) were examined by means of a confocal laser scanning microscope (TCS-SP2; Leica, Bensheim, Germany). For this purpose, ca. 50 μL of every sample was transferred to poly-dimethyl-diallyl-ammonium chloride (PDDA)-coated slides and incubated in moist chambers for 60 min at 37 °C. The slides were fixed with 2% paraformaldehyde for 10 min. After 30 min drying, slides were covered with aluminum foil and stored at − 20 °C until further examination.
For staining the immune cells, the slides were incubated with the unlabeled monoclonal antibodies anti-avian CD8α and anti-avian BU1 (see Table 2) for 30 min at room temperature and subsequently stained with an anti-IgG1 antibody labeled with Alexa Fluor 546 (Invitrogen, Karlsruhe, Germany). Finally, the samples were counterstained with 1 μg/mL DAPI for DNA staining (Sigma, Taufkirchen, Germany) and mounted with ProLong® Gold anti-Fade reagent (Invitrogen, Karlsruhe, Germany).
Light microscopy (experiment 2)
For light microscopy, cells were placed on the slides as described above, fixed with methanol for 10 min and air-dried.
For staining of parasites, slides were incubated with a polyclonal anti-Toxoplasma gondii rabbit antibody-FITC (PA1-7253; Thermo Fisher Scientific, Schwerte, Germany) and the parasites visualized using a goat anti-rabbit immunoglobulin labeled with horseradish peroxidase (Dako, Hamburg, Germany). The enzyme-linked antibody was detected by its reaction with 3,3-diaminobenzidine (DAB; Dako, Hamburg, Germany). Cells were counterstained with hematoxylin (Carl Roth, Karlsruhe, Germany) and mounted with Canada balsam (Riedel de Haen AG, Seelze-Hannover, Germany). As a negative control, the primary antibody was replaced with phosphate-buffered saline (PBS).
Statistical methods
Statistical evaluation of the parasite replication and CNRQ values of cytokine mRNA expression was performed by using IBM SPSS Statistics v.22 (IBM, New York, USA). Data was analyzed for normal distribution by Kolmogorov–Smirnov test. Because of non-normal distribution, group differences were tested by Kruskal–Wallis and successive Mann–Whitney U test. Flow cytometry data was analyzed by BD FACSDIVA™ software (BD Biosciences, Heidelberg, Germany) and statistically evaluated by one- or two-way ANOVA. P values < 0.05 were considered as statistically significant.
Results
Parasite replication (experiment 1)
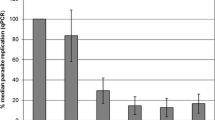
All samples were measured in triplicates by qPCR at the given four time points up to 48 h p.i. There was no parasite replication in the uninfected, NC wells at any sampling time point. The infection wells showed a decrease of parasite DNA copy numbers from 1 h p.i. onwards (see Fig. 1), which was statistically significant from 1 h p.i. to 24 h p.i. (p = 0.01, Mann–Whitney U test) and to 48 h p.i. (p = 0.015, Mann–Whitney U test). Compared to parasite numbers at 1 h p.i. (= 100%), median number of parasites was about 51% at 12 h p.i. and 36.7% at 24 h p.i. This was followed by a moderate decrease to a median relative copy number of 35.1% (compared to 1 h p.i.) until 48 h p.i. However, at the end of the experiment, half of the individual chicken blood samples (n = 4) showed an increase of parasite numbers compared to 24 h p.i. between 19% and 63.3%.

Parasite replication in individual chicken PBMC cultures (1.25 × 106 cells per well) following infection with T. gondii (ME49; experiment 1) at a multiplicity of infection (MOI) of 1 tachyzoite per blood cell. Data points of individual chicken (n = 8) and the median per sampling time point (1 h/12 h/24 h/48 h p.i.) are presented (**p = 0.01, *p = 0.015; Mann–Whitney U test)
Cytokine and iNOS expression profile (experiment 1)
Cytokine and iNOS expression in infected wells were assessed as CNRQs for each sampling time point compared to the mRNA expression levels in uninfected, NC wells. CNRQs of iNOS, IL-10, and TNF-α mRNA were compared between infected and NC wells at all sampling time points (see Fig. 2a–c). Additionally, CNRQs were statistically compared for infected wells between different sampling time points. The fold change was calculated for infected wells compared to NC wells by setting NC values to 1. Statistical evaluation of IFN-γ levels was not possible due to rare detection of IFN-γ mRNA above identification threshold. The displayed p values were calculated by Mann–Whitney U test.

Mean calibrated normalized relative quantities (CNRQ) of iNOS (a), IL-10 (b), and TNF-α (c) inclusive standard deviation at all sampling time points compared between NC (uninfected) and infected group (*1p < 0.001, *2p < 0.01; Mann–Whitney U test; experiment 1)
iNOS
Infected wells showed a significantly higher iNOS mRNA expression at 12 h p.i. (p < 0.001; fold change 1.83), 24 h p.i. (p < 0.001; fold change 2.81), and 48 h p.i. (p < 0.001; fold change 10.41) compared to uninfected, NC wells (Fig. 2a). Over the course of time, we detected statistically significant increases of iNOS levels from 1 h p.i. to 48 h p.i. (p = 0.001), 12 h p.i. to 24 h p.i. (p = 0.001), 12 h p.i. to 48 h p.i. (p < 0.001), and from 24 h p.i. to 48 h p.i. (p < 0.01).
IL-10
Infected wells showed higher IL-10 levels at 1 h p.i. (p < 0.01; fold change 3.42) and 24 h p.i. (p > 0.05; fold change 1.63; Fig. 2b). Statistical evaluation at 48 h p.i. was not possible due to only one detectable value in the NC wells. Moreover, we detected a significant decrease in IL-10 mRNA expression in the infected wells from 1 h p.i. to 12 h p.i. (p < 0.05).
TNF-α
We detected significantly higher TNF-α levels at 48 h p.i. (p < 0.001; fold change 2.13) in infected wells compared to NC wells (Fig. 2c). When comparing different sampling time points in infected wells, we found significant decreases in TNF-α mRNA expression from 1 h p.i. to 24 h p.i. (p < 0.05) and 12 h p.i. to 24 h p.i. (p < 0.01). In contrast, TNF-α levels significantly increased from 12 h p.i. (p = 0.02) and 24 h p.i. (p < 0.001) to 48 h p.i.
Characterization of host cell culture by flow cytometry, confocal laser scanning microscopy, and light microscopy (experiment 2)
By flow cytometry, the percentage of dead leukocytes was 5.47 ± 0.26% in the control culture and 21.7 ± 1.00% in the infected culture (p > 0.05; Fig. 3). The proportion of T. gondii-positive immune cells after 24 h of cultivation in infected and NC culture is displayed in Fig. 4a. In the infected culture, 73.0% of the monocytes/macrophages and 11.2% of B cells and/or monocytes/macrophages were T. gondii-positive (Fig. 4a). To a very low extent, T. gondii-positive cells were also found within the CD4+ (3.13%), CD8α+ (1.12%), and γδ (1.63%) T cells (Fig. 4a).

Flow-cytometric analysis of the percentage of dead cells within the CD45+ leukocyte population (mean, standard deviation) of non-infected (Control) and T. gondii-infected PBMCs after 24 h of cultivation (n = 3; experiment 2)

Flow cytometric analyses of control and T. gondii-infected PBMC (RH-GFP) after 24 h of cultivation (experiment 2; mean, standard deviation). a Percentage of T. gondii-positive immune cell subsets (CD4+, CD8α+, γδ T cells, B cells, and monocytes/macrophages) of control and T. gondii-infected PBMCs after 24 h of cultivation (*p < 0.05; ANOVA). b Comparison percentages of immune cell subsets (CD4+, CD8α+, γδ T cells, B cells, and monocytes/macrophages) within CD45+ leukocytes between control and T. gondii-infected PBMCs after 24 h of cultivation
The percentage of T. gondii-positive cells within the monocytes/macrophages in the infected culture is significantly higher than in all other immune cell subsets (p < 0.05). By comparing the infected and control cultures, a statistically significant difference was detected only in the proportion of the monocytes/macrophages subset (p < 0.05; Fig. 4a).
The proportion of immune cell subsets within CD45+ leukocytes of T. gondii-infected and control culture is shown in Fig. 4b. CD4+ T cells were increased in the infected culture, whereas proportions of CD8α+ T cells and γδ T cells were similar in infected and control culture. In contrast, B cells and monocytes/macrophages were slightly reduced in infected culture. No statistically significant differences were detected between infected and control culture.
Using CLSM for DAPI stained samples at 24 h p.i., mainly free T. gondii tachyzoites were seen (Fig. 5a). Intracellular tachyzoites were detected in monocytes close to the host cell nucleus (Fig. 5b).

CLSM picture of infected PBMC culture 24 h p.i. stained with DAPI (blue nuclei). Merge of transmitted and ultraviolet light (excitation with 488 and 405 nm laser; experiment 2). a Free T. gondii tachyzoites (green) located between nuclei (blue) of different PBMC. b Monocyte with intracellular tachyzoite (green; arrow)
Staining with BU1 (Fig. 6a, b) and CD8α (Fig. 7) revealed co-localization of cells and tachyzoites only occasionally. In single cases, T. gondii-infected B cells and/or monocytes/macrophages and cytotoxic T cells were detected.

CLSM picture of infected PBMC culture 24 h p.i. stained with BU1 and an anti-IgG1 antibody labeled with Alexa Fluor 546. Counterstaining with DAPI (experiment 2). a B cell (red) with tachyzoite (green; arrow). Larger green round structures may represent fixation artifacts. b B cell (red) with tachyzoite (green; arrow) and B cell (red) in close contact with tachyzoite (green; arrowhead)

CLSM picture of infected PBMC culture 24 h p.i. stained with CD8α and an anti-IgG1 antibody labeled with Alexa Fluor 546. Counterstaining with DAPI. Cytotoxic T cell (red) with tachyzoite (green; arrow) (experiment 2)
Light microscopy of slides stained with hematoxylin and sequentially incubated with an FITC-conjugated anti-T. gondii rabbit antibody and a horseradish peroxidase-conjugated anti-rabbit IgG led to detection of mainly free tachyzoites and several indications of attached or intracellular parasites in monocytes or lymphocytes (Fig. 8).

Light-microscopic picture of infected PBMC culture 24 h p.i. using a polyclonal anti-Toxoplasma gondii rabbit antibody-FITC and goat anti-rabbit immunoglobulin labeled with horseradish peroxidase. Counterstaining with hematoxylin. Nuclei (blue) of PBMC and intracellular tachyzoite (brown; arrow) (experiment 2)
Discussion
Recently, investigations concerning interactions between T. gondii and monocultures of different chicken blood cells like monocytes, erythrocytes, and thrombocytes were published (Malkwitz et al. 2017, 2018). However, to the best of our knowledge, this is the first experiment in a mixed chicken blood cell culture following T. gondii infection in vitro.
In the present study, we examined parasite replication and expression of iNOS and several cytokines at different time points after infection with T. gondii tachyzoites (ME49, experiment 1). Additionally, by using genetically modified RH-GFP T. gondii in experiment 2, we aimed for visualizing infected host cells in a PBMC culture. It has to be considered that the use of two different T. gondii strains does not allow direct comparison between both experiments. Nevertheless, we chose T. gondii strain ME49 in experiment 1 because type II strain infections are most common in chicken and other animal species in Europe (Dubey et al. 2005; Schlüter et al. 2014) and mimic field conditions as a cyst-forming strain more precisely.
During the first 24 h after infection, we observed a massive decrease of the parasitic stage number to 36.7% of the initial amount (1 h p.i.), followed by an approximately plateau phase up to 48 h p.i. This is in line with findings from Malkwitz et al. (2017) in separate, T. gondii-infected chicken cell cultures of erythrocytes or thrombocytes and proves the lacking suitability of these cell types to host T. gondii. However, we observed an increased replication of T. gondii at 48 h p.i. in individual cell cultures obtained from single chickens. This may be explained by a varying composition of the isolated blood cell cultures obtained from different chickens. Since the different blood cell populations and their interaction are essential for innate and adaptive immune function to control the parasite population, the efficacy of signaling pathways may depend on the proportions of different immune cells available at 48 h p.i. A similar variation in individual chicken PBMC culture composition has been demonstrated before by Fair et al. (2008). Fair et al. (2008) examined blood samples from 284 adult chickens by flow cytometry and detected a large range in different lymphocyte subpopulations like CD3+, CD4+, or CD8+. The author assumed that this variability may be attributed to individual health or condition variables or stress due to environmental factors. Besides, some cell types are already known to serve as important host cells for T. gondii in chicken like monocyte-derived macrophages (Quéré et al. 2013; Malkwitz et al. 2013, 2017) which form about 48 to 72 h after blood cell isolation. Other cell types such as thrombocytes or erythrocytes seem not to hold a relevant host cell function in chicken (Malkwitz et al. 2017) thus the exact proportion of blood cells available in the individual cultures at 48 h p.i. may play a crucial role for the variation observed.
Previous long-term studies in our laboratory with pure monocyte-derived macrophages cultures infected with T. gondii strain ME49 (type II) or NED (type III) indicated a decrease of parasite stages until 12 h p.i., followed by an increase until 36 h (ME49) or 48 h p.i. (NED) and a subsequent distinct decrease of parasite stages until 72 h p.i. (Malkwitz et al. 2018). However, differences to our experiment may be explained by the cultivation of mixed chicken blood cells instead of monocultures, a different MOI, and a slightly changed cultivation procedure.
Since a clear decrease of parasite stage numbers was obvious in our mixed chicken blood cell culture, we conclude that chicken blood is not a major site of parasite replication but serves predominantly for parasite transport only. In addition, monocyte-derived macrophages may function as Trojan horses for entry of and later replication in tissues (Da Gama et al. 2004; Courret et al. 2006; Unno et al. 2008).
In our experiment, we examined mRNA expression of different cytokines as representatives for Th1 (IFN-γ, TNF-α) and Th2 (IL-10) immune response and iNOS produced by monocytes/macrophages. We observed mainly significantly increased iNOS mRNA expression from 12 h to 48 h p.i. in T. gondii-infected cells compared to uninfected control cells with an approximately 10-fold higher level at 48 h p.i. in comparison to 1 h p.i. Upregulation of iNOS expression in chicken monocyte-derived macrophage culture following T. gondii infection has also been reported lately by Malkwitz et al. (2018). In contrast, DaMatta et al. (2000) did not detect NO production in chicken macrophages infected with T. gondii. Inhibition of NO production is also described in chicken macrophage cell lines MQ-NCSU and HD11 during T. gondii infection (Guillermo and DaMatta 2004). Similar results were found in murine macrophages (Seabra et al. 2002; Lüder et al. 2003). These differences to our findings may be explained by usage of type I strain (RH) of T. gondii in most of these studies in contrast to type II strain (ME49) in our experiment. However, Lüder et al. (2003) used also T. gondii type II strain (NTE) for infection and examined a downregulation of iNOS mRNA levels by T. gondii in IFN-γ and IFN-γ/LPS-treated RAW264.7 cells. At the same time, NO production was dose-dependently reduced by T. gondii or only marginally detectable. Nonetheless, it has to be taken into consideration that it cannot necessarily be assumed that iNOS mRNA expression correlates with NO production. Moreover, it can be speculated that T. gondii may inhibit NO production at a later stage after initial mRNA formation, while iNOS mRNA expression itself is not altered. Nevertheless, it is known that NO plays an important role in inhibition of T. gondii replication (Adams et al. 1990; Zhao et al. 2009).
Regarding Th1 immune response, we detected significantly increased TNF-α mRNA expression 48 h p.i. in the infected cultures compared to control cells. Over time, an initial decrease (1 h to 24 h p.i.) of mRNA expression followed by a statistically significant increase (24 h to 48 h p.i.) could be observed. Robben et al. (2004) described increased TNF-α levels in murine macrophages following T. gondii type II infection. Similar findings are reported regarding serum measurements in T. gondii-infected mice (Dias et al. 2014) and mRNA expression levels in spleens of the fat-tailed dunnart (Sminthopsis crassicaudata) experimentally infected with T. gondii (Donahoe et al. 2017). Strikingly, Butcher and Denkers (2002) described inhibition of TNF-α production in murine macrophages, but used T. gondii type I for infection. This fact illustrates that parameters like T. gondii strain, host species, or infection dose influence the course of immunological pathways, like also reported by Kim et al. (2006) concerning influence of T. gondii genotype on IL-12 regulation. Surprisingly, we did not detect increased IFN-γ mRNA expression in infected cells. As NK cells and T lymphocytes are main sources for IFN-γ (Hunter et al. 1994; Dupont et al. 2012), we assume a low amount of these cells in our culture, leading to a low sensitivity in detecting this kind of response, or an effective inhibition via the Th2 immune response.
In respect to the measured Th2 response, higher IL-10 levels in infected cells compared to control cells reflect the initiation of counter-regulatory mechanisms. Robben et al. (2004) also described elevated IL-10 levels in T. gondii-infected murine macrophages. Similar findings are reported by Khan et al. (1995) in splenocytes of infected mice. T. gondii is known to trigger production of anti-inflammatory cytokines such as IL-10 or transforming growth factor-β (Gazzinelli et al. 1996) for achieving an adequate balance between immune-regulating mechanisms.
Differentiation of avian leukocytes by flow cytometry has already been described for thrombocytes (Viertlboeck and Göbel 2007), lymphocyte subpopulations (Fair et al. 2008), and monocytes/macrophages (Mast et al. 1998).
By using T. gondii tachyzoites expressing GFP for infection of our chicken PBMC culture, the generated flow cytometric data confirm monocytes/macrophages as main target cells for T. gondii.
About 11.2% of B cells and/or monocytes/macrophages (BU1) were T. gondii positive. This suggests that B cells are also capable to host tachyzoites; however, it has to be considered that the marker BU1 also slightly stained monocytes/macrophages. Different T-cell subpopulations like CD8α+-, CD4+-, or γδ T cells seem to be targeted by T. gondii only to a very low extent (≤ 3.13%).
We observed a slight decrease of monocytes/macrophages and B cells in the infected cell culture compared to uninfected cells, although this was not statistically significant. It can be assumed that besides holding host cell and shuttle function (Courret et al. 2006; Unno et al. 2008; Quéré et al. 2013; Malkwitz et al. 2013, 2017) or direct parasite killing by macrophages (Anderson and Remington 1974; Jones et al. 1975), a proportion of these cells may undergo apoptosis or necrosis due to intracellular parasites. It is known that T. gondii considerably modulates apoptosis of infected as well as uninfected host cells (Lüder and Gross 2005). Interestingly, by triggering (Khan et al. 1996; Gavrilescu and Denkers 2001; Mordue et al. 2001) or inhibiting cell death program (Nash et al. 1998; Goebel et al. 1999; Payne et al. 2003), T. gondii is able to facilitate its intracellular development and persistence in the host.
Visualization of the infected cell cultures by CLSM confirmed monocytes/macrophages as host cells for T. gondii. Additional staining with BU1 revealed positive B cells and/or monocytes/macrophages that corroborates flow cytometry results. T. gondii-positive cytotoxic T cells were detected only occasionally by using CD8α staining. It could be possible that the found co-localizations between cytotoxic T cells and T. gondii have happened by chance, as the obtained flow cytometry results are also very low (1.12%). In contrast to Malkwitz et al. (2017), who detected T. gondii replication in monocyte/macrophages culture, we did not observe parasite replication within any host cell at 24 h p.i. Visual observation was only carried out at 24 h p.i., which is a weakness of this experiment. It can be assumed that sampling at additional time points until 48 h p.i. may have revealed a different picture.
In conclusion, we demonstrated by using flow cytometry and CLSM of mixed chicken blood cell culture that monocytes/macrophages and to lesser extent B cells are susceptible for T. gondii infection. Indications of T. gondii-positive cytotoxic T cells have been found only rarely. However, monocytes/macrophages could be confirmed as main target cells. Decrease of parasite stages until 48 h p.i. may be explained due to immediate and effective immune response. Seemingly, avian blood is not a preferred replication medium for T. gondii. Results of our investigations concerning cytokine expression indicate that the innate cellular immune response is dominant, represented by an activation of monocytes/macrophages and up-regulation of iNOS mRNA expression. We conclude that besides holding a host cell function, monocytes/macrophages play a crucial role for triggering immune response and host defense mechanisms to achieve inhibition of T. gondii replication. Further studies are required to reveal more information regarding parasite–host cell interaction in the complex medium blood.
References
Adams LB, Hibbs JB Jr, Taintor RR, Krahenbuhl JL (1990) Microbiostatic effect of murine-activated macrophages for Toxoplasma gondii. Role for synthesis of inorganic nitrogen oxides from L-arginine. J Immunol 144(7):2725–2729
Anderson SE Jr, Remington JS (1974) Effect of normal and activated human macrophages on Toxoplasma gondii. J Exp Med 139(5):1154–1174
Bouchot A, Millot JM, Charpentier S, Bonhomme A, Villena I, Aubert D, Pinon JM (2001) Membrane potential changes after infection of monocytes by Toxoplasma gondii. Int J Parasitol 31:1114–1120
Butcher BA, Denkers EY (2002) Mechanism of entry determines the ability of Toxoplasma gondii to inhibit macrophage proinflammatory cytokine production. Infect Immun 70(9):5216–5224
Channon JY, Seguin RM, Kasper LH (2000) Differential infectivity and division of Toxoplasma gondii in human peripheral blood leukocytes. Infect Immun 68(8):4822–4826
Courret N, Darche S, Sonigo P, Milon G, Buzoni-Gâtel D, Tardieux I (2006) CD11c- and CD11b-expressing mouse leukocytes transport single Toxoplasma gondii tachyzoites to the brain. Blood 107(1):309–316
Da Gama LM, Ribeiro-Gomes FL, Guimarães U Jr, Arnholdt AC (2004) Reduction in adhesiveness to extracellular matrix components, modulation of adhesion molecules and in vivo migration of murine macrophages infected with Toxoplasma gondii. Microbes Infect 6(14):1287–1296
DaMatta RA, Seabra SH, Manhães L, de Souza W (2000) Nitric oxide is not involved in the killing of Trypanosoma cruzi by chicken macrophages. Parasitol Res 86(3):239–243
De Boever S, Vangestel C, De Backer P, Croubels S, Sys SU (2008) Identification and validation of housekeeping genes as internal control for gene expression in an intravenous LPS inflammation model in chickens. Vet Immunol Immunopathol 122(3–4):312–317
Dias RR, Carvalho EC, Leite CC, Tedesco RC, Calabrese Kda S, Silva AC, DaMatta RA, de Fatima Sarro-Silva M (2014) Toxoplasma gondii oral infection induces intestinal inflammation and retinochoroiditis in mice genetically selected for immune oral tolerance resistance. PLoS One 9(12):e113374
Donahoe SL, Phalen DN, McAllan BM, O'Meally D, McAllister MM, Ellis J, Šlapeta J (2017) Differential gamma interferon- and tumor necrosis factor alpha-driven cytokine response distinguishes acute infection of a Metatherian host with Toxoplasma gondii and Neospora caninum. Infect Immun 85(6):e00173–e00117
Dubey JP (2008) The history of Toxoplasma gondii—the first 100 years. J Eukaryot Microbiol 55:467–475
Dubey JP (2010) Toxoplasma gondii infections in chickens (Gallus domesticus): prevalence, clinical disease, diagnosis and public health significance. Zoonoses Public Health 57:60–73
Dubey JP, Edelhofer R, Marcet P, Vianna MC, Kwok OC, Lehmann T (2005) Genetic and biologic characteristics of Toxoplasma gondii infections in free-range chickens from Austria. Vet Parasitol 133(4):299–306
Dupont CD, Christian DA, Hunter CA (2012) Immune response and immunopathology during toxoplasmosis. Semin Immunopathol 34(6):793–813
Edvinsson B, Lappalainen M, Evengård B (2006) ESCMID Study Group for Toxoplasmosis. Real-time PCR targeting a 529-bp repeat element for diagnosis of toxoplasmosis. Clin Microbiol Infect 12:131–136
Fair JM, Taylor-McCabe KJ, Shou Y, Marrone BL (2008) Immunophenotyping of chicken peripheral blood lymphocyte subpopulations: individual variability and repeatability. Vet Immunol Immunopathol 125(3–4):268–273
Filisetti D, Candolfi E (2004) Immune response to Toxoplasma gondii. Ann Ist Super Sanita 40(1):71–80
Gavrilescu LC, Denkers EY (2001) IFN-gamma overproduction and high level apoptosis are associated with high but not low virulence Toxoplasma gondii infection. J Immunol 167(2):902–909
Gazzinelli RT, Hakim FT, Hieny S, Shearer GM, Sher A (1991) Synergistic role CD4+ and CD8+ T lymphocytes in INF-gamma production and protective immunity induced by an attenuated Toxoplasma gondii vaccine. J Immunol 146(1):286–292
Gazzinelli RT, Denkers EY, Sher A (1993) Host resistance to Toxoplasma gondii: model for studying the selective induction of cell-mediated immunity by intracellular parasites. Infect Agents Dis 2:139–149
Gazzinelli RT, Amichay D, Sharton-Kersten T, Grunwald E, Farber JM, Sher A (1996) Role of macrophage-derived cytokines in the induction and regulation of cell-mediated immunity to Toxoplasma gondii. Curr Top Microbiol Immunol 219:127–139
Goebel S, Lüder CG, Gross U (1999) Invasion by Toxoplasma gondii protects human-derived HL-60 cells from actinomycin D-induced apoptosis. Med Microbiol Immunol 187(4):221–226
Guillermo LV, Damatta RA (2004) Nitric oxide inhibition after Toxoplasma gondii infection of chicken macrophage cell lines. Poult Sci 83:776–782
Hiob L, Koethe M, Schares G, Goroll T, Daugschies A, Bangoura B (2017) Experimental Toxoplasma gondii and Eimeria tenella co-infection in chickens. Parasitol Res 116(11):3189–3203
Hong YH, Lillehoj HS, Lee SH, Dalloul RA, Lillehoj EP (2006) Analysis of chicken cytokine and chemokine gene expression following Eimeria acervulina and Eimeria tenella infections. Vet Immunol Immunopathol 114:209–223
Hunter CA, Subauste CS, Van Cleave VH, Remington JS (1994) Production of gamma interferon by natural killer cells from Toxoplasma gondii-infected SCID mice: regulation by interleukin-10, interleukin-12, and tumor necrosis factor alpha. Infect Immun 62(7):2818–2824
Jones TC, Len L, Hirsch JG (1975) Assessment in vitro of immunity against Toxoplasma gondii. J Exp Med 141:466–482
Khan IA, Matsuura T, Kasper LH (1995) IL-10 mediates immunosuppression following primary infection with Toxoplasma gondii in mice. Parasite Immunol 17(4):185–195
Khan IA, Matsuura T, Kasper LH (1996) Activation-mediated CD4+ T cell unresponsiveness during acute Toxoplasma gondii infection in mice. Int Immunol 8(6):887–896
Kim L, Butcher BA, Lee CW, Uematsu S, Akira S, Denkers EY (2006) Toxoplasma gondii genotype determines MyD88-dependent signaling in infected macrophages. J Immunol 177(4):2584–2591
Lambert H, Barragan A (2010) Modelling parasite dissemination: host cell subversion and immune evasion by Toxoplasma gondii. Cell Microbiol 12(3):292–300
Lambert H, Hitziger N, Dellacasa I, Svensson M, Barragan A (2006) Induction of dendritic cell migration upon Toxoplasma gondii infection potentiates parasite dissemination. Cell Microbiol 8(10):1611–1623
Lambert H, Vutova PP, Adams WC, Loré K, Barragan A (2009) The Toxoplasma gondii-shuttling function of dendritic cells is linked to the parasite genotype. Infect Immun 77(4):1679–1688
Lüder CG, Gross U (2005) Apoptosis and its modulation during infection with Toxoplasma gondii: molecular mechanisms and role in pathogenesis. Curr Top Microbiol Immunol 289:219–237
Lüder CG, Algner M, Lang C, Bleicher N, Gross U (2003) Reduced expression of the inducible nitric oxide synthase after infection with Toxoplasma gondii facilitates parasite replication in activated murine macrophages. Int J Parasitol 33(8):833–844
Malkwitz I, Berndt A, Daugschies A, Bangoura B (2013) Long-term investigations on Toxoplasma gondii-infected primary chicken macrophages. Parasitol Res 112(9):3115–3122
Malkwitz I, Berndt A, Zhang R, Daugschies A, Bangoura B (2017) Replication of Toxoplasma gondii in chicken erythrocytes and thrombocytes compared to macrophages. Parasitol Res 116(1):123–131
Malkwitz I, Berndt A, Daugschies A, Bangoura B (2018) Characterisation of susceptibility of chicken macrophages to infection with Toxoplasma gondii of type II and III strains. Exp Parasitol 187:22–29
Mast J, Goddeeris BM, Peeters K, Vandesande F, Berghman LR (1998) Characterisation of chicken monocytes, macrophages and interdigitating cells by the monoclonal antibody KUL01. Vet Immunol Immunopathol 61(2–4):343–357
Mordue DG, Monroy F, La Regina M, Dinarello CA, Sibley LD (2001) Acute toxoplasmosis leads to lethal overproduction of Th1 cytokines. J Immunol 167(8):4574–4584
Munoz M, Liesenfeld O, Heimesaat MM (2011) Immunology of Toxoplasma gondii. Immunol Rev 240(1):269–285
Nang NT, Lee JS, Song BM, Kang YM, Kim HS, Seo SH (2011) Induction of inflammatory cytokines and toll-like receptors in chickens infected with avian H9N2 influenza virus. Vet Res 42:64
Nash PB, Purner MB, Leon RP, Clarke P, Duke RC, Curiel TJ (1998) Toxoplasma gondii-infected cells are resistant to multiple inducers of apoptosis. J Immunol 160(4):1824–1830
Ong YC, Boyle JP, Boothroyd JC (2011) Strain-dependent host transcriptional responses to Toxoplasma infection are largely conserved in mammalian and avian hosts. PLoS One 6(10):e26369
Park SS, Lillehoj HS, Allen PC, Park DW, FitzCoy S, Bautista DA, Lillehoje EP (2008) Immunopathology and cytokine responses in broiler chickens coinfected with Eimeria maxima and Clostridium perfringens with the use of an animal model of necrotic enteritis. Avian Dis 52:14–22
Payne TM, Molestina RE, Sinai AP (2003) Inhibition of caspase activation and a requirement for NF-kappaB function in the Toxoplasma gondii-mediated blockade of host apoptosis. J Cell Sci 116(Pt 21):4345–4358
Persson EK, Agnarson AM, Lambert H, Hitziger N, Yagita H, Chambers BJ, Barragan A, Grandien A (2007) Death receptor ligation or exposure to perforin trigger rapid egress of the intracellular parasite Toxoplasma gondii. J Immunol 179(12):8357–8365
Persson CM, Lambert H, Vutova PP, Dellacasa-Lindberg I, Nederby J, Yagita H, Ljunggren HG, Grandien A, Barragan A, Chambers BJ (2009) Transmission of Toxoplasma gondii from infected dendritic cells to natural killer cells. Infect Immun 77(3):970–976
Quéré P, Pierre J, Hoang MD, Esnault E, Domenech J, Sibille P, Dimier-Poisson I (2013) Presence of dendritic cells in chicken spleen cell preparations and their functional interaction with the parasite Toxoplasma gondii. Vet Immunol Immunopathol 153(1–2):57–69
Robben PM, Mordue DG, Truscott SM, Takeda K, Akira S, Sibley LD (2004) Production of IL-12 by macrophages infected with Toxoplasma gondii depends on the parasite genotype. J Immunol 172(6):3686–3694
Schlüter D, Däubener W, Schares G, Groß U, Pleyer U, Lüder C (2014) Animals are key to human toxoplasmosis. Int J Med Microbiol 304(7):917–929
Schupp E, Michel R, Raether W, Niemeitz H, Uphoff M (1978) Invasion of erythrocytes by Toxoplasma gondii. Z Parasitenkd 55:189–193
Seabra SH, de SW, Damatta RA (2002) Toxoplasma gondii partially inhibits nitric oxide production of activated murine macrophages. Exp Parasitol 100:62–70
Sher A, Oswald IP, Hieny S, Gazzinelli RT (1993) Toxoplasma gondii induces a T-independent IFNgamma response in natural killer cells that requires both adherent accessory cells and tumor necrosis factor-alpha. J Immunol 150(9):3982–3989
Sibley LD (2004) Intracellular parasite invasion strategies. Science 304(5668):248–253
Suzuki Y, Orellana MA, Schreiber RD, Remington JS (1988) Interferon-gamma: the major mediator of resistance against Toxoplasma gondii. Science 240(4851):516–518
Tanabe K, Kimata I, Takada S (1980a) Penetration of chick embryo erythrocytes by Toxoplasma gondii tachyzoites in simplified incubation media. J Parasitol 66:240–244
Tanabe K, Kimata I, Takada S (1980b) Multiplication of Toxoplasma gondii in maturing erythroid cells. J Gen Microbiol 118(1):45–49
Unno A, Suzuki K, Xuan X, Nishikawa Y, Kitoh K, Takashima Y (2008) Dissemination of extracellular and intracellular Toxoplasma gondii tachyzoites in the blood flow. Parasitol Int 57(4):515–518
Viertlboeck BC, Göbel TW (2007) Chicken thrombocytes express the CD51/CD61 integrin. Vet Immunol Immunopathol 119(1–2):137–141
Zhao Y, Ferguson DJ, Wilson DC, Howard JC, Sibley LD, Yap GS (2009) Virulent Toxoplasma gondii evade immunity-related GTPase-mediated parasite vacuole disruption within primed macrophages. J Immunol 182(6):3775–3781
Zintl A, Halova D, Mulcahy G, O’Donovan J, Markey B, DeWaal T (2009) In vitro culture combined with quantitative TaqMan PCR for the assessment of Toxoplasma gondii tissue cyst viability. Vet Parasitol 164:167–172
Acknowledgements
The authors would like to thank all members of the Institute of Parasitology, University Leipzig, especially Tina Goroll, for the consistent technical and practical support. We acknowledge the excellent technical assistance of Katrin Schlehahn. We deeply thank Prof. Soldati-Favre for providing the RH-GFP strain.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
Blood sampling from chicken has been conducted in compliance with the current European and national legal requirements under registration and permission by the responsible authority (Landesdirektion Leipzig, file no. V 10/14).
Additional information
Handling Editor: Julia Walochnik
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Hiob, L., Berndt, A., Daugschies, A. et al. Host–pathogen interaction in Toxoplasma gondii-infected mixed chicken blood cell cultures. Parasitol Res 118, 1479–1491 (2019). https://doi.org/10.1007/s00436-019-06265-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-019-06265-2