
Abstract
Transmembrane potassium (K) gradients are key determinants of membrane potential that can modulate action potentials, control muscle contractility, and influence ion channel and transporter activity. Daily K intake is normally equal to the amount of K in the entire extracellular fluid (ECF) creating a critical challenge — how to maintain ECF [K] and membrane potential in a narrow range during feast and famine. Adaptations to maintain ECF [K] include sensing the K intake, sensing ECF [K] vs. desired set-point and activating mediators that regulate K distribution between ECF and ICF, and regulate renal K excretion. In this focused review, we discuss the basis of these adaptions, including (1) potential mechanisms for rapid feedforward signaling to kidney and muscle after a meal (before a rise in ECF [K]), (2) how skeletal muscles sense and respond to changes in ECF [K], (3) effects of K on aldosterone biosynthesis, and (4) how the kidney responds to changes in ECF [K] to modify K excretion. The concepts of sexual dimorphisms in renal K handling adaptation are introduced, and the molecular mechanisms that can account for the benefits of a K-rich diet to maintain cardiovascular health are discussed. Although the big picture of K homeostasis is becoming more clear, we also highlight significant pieces of the puzzle that remain to be solved, including knowledge gaps in our understanding of initiating signals, sensors and their connection to homeostatic adjustments of ECF [K].
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Eukaryotes evolved to possess steep transmembrane gradients of potassium (K) and sodium (Na), created and maintained by the ubiquitous Na,K-ATPase (NKA, the sodium pump). By actively pumping K into the intracellular fluid (ICF) and Na out of the cell into the extracellular fluid (ECF), this system supports a number of critical life-promoting functions: high ICF K facilitates protein synthesis and cell volume regulation, the Na and K gradients are “storehouses” of potential energy used to transport electrolytes and substrates across membranes, and the transmembrane K gradients are key determinants of membrane potential that can modulate action potentials, control muscle contractility, and influence ion channel and transporter activity [111]. As a result of this dynamic, the ICF K pool is large (~ 3500 mEq) and the ECF K pool is small (~ 70 mEq). In mammals, the ECF and ICF [K] are 3.8–5, and 120–140 mM, respectively. This system evolved to accommodate prevalent K-rich food sources; in fact, high dietary K intake is beneficial as it can lower blood pressure, blunt the negative effects of dietary NaCl, and reduce the risk of kidney stones and bone loss, benefits that are probably also impacted by higher organic anions like citrate in high K diets [34, 72, 116]. Among the major electrolytes, K has the highest ratio of daily intake (70–120 mEq) to extracellular pool size (70 mEq), and as such is a significant daily homeostatic challenge. Since changes in the ECF [K] can provoke life-threatening disruptions in membrane potential, the system evolved multilayered complex mechanisms of homeostatic control to minimize changes in ECF [K] brought about by the significant challenge of a simple meal [73].
The numerous components of the K homeostatic system are still being elucidated but can be categorized into (1) sensing the K intake, (2) sensing ECF [K] and comparing it to the desired set point, (3) regulating K distribution between ECF and ICF, and (4) regulating K excretion (Fig. 1). For illustration, let us consider the response to a simple meal of an avocado sandwich, tomato soup, yogurt with blueberries and black coffee. Containing ~ 35 mEq (1.4 mg) K, the meal has the potential to raise the ECF K pool 50%. Studies in rodents and humans suggest that the gastrointestinal system senses the incoming K [102, 164] (and glucose with it) and, before any detectable increase in ECF K, sends anticipatory signals to various targets to prepare to blunt the rise in ECF K. Once absorbed, the ECF K concentration ([K]) increases, a classic signal to shift K from ECF to ICF. ECF K is first transiently shifted into liver [7], then persistently into muscle, facilitated by the glucose in the meal driving a rise in insulin that is known to activate the NKA via an insulin receptor–mediated increase in the affinity of sodium pumps which increase net K uptake into ICF [25]. In parallel, the rise in ECF [K] directly activates sensors in the kidney to secrete and excrete K, and in the adrenal gland to synthesize aldosterone, which further stimulates K excretion from the kidney (as well as from colon, salivary and sweat glands). These adaptations accomplish the goal of minimizing postprandial increases in ECF [K]. This review aims to cover recent advances in understanding systemic control of whole-body K homeostasis (Fig. 1), and to identify key knowledge gaps that remain: (1) sensing [K] and comparing it to the desired set point in various cells and tissues, including sensing and responding to depressed ECF K; (2) mediators of the adaptations including K per se, aldosterone, and local modulators; (3) target transporter responses across tissues (extrarenal, intrarenal, importance of Na–K crosstalk); (4) therapeutic opportunities to use what we know about K homeostasis to treat chronic disease (e.g. hypertension). The readers are further referred to many excellent comprehensive reviews of various aspects of K homeostasis [7, 24, 43, 46, 73].

Mechanisms to maintain potassium (K) homeostasis after a K-rich meal. A K-rich meal stimulates the intestine to release an unidentified “gut factor” that potentially targets the pituitary to release a mediator that targets skeletal muscle and liver to take up K from ECF to ICF, in addition to stimulating the renal nephron to excrete K before a rise in plasma [K]. Additionally, the meal stimulates the pancreas to release insulin, a mediator that activates muscle Na,K-ATPase independent of a rise in plasma [K]. These feedforward mechanisms are the initial homeostatic response to buffering ECF [K] in response to K intake. As the meal is absorbed, the rise in plasma [K] is a power signal that targets (1) Skeletal muscle Na,K-ATPase alpha 2 isoform, which is kinetically activated by rising [K], (2) Kidney basolateral membrane K-channel sensors Kir4.1/5.1 and potentially Kir4.2/5.1, which rapidly stimulate signaling cascades that mediate increased K secretion and excretion and depress ammoniagenesis, and (3) Adrenal glomerulosa cell K channels GIRK-4, TASK-1, and TASK-3 which change membrane potential and cell [Ca] and stimulate biosynthesis and release of the mediator aldosterone. Aldosterone targets mineralocorticoid receptors in the renal distal nephron, colon, sweat and salivary glands to secrete and excrete K. Together, stimulation of K secretion and excretion match K output to input, forming complex feedforward plus feedback loops that are needed to tightly maintain ECF [K]. Figure created with BioRender.com
Rapid buffering of K intake — timing is everything
K homeostasis is, ultimately, a function of balancing K output to K intake [46]. Most recent research attention has focused on discoveries about the kidney K excretion arm of adaptation (see Potassium handling by kidney section below). Nonetheless, Bia and DeFronzo’s classic 1981 review, Extrarenal Potassium Homeostasis [7], lays out the importance of the postprandial shift in K from ECF to ICF and highlights that during the first 3–6 h after a K load (whether a meal or intravenous, and across species), only about ½ of the K is excreted in the urine. This observation was confirmed in rats [106] and humans [66]. If the remaining 50% of the K load were retained within the ECF, the [K] would rise to potentially dangerous levels. This does not occur because nearly all the retained K is rapidly translocated into the ICF. Between meals, this excess K, mainly stored in muscle ICF, moves into the ECF as a function of muscle activity, and is filtered into the kidneys to be excreted to match K intake over the next 10 h. Whether ECF [K] remains elevated has not been measured in parallel, but plasma aldosterone remains elevated [106, 123]. This remarkable multilayered acute adaptation occurs with each K-containing meal over a lifetime (Fig. 1).
Given the importance of preventing large fluctuations in ECF [K], it is not surprising that feedforward regulation, specifically, responses that operate independently of a change in the ECF [K] are key (Fig. 1). Proposed by Rabinowitz [103], and reviewed more recently by Youn [164], evidence suggests that K is “sensed” in the GI tract and signals are sent to the periphery and the kidneys to shift K from ECF to ICF and to increase K excretion — prior to any change in ECF [K]. In a meal-fed sheep model, Rabinowitz provided evidence for feed-forward sensing of K intake in the splanchnic bed that signaled kidneys to excrete K, independent of changes in plasma K or aldosterone [104]. Followup studies by Morita et al. provided evidence for involvement of hepatic afferent nerve activity in the sensing of both Na and K intake, attenuated by inhibitors of the Na,K,2Cl transporter (NKCC) [77]. A followup study reported that high-NaCl or high-KCl diets decreased liver NKCC1 expression, and potentially reduced hepatic sodium- and potassium-receptor sensitivity [139]. Further investigations by Youn and colleagues indicated that a number of GI hormones, including guanylin peptides and incretin hormones were not requisite mediators of this signal [87]. Their more recent investigation of feedforward signal(s) pursued the idea that the brain, specifically the pituitary, receives K intake signals from the gut. Carefully controlling for light/dark cycles, they observed that normal nighttime feeding of rats with a control 1% K diet was associated with a 0.2 mEq rise in plasma K in sham rats, while hypophysectomized rats exhibited a fivefold higher 1 mEq rise in plasma K, evidence of K intolerance and supporting a role of central regulation [88]. While the specific sensors and signals of gut sensing feedforward regulation remain elusive, there is evidence for such mechanisms in humans. A study by Preston and colleagues provided 32 individuals with either a complex meal that was deficient in Na and K, a 35 meq oral K load, or both [52, 102]. The oral K load alone did not provoke insulin release but did increase serum [K] by 0.5 mM and raised urine K excretion fivefold within 1 h. Adding the meal to the oral K load prevented any change in serum K, and blunted the acute rise in urine K excretion to twofold. Using mineralocorticoid blockers, they demonstrated the gut-kidney-kaliuresis axis functions independent of any change in mineralocorticoids or serum [K] [102].
The role of the postprandial rise in insulin as a driver of the cellular uptake of both glucose and K, (independent of a rise in ECF [K]) warrants discussion as a parallel feed-forward mechanism. The Youn and McDonough labs developed and implemented a hyperinsulinemic glucose and K clamp technique in conscious rats with indwelling tail catheters in which, during insulin administration, the incremental infusions of glucose (Ginf) and K (Kinf) necessary to maintain constant plasma concentrations of both substrates were quantified, i.e., the cellular uptake of glucose and K in response to insulin/time can be estimated from the rate of infusion of these substrates [23]. The technique was applied to rats fed a high fat diet, which is known to provoke insulin resistance to glucose uptake, in order to assess the impact on insulin-sensitive cellular uptake of K [22]. After normalizing dietary K intake between rat groups fed control and high fat diet, we observed the expected insulin resistance to glucose uptake in the high-fat fed group, but no decrement in insulin sensitivity to cell K uptake. That is, insulin sensitivity of cell K uptake was preserved despite insulin resistance to glucose uptake during high fat diet. Nguyen and Moe extended these findings to humans with type 2 diabetes (also implementing a hyperinsulinemic euglycemic clamp) [85]: glucose cellular disposal was significantly lower with increasing body mass index, while cellular K uptake was independent of body mass index, that is, postprandial K uptake is preserved in type 2 diabetes in spite of insulin resistance to cell glucose uptake.
Does postprandial cellular K uptake adapt in response to consuming a K-deficient meal or diet? As described above, a K-deficient meal alone stimulated a rise in insulin along with a fall in serum [K] (by 0.5 mM over 90 min) without a rise in urinary K excretion [102]. To determine whether dietary K intake impacts insulin-stimulated cell K uptake (or whether it is solely determined by insulin), Choi et al. used the “hyperinsulinemic K- and glucose-clamp” described above. In control rats on a normal diet, both Kinf and Ginf increased in a dose-dependent manner with insulin infusion, as expected. In rats fed a K-deficient diet for 2 days (plasma [K] fell from 4.2 to 3.8 mM), then infused with insulin, Ginf remained at control levels, and Kinf fell 80%, providing evidence for the development of a novel variety of insulin resistance to cellular K uptake when dietary K is limiting [23]. A role for the fall in ECF [K] was ruled out in a subsequent study in which rats were fed a diet with K reduced from 1 to 0.33% in which plasma [K] remained unchanged from baseline levels [20]: this diet reduced both urine K excretion (80%) and insulin-stimulated Kinf (50%) indicating that these feedforward responses were independent of plasma [K]. Clearly, these adaptations serve to minimize fluctuations in ECF [K]. But what are the mechanisms of this insulin resistance? One hypothesis is that the feedforward signal(s) from the gut are permissive for insulin-sensitive cellular K uptake and for anticipatory K excretion in the urine.
Maintaining plasma [K] in feast and famine — buffering by muscle K stores
A 2021 review of Regulation of Muscle Potassium by Lindinger and Cairns [65] lists K transport proteins in skeletal muscle including NKA α1 and α2 isoform sodium pumps which create and maintain the transmembrane Na and K gradients and respond to catecholamines and insulin stimulation; the co-transporters NKCC1 and KCC1, which stabilize cell volume and membrane potential; ATP-sensitive K channels (Kir 6.2) which open when ATP falls; KCa 1.1, a voltage-gated, Ca-sensitive delayed rectifier K channel which releases ICF K during stimulation; and IKir 2.1 and IKir 2.2 which re-capture K from the T-tubules after stimulation [31]. DiFranco and Heiny compared properties of the two skeletal muscle NKA (sodium pump) isoforms and provided evidence that they operate over different ranges of ECF [K] such that the α2 isoform can serve as an ECF K–sensor in skeletal muscles [30]. In brief, both NKA isoforms are rapidly stimulated by a rise in ECF K through occupancy of an ECF facing K binding site. At baseline, the α1 isoform, localized to the muscle outer sarcolemma, works above its K1/2 for K (1–2 mM) and maintains the resting membrane potential and transmembrane Na and K gradients (as in most other cells). In contrast, the more abundant muscle α2 NKA, localized to the T-tubules, operates below its K1/2 (~ 4.3 mM) at rest, but is rapidly activated when ECF [K] rises. That is, the K affinity of the NKA α2 isoform is much lower than that of the α1 isoform, but active K uptake into muscle ICF is rapidly increased in proportion to the T-tubular ECF [K] in the physiological range (4–40 mM), demonstrating K-sensor properties of the NKA α2 isoform (Fig. 1).
While this K sensing by α2 was defined as a function of muscle activity [30], the properties are applicable to challenges of feast (K rich meal) and famine (low K meal). The DiFranco and Heiny results suggest that after a K-rich meal, when ECF creeps above 4 mM, NKA α2 is immediately activated to pump K from ECF to ICF, a feedback loop that is independent of — yet amplifies — the feed-forward gut factor and insulin signals arising from K intake. Although not tested experimentally, the kinetic properties of NKA α2 may also explain the blunted response to insulin after a low K meal: without a rise in ECF [K] the feed-forward response could, potentially, be limited to insulin stimulation of the small pool of muscle NKA α1. McKenna and Clausen investigated the activation of NKA α2 by insulin (100 mU/ml) in isolated soleus [75] and found uptake of 86 Rb (a K surrogate) increased rapidly by 23%, while ouabain binding (a surrogate for plasma membrane NKA) did not change, indicating that insulin can stimulate NKA-mediated K uptake independent of translocation of NKA.
In response to a K deficient diet, skeletal muscle altruistically donates K from ICF to buffer the fall in ECF [K]. This fall in ECF [K] occurs secondary to the persistent non-zero K secretion and excretion from kidney, colon, saliva, and sweat glands. We can now predict that as ECF [K] falls, especially below the NKA α2 K1/2 of 4.3 mM, K influx via this isoform will fall and the equilibrium will be shifted to greater net K efflux (e.g. via KCa 1.1) vs. K influx during muscle activity. With prolonged low K diet, the pool size of NKA α2 protein is (post-translationally) reduced [132, 133], further contributing to net K efflux. We recently examined the coordinate adaptations of skeletal muscle and kidney to a K deficient diet in mice [74]. As previously determined in rats [20, 23, 132], plasma [K] fell from 4.5 to 2 mM despite renal K conservation (K excretion fell from 200 to 3 µmol/overnight collection). Compartmental modeling revealed that the amount of K lost in the urine over 10 days (48 µmol) was equivalent to the amount leaving the muscle during the same time (47 µmol). Despite this correlation, ECF K mass fell from 26 to 11 µmol, likely an important error signal to continue the redistribution of K from ICF to ECF until ECF [K] is corrected with sufficient intake. Muscle [K] fell by 28 mM and [Na] increased by 27 mM in the same study; this doubling of tissue Na was not accompanied by any evidence of inflammation, contrary to studies suggesting that tissue Na accumulation is proinflammatory [58, 60, 136]. At the transporter level, NKA α2 and β2 isoform subunits fell 25 and 50%, respectively (NKA α1-β1 subunits were unchanged). Interestingly, during K deficiency, both muscle and kidney exhibited increased pools of phosphorylated STE20/SPS1-related proline/alanine-rich kinase and oxidative stress–responsive kinase 1 (SPAK and OSR1), the kinases that activate Slc12 cotransporters. In the muscle, this was associated with elevated phosphorylation of NKCC1, a cell volume regulator, but no activation of KCC, likely a response to contracted muscle cell volume. In the kidney, SPAK and OSR1 activation are correlated with elevated Na-Cl cotransporter activity, (discussed under Potassium handling by kidney section).
What do we know about how the system restores basal muscle stores in the transition from famine to feast? When K intake is restored (e.g., the tiger finally catches a boar) the K-rich meal raises ECF [K] and activates feedforward and direct kinetic stimulation of NKA α2 in muscle which rapidly re-fill the depleted ICF stores; the renal adaptations to retain K during hypokalemia would exhibit a lag until transporters are re-adjusted. Note that hypokalemia that is secondary to renal K excretion also provokes K donation from the muscle ICF to ECF (e.g., aldosterone excess, loop diuretics). Veiras et al. [143] showed that angiotensin II infusion in rats fed a normal K-containing diet provokes hypokalemia attributable to increased activation of collecting duct ENaC and Na,K-ATPase which drives K secretion and excretion. Providing these rats a single K-rich meal acutely increased ECF [K] by 1 mM but did not increase K excretion, suggesting that the ingested K was preferentially routed to replenish ICF stores, stimulated by feedforward responses and NKA α2 activation. In humans with K-wasting phenotypes, this is likely the restorative response to K-supplementation.
Potassium regulation of aldosterone biosynthesis
Plasma aldosterone increases two–sixfold as early as 30 min after an acute K intake challenge (gavage, feeding, venous infusion) [106, 123], a classic feedback response triggered when adrenal zona glomerulosa cells sense rising ECF [K] and initiate aldosterone biosynthesis. The K-sensors in the glomerulosa cells include TWIK-related acid-sensitive K channels (TASK-1 and TASK-3), which are constitutively open K leak channels that maintain cell membrane potential (Fig. 1). A small rise in ECF [K] depolarizes the membrane potential and triggers Ca influx via voltage dependent L- and T-type Ca channels, which sets off a signaling cascade culminating in increased biosynthesis of aldosterone synthase (CYP11B2) and release of aldosterone into the ECF [3, 45, 47]. Once secreted, aldosterone binds to the mineralocorticoid receptor (MR) in target tissues, including kidney, colon, salivary and sweat glands, where it clearly contributes to increased Na reabsorption, K excretion, and restoration of K homeostasis (Fig. 1) [111].
Does a rise in plasma aldosterone in response to elevated ECF [K] play a role in stimulating cellular K uptake, e.g., into skeletal muscle? MuscleDB, a database of muscle tissue gene expression from rats and mice [131], indicates that the MR (NR3C2) is expressed in skeletal muscles at 5–10 Fragments Per Kilobase per Million reads (FPKM). Evidence for physiologically relevant activity of MR in muscle comes from studies in mouse models of muscular dystrophy showing that antagonizing MR activity in myotubes alters expression of known targets [17]. However, expression of 11 β OH steroid dehydrogenase 2 (HSD11B2) is not significantly detected (< 0.2 FPKM) in rat or mouse muscles [131]. HSD11B2 confers aldosterone specificity to the MR by degrading cortisol (which can also activate MR) [41], suggesting that skeletal muscle MR may be a receptor for cortisol, rather than for aldosterone. Nevertheless, can aldosterone increase cellular K uptake? While it is challenging to assign an aldosterone-driven drop in plasma K to muscle K uptake versus kidney K excretion, a classic study by Bia and DeFronzo attempted to control kidney variables [8]. Rats were adrenalectomized, repleted with the glucocorticoid dexamethasone and saline drinking water, anesthetized, acutely nephrectomized, then infused with KCl for 60 min. Plasma [K] increased from 5.3 to 8.9 mEq (ΔK of 3.6 mEq). In sham-treated control rats, the same K infusion increased plasma [K] less from 4.4 to 7 mEq (ΔK of 2.6 mEq). When the adrenalectomized + nephrectomized rats were repleted with aldosterone along with dexamethasone, both baseline and ΔK values were indistinguishable from controls, consistent with a role for aldosterone in lowering plasma K during K infusion. However, a role for the other aldosterone sensitive K-secreting epithelial tissues (colon, salivary and sweat glands) in lowering K cannot be ruled out. Nowadays, investigators could address this issue with skeletal muscle–specific knockdown of MR, which has not been reported to our knowledge [26]. In any case, we can assume that the “heavy lifting” of moving K from the ECF to the ICF in response to a K-load can be accomplished by the feedback kinetic activation of the NKA α2 in the T-tubules of the muscles as ECF [K] increases.
Aldosterone-producing adenomas (APA) are the most prevalent cause of secondary hypertension characterized by arterial hypertension and hypokalemia. Somatic mutations leading to the APAs have been defined over the past decade [6, 115, 117]. The G protein–gated, inwardly rectifying potassium channel GIRK (KCNJ5) is abundant in adrenal cortex (Fig. 1), and mutations, accounting for 40% of APAs, cause the loss of K selectivity and provoke membrane depolarization that initiates the signaling cascade that sets off chronic activation of CYP11B2, aldosterone biosynthesis and release. This is an example of an adenoma provoked by K-sensor malfunction. Somatic mutations in zona glomerulosa cell plasma membrane ATPases can also cause APAs: NKA mutations (ATP1A1, 5% of APAs) can cause loss of K affinity and ATPase activity associated with membrane depolarization, and the Ca-ATPase mutations (ATP2B3, 1.6% of APAs) can reduce Ca efflux from the cytoplasm and raise cell Ca; both chronically activate CYP11B2, increase aldosterone release and provoke arterial hypertension and hypokalemia [6].
Potassium handling by the kidney — bulk reclamation along proximal nephron coupled to decision-making in the distal nephron
Several recent excellent reviews cover the regulation of renal K handling in great detail, [16, 51, 99, 101, 152] so herein, only an overview of the most recent developments is provided. Independent of the plasma [K], the majority of K freely filtered at the glomerulus is reabsorbed by the proximal tubule (PT) and the thick ascending limb of Henle’s loop (TAL). Reabsorption of K in the PT occurs passively via a paracellular pathway (Fig. 2). This reabsorption is secondary to the reabsorption of Na and water (predominantly by the Na/H exchanger NHE3 and Aquaporin 1, respectively), and in later portions of PT further enhanced by the lumen positive potential difference produced by Cl− reabsorption [153]. An unanswered question regarding PT K reabsorption is whether there is a role of claudins. For example, although the small cation selective claudin-2 is expressed in the PT, genetically modified mice lacking this channel appear to have no abnormalities in renal K handling [5, 92]. In the TAL, secondary active transport via the Na+-K+-2Cl−-cotransporter (NKCC2) provides a transcellular route for K reabsorption, which is coupled to K recycling through the renal outer medullary K channel (ROMK). This recycling of K increases the lumen-positive potential difference, driving the paracellular reabsorption of K (Fig. 2).

Basics of renal K handling. The different segments of the renal tubule involved in K reabsorption and secretion are highlighted along with some of the main apical plasma membrane transport pathways. Some of the mediators of K transport are also shown (? indicates potential role). Reabsorption of K in the proximal tubule occurs passively via a paracellular pathway secondary to the reabsorption of Na. In the TAL, secondary active transport provides a transcellular route for K reabsorption and a lumen-positive potential difference drives paracellular K reabsorption. Cells of the late DCT, CNT, and CCD are important for K secretion by K chloride cotransporters (KCC), the renal outer medullary K channel (ROMK), and “Big” K (BK) channels. Distal delivery of Na alters electrogenic Na reabsorption by the epithelial Na+ channel (ENaC) and subsequent K secretion by ROMK, whereas increased tubular flow stimulates K secretion by BK channels. Reabsorption of K through the HK-ATPase in type A intercalated cells can also occur. Abbreviations: DCT, distal convoluted tubule; CNT, connecting tubule; CCD, cortical collecting duct; NHE3, sodium-hydrogen exchanger 3; NKCC2, Na+-K+-Cl.− cotransporter 2. Figure created with BioRender.com
As the PT and the TAL reabsorb ~ 90% of the filtered K, the enormous capacity of the mammalian kidney to alter urinary K excretion is reliant on the “distal nephron,” comprised of the late distal convoluted tubule (DCT), the connecting tubule (CNT), and the cortical collecting duct (CCD) (Fig. 2). Except under extreme K depletion, when the actions of the H,K-ATPase in α-intercalated cells can promote K reabsorption [46], the distal nephron is almost exclusively responsible for K secretion, the magnitude of which is determined by the intracellular and tubular K concentrations and pH, the voltage difference across the apical plasma membrane, and the permeability of this membrane for K. Several of these parameters are altered by aldosterone effects on the MR; hence, this region is often named the aldosterone-sensitive distal nephron (ASDN) [118]. An electroneutral K secretory pathway is present in the ASDN [144] and aldosterone-regulated flow-dependent K secretion can occur via big-K channels (BK, or “maxi-K” channels) [108, 156, 158]. However, the predominant mode of K secretion is electrogenic, primarily occurring through the renal outer medulla K channel ROMK (Kir1.1) subsequent to active K uptake via the basolateral Na,K-ATPase. Na reabsorption via apical ENaC and thus apical membrane depolarization provides the electrical driving force for this K secretion (Fig. 2) [39, 147]. ROMK and ENaC activity are altered by dietary K [39, 62, 64, 74, 84, 147], with recent studies suggesting that the late DCT and early CNT have high baseline ROMK and ENaC activity that are important for regulating K secretion during dietary K deficiency in an aldosterone-independent manner, whereas the latter portions of the CNT and/or CCD play a greater role in K secretion during increased K intake [83, 163].
In addition to the effects of aldosterone on tubular transport, urinary K excretion is increased when distal delivery of Na is increased. In the following sections, we initially discuss how K-mediated changes in Na handling by the PT and the TAL likely contribute to alterations in distal Na delivery [155]. Subsequently, we provide more extensive insight into the role of the DCT as a K sensor, a topic that has recently gained enormous attention due to the potential impact on blood pressure (BP, see later): specifically, how the DCT “senses” hyperkalemia and responds by reducing the activity of the main NaCl transport pathway in this segment, the thiazide-sensitive sodium chloride cotransporter (NCC).
Modulation of distal sodium delivery by the PT and TAL
Mathematical modeling has indicated that, despite potentially attenuated tubuloglomerular feedback (TGF), Na delivery to the ASDN can be influenced by reduced Na reabsorption in the PT and TAL [154]. K loads decrease PT fluid reabsorption and increase Na delivery to the DCT [159] (accompanied by a TGF-mediated reduction in GFR) [155], whereas low K diets can decrease intracellular pH and increase NHE3 activity [2, 38, 122], consistent with reduced Na delivery to the ASDN during K depletion. K channels also play a central role in modulating PT ammoniagenesis [9, 146], which can also impact K secretion. Potassium depletion is associated with an increase in PT ammoniagenesis and decreased K secretion, whereas decreased ammoniagenesis increases K secretion. The mechanisms responsible for this are not fully elucidated, but it is likely due to alterations in delivery of NH4+ to the CNT and CCD, with subsequent changes in cell pH altering ENaC and ROMK conductance [155]. Whether changes in PT ammoniagenesis also contribute to the hypokalemia observed in individuals with mutations in KCNJ16 (encoding Kir5.1) warrants consideration [113]. K loading also leads to a reduction in the active phosphorylated form of NKCC2 [106] and decreased NaCl reabsorption in the TAL [140], which would increase the delivery of Na to the ASDN to promote K secretion. In summary, it is likely that all segments proximal to the ASDN work together to modulate the amount of Na delivered to the K secreting segments of the ASDN. How the K sensors drive altered transporter activity along the PT and TAL is not as well defined as the signaling cascades elucidated along the distal nephron, providing an important area for further study.
Altering DCT sodium reabsorption influences kaliuresis
Adequate homeostatic kaliuresis can only occur if the activity of apical cation transporters in the ASDN is modulated appropriately. The majority of NaCl reabsorption in the DCT is electroneutral via NCC [32], with a contribution by ENaC in later portions [83, 163]. In addition to Na balance, the importance of NCC activity for K homeostasis is highlighted by the urinary K wasting and hypokalemia in patients with Gitelman syndrome due to loss of NCC function [121]. Phosphorylation of threonine (Thr) and serine (Ser) residues in the amino-terminal domain of NCC can be considered as the master regulator of NCC function, as they increase NCC transport capacity and decrease NCC endocytosis from the DCT plasma membrane [90, 109, 110]. The intricate details of the signaling mechanisms underlying NCC phosphorylation are covered in other excellent reviews [16, 51, 152], but as the molecular basis of this pathway is key to understanding how the DCT senses K to alter NCC activity, a simplistic overview is provided here (Fig. 3). In low K states, NCC is phosphorylated primarily by the Ste-20-related proline alanine-rich protein kinase (SPAK) [44, 71, 107, 145], which itself is phosphorylated and activated by a member of the With No Lysine (K) kinases family WNK4, aided by WNK1 [80]. WNK4 is activated by autophosphorylation, a process that is prevented when a Cl− ion binds to the kinases active site [97]. Hence, WNK4 senses changes in the intracellular chloride concentration [Cl−]i: low cell [Cl−]i enhances WNK4 activity and subsequently SPAK and NCC phosphorylation, and high cell [Cl−]i inhibits WNK4 activity and ultimately NCC phosphorylation and activation [4]. WNKs are also targeted for proteasomal degradation by the Cul3-KLHL3-Ring E3 ubiquitin ligase complex [13, 69, 89]. The syndrome of pseudohypoaldosteronism type II (PHAII, also called familial hyperkalemic hypertension or Gordon syndrome), presents in patients as increased NCC activity, hyperkalemia and hypertension, and is a result of mutations in the WNK1, WNK4, CUL3, or KLHL3 genes [16], underscoring the importance of this signaling network for K balance. Importantly, dephosphorylation of NCC virtually eliminates NCC activity [110], but discussion remains regarding the responsible phosphatase [42, 78, 94, 98, 100].

Effects of low and high dietary K intake on NCC-mediated Na reabsorption in the DCT. During hypokalemia, Kir4.1/5.1 is activated leading to a reduction in intracellular [K] and hyperpolarization of the basolateral membrane, thus creating a driving force for Cl− efflux via ClC-Kb. The subsequent reduction in intracellular Cl− relieves the inhibition of WNK4 autophosphorylation and allows the WNK-SPAK pathway to phosphorylate and activate NCC. This leads to greater NaCl reabsorption in the DCT, less Na delivery to the distal tubule and reduced K secretion. During high K intake, high ECF [K] inhibits Kir4.1/5.1 leading to membrane depolarization and inhibition of Cl− efflux via ClC-Kb. The increase in intracellular [Cl−] inhibits WNK4 resulting in reduced SPAK and NCC phosphorylation. The influence of the SPAK pathway is also reduced by enhanced proteasomal degradation of WNK4. High ECF [K] also causes NCC dephosphorylation and degradation. The reduction in NCC activity increases distal Na delivery and flow which promotes kaliuresis. Figure created with BioRender.com
So, how does the DCT cell sense changes in K in order to set off the signaling cascade that targets NCC activity? A key observation to answering this question came from the clinical presentation of patients with EAST syndrome due to mutations in the KCNJ10 gene (encodes the K channel Kir4.1). These patients suffer from hypokalemia, salt-wasting, hypomagnesaemia and hypocalciuria [10], symptoms very similar to those observed in Gitelman syndrome, suggesting that the activity of Kir4.1 could play a crucial role in regulating NCC [10, 114]. Kir4.1 is present in the DCT basolateral membrane where it interacts with Kir5.1 (encoded by KCNJ16) to form a 40 pS heterotetrameric K channel [70, 91, 126] (Fig. 3). K efflux through Kir4.1 (e.g., when ECF K is low), increases the driving force for Cl efflux through basolateral ClC-Kb channels, lowering intracellular [Cl] and Cl-mediated WNK inhibition which ultimately increases NCC phosphorylation via the WNK-SPAK cascade [4, 19, 27, 50, 59, 97, 130, 148]. Indeed, mouse models with disruption of Kir4.1 suffer from hypokalemia and present with decreased SPAK and NCC phosphorylation [27, 149, 165]. Supporting the idea that Kir4.1 (together with Kir5.1) is a critical K sensor that influences intracellular Cl levels and the WNK-NCC pathway, kidney-specific KCNJ10 knockout mice have a depolarized membrane potential not influenced by the altered plasma K levels accompanying either a low or high K diet [27, 148]. Deletion of ClC-Kb in mice also leads to a marked downregulation of NCC [50] and these mice do not increase NCC phosphorylation on a low K diet [86]. The WNK mediating these effects is likely WNK4, as WNK4 knockout mice (or knock in mice with a Cl− insensitive WNK4) do not increase NCC phosphorylation during low K diet [19, 128]. An intriguing hallmark of dietary K restriction is the appearance of discrete and membrane-lacking foci in the DCT cytosol called “WNK bodies” that are thought to be potentially important for signal amplification [11, 15, 134, 135]. In summary, in response to low ECF K, K efflux through Kir4.1 increases Cl− efflux through ClC-Kb resulting in reduced intracellular Cl. Cl-mediated inhibition of WNK4 is relieved, allowing phosphorylation of SPAK and NCC, which increases Na+ reabsorption in the DCT and reduces distal Na+ delivery to limit kaliuresis (Fig. 3, left panel).
In contrast to the well-defined signaling cascades responsible for increasing NCC activity in response to a low K diet, the mechanisms for reducing NCC activity to increase renal K secretion following a high K diet are less well-defined. As reviewed in an earlier section, adaptation to high K intake involves feed-forward signaling that increases urinary K secretion and excretion before any measurable changes in ECF [K] [164]; the rapid signaling events in kidney remain to be characterized. Additionally, half of the ingested K is rapidly taken into the muscle ICF and then released over hours into the ECF (as a function of muscle activity) and K secretion and excretion continue to be elevated over hours to match output to intake. Renal adaptation to high K feeding involves a requirement to “turn off” the Kir4.1-WNK4-SPAK cascade, supported by: blunting of the inhibitory effects of a high K diet on NCC in Kir4.1 or Kir5.1 knockout mice [148, 160], and the absence of the acute K loading effects on NCC phosphorylation in Cl-insensitive WNK4 mice [19] (Fig. 3. Right panel). Other mediators that play a role include K-dependent alterations in Cul3-KLHL3–driven WNK degradation [54, 55, 79, 119], alteration in Kir4.1/Kir5.1 activity by Nedd4-2 (neural precursor cell expressed developmentally downregulated 4–2) [161], and K-mediated ubiquitylation and degradation of NCC [61]. Furthermore, the ability of high K to rapidly reduce (within minutes) NCC phosphorylation in vivo and in vitro strongly suggests that high K increases NCC dephosphorylation [56, 93, 120, 124]. As NCC dephosphorylation can occur by raising plasma K by intravenous K infusion [106] or by using amiloride [40, 130], the dephosphorylating effect of high K on NCC does not require feed-forward gastrointestinal kaliuretic signaling. Characterizing the DCT responses to a K load during the window of time when urinary K increases before plasma K increases remains to be determined [102, 164]. Despite all the evidence for rapid NCC dephosphorylation, the phosphatase responsible for mediating the effects after an acute high K load is still unclear as results with various phosphatase inhibitors are inconsistent [78, 93, 100, 120, 124]. In summary, in response to high extracellular K (Fig. 3. Right panel), the Kir4.1-WNK4-SPAK cascade is inhibited, which, in combination with rapid NCC dephosphorylation, results in reduced DCT Na reabsorption, increased distal Na delivery and kaliuresis. These mechanisms, coupled to the observations that chronic aldosterone infusion or high K diet decreases NCC expression and membrane abundance [39, 150] lead to the conclusion that the changes in NCC when aldosterone is elevated are due to aldosterone-mediated ENaC activation driving K secretion and excretion, rather than aldosterone effects on the MR in the DCT. Further supporting this concept, NCC remains functional in kidney-specific MR knockout mice [14, 129], and NCC abundance and phosphorylation are essentially the same in MR-negative versus MR-containing DCT cells [28]. K-induced NCC dephosphorylation is also aldosterone- and MR-independent [124]. The regulation of NCC via plasma K also explains how aldosterone can cause Na retention without K secretion (hypovolemia), but also K secretion without Na retention (hyperkalemia) [51]. Of note, aldosterone can increase NCC phosphorylation acutely (within minutes) in the DCT independent of changes in plasma K [21] but the physiological significance of this response is unclear.
Role of the CNT and CD
K handling in the CNT and CD is covered extensively in recent reviews [16, 51, 99, 101, 105, 152]. Principal cells of the CNT and CD are the main K-secreting cells of the renal tubule (Fig. 2). Classically, their ability to secrete K depends on the delivery of Na and the actions of aldosterone on ENaC and ROMK [141]. However, peritubular K can also acutely regulate ENaC and ROMK in the CNT and CCD by mechanisms most likely dependent on basolateral K channels [37, 95, 125, 138] and subsequent to changes in intracellular [Cl]; mechanisms similar to those observed in the DCT. CD intercalated cells also play a role in K homeostasis [99], with BK channels activated by high dietary K, volume flow, and the respective ENaC activity in surrounding principal cells [12, 67, 151]. Additionally, Type A ICs express an apical H+, K-ATPase that can reabsorb K in exchange for H+ during a low K diet in a mechanism requiring TRPV4 activity [137]. The Cl−/HCO3− exchanger pendrin in Type B ICs also plays a role in K homeostasis by modulating the activity of principal cell ENaC [68, 96].
K handling in females versus males. Adaptation to reproduction
Recent comparative studies in female and male rodents demonstrate sex-specific differences in renal K handling. Females exhibit less Na reabsorption via NHE3 along the PT, more Na reabsorption along the distal nephron and lower fasting plasma [K]. The female anatomy and lower PT Na reabsorption in females expedite excretion of a saline load and enhances NCC and ENaC activity, facilitating K secretion and consequently lowering the fasting plasma [K] set point relative to males [53, 142]. Interestingly, this pattern in females is similar to that of male rodents fed a high K diet, discussed above, which also facilitates K excretion. Consistent with NCC differences in mice at baseline, females exhibit greater thiazide-induced K excretion than males [64]. Under high K diet, males exhibit decreased NHE3, and larger increments in thiazide sensitive K excretion than females, thus coming to resemble the female transporter profile [64]. In response to low K feeding for 1 week, NCC abundance and activation are doubled in males with only a minor effect in females, and plasma K is reduced more in females (to 2.7 mEq) than males (to 3.3 mEq) [162]. Overall, the results suggest more robust responses to high- and low-K diets in males than females. Related to this issue, sexual dimorphisms in adrenal regulation of aldosterone production have been reported that warrant future investigation [33, 48].
Sexual dimorphisms in nephron operation likely evolved in females to facilitate reproductive transitions, as pregnancy and lactation involve dramatic increases in Na and K retention by the mother [29]. An analysis of kidney and colonic K transporters in pregnant rats identified the following adaptations: increased active K reabsorption by H,K-ATPase α2 in CD and colon, and decreased K secretion via collecting duct ROMK and BK. This K-conserving pattern is observed in males and non-pregnant females fed a K-deficient diet, thus, the pattern in pregnancy is consistent with requisite K retention for the fetal-placental unit [157]. Whether there are extrarenal (skeletal muscle) adaptation to facilitate K retention in muscle stores during increased K intake during pregnancy has not been examined.
K-induced natriuresis, diuresis, and blood pressure
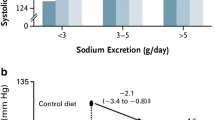
K-induced natriuresis is a mechanism that promotes kaliuresis independent of the actions of aldosterone [49]. Dietary K also acts as a genuine diuretic by altering the sensitivity of the CD to vasopressin [127]. These observations are of relevance when considering that urinary K excretion negatively correlates with BP [76, 81]. The fact that a low K diet activates NCC provides a link between dietary K and salt-sensitive hypertension and explains why dietary K depletion increases BP even in non-hypertensive subjects [63]. Conversely, diets rich in K are associated with lower BP in hypertensive and normotensive individuals [1, 36, 72, 112]. Furthermore, a recent study with > 20,000 participants demonstrated that in older patients with hypertension, substituting 25% of dietary table NaCl with KCl lowered SBP by ~ 3.5 mmHg and significantly reduced the rates of stroke and cardiovascular events [82]. Although there are several possible explanations for the ability of increased dietary K to lower BP, K-induced natriuresis through NCC inhibition provides a valid mechanism, with the anti-hypertensive effects of high K similar to those obtained with NCC inhibiting thiazide diuretics. Thus, increasing dietary K intake may be a low-cost and effective measure to curtail the global epidemic of hypertension. However, too much dietary K is not always beneficial; urinary K excretion ≥ 1 g/day in DASH (dietary approach to stop hypertension) trial participants consuming their regular diet [18] was not associated with lower SBP. Furthermore, a “U-shaped” association between K intake and BP was uncovered in a large meta-analysis of randomized-controlled trials (duration ≥ 4 weeks), with elevated SBP both when dietary K intake was under 30 or over 80 mmol/day [35].
Consumption of a fruit and vegetable enriched “Paleolithic” diet [57] containing a surfeit of K is a commonsense recommendation that would likely reduce the global burden of hypertension and cardiovascular disease. This review has discussed the molecular mechanisms that can account for the benefits of a K-rich diet to maintain cardiovascular health. However, further studies are needed to understand if raising dietary K will benefit those with kidney disease or diabetes. Whether raising K intake by KCl-enriched table salt or a DASH-style diet, the current challenge is society-wide implementation. Strategies for consideration include raising the K/Na ratio in processed and fast foods and subsidizing the costs of K-rich fruits and vegetables.
References
Aburto NJ, Hanson S, Gutierrez H, Hooper L, Elliott P, Cappuccio FP (2013) Effect of increased potassium intake on cardiovascular risk factors and disease: systematic review and meta-analyses. BMJ 346:f1378
Amemiya M, Tabei K, Kusano E, Asano Y, Alpern RJ (1999) Incubation of OKP cells in low-K+ media increases NHE3 activity after early decrease in intracellular pH. Am J Physiol 276:C711–C716
Bandulik S, Tauber P, Lalli E, Barhanin J, Warth R (2015) Two-pore domain potassium channels in the adrenal cortex. Pflugers Arch 467:1027–1042
Bazua-Valenti S, Chavez-Canales M, Rojas-Vega L, Gonzalez-Rodriguez X, Vazquez N, Rodriguez-Gama A, Argaiz ER, Melo Z, Plata C, Ellison DH, Garcia-Valdes J, Hadchouel J, Gamba G (2015) The effect of WNK4 on the Na+-Cl- cotransporter is modulated by intracellular chloride. J Am Soc Nephrol 26:1781–1786
Beggs MR, Young K, Pan W, O’Neill DD, Saurette M, Plain A, Rievaj J, Doschak MR, Cordat E, Dimke H, Alexander RT (2021) Claudin-2 and claudin-12 form independent, complementary pores required to maintain calcium homeostasis. Proc Natl Acad Sci U S A 118
Beuschlein F, Boulkroun S, Osswald A, Wieland T, Nielsen HN, Lichtenauer UD, Penton D, Schack VR, Amar L, Fischer E, Walther A, Tauber P, Schwarzmayr T, Diener S, Graf E, Allolio B, Samson-Couterie B, Benecke A, Quinkler M, Fallo F, Plouin PF, Mantero F, Meitinger T, Mulatero P, Jeunemaitre X, Warth R, Vilsen B, Zennaro MC, Strom TM, Reincke M (2013) Somatic mutations in ATP1A1 and ATP2B3 lead to aldosterone-producing adenomas and secondary hypertension. Nat Genet 45:440–4, 444e1–2
Bia MJ, DeFronzo RA (1981) Extrarenal potassium homeostasis. Am J Physiol 240:F257–F268
Bia MJ, Tyler KA, DeFronzo RA (1982) Regulation of extrarenal potassium homeostasis by adrenal hormones in rats. Am J Physiol 242:F641–F644
Bignon Y, Pinelli L, Frachon N, Lahuna O, Figueres L, Houillier P, Lourdel S, Teulon J, Paulais M (2020) Defective bicarbonate reabsorption in Kir4.2 potassium channel deficient mice impairs acid-base balance and ammonia excretion. Kidney Int 97:304–315
Bockenhauer D, Feather S, Stanescu HC, Bandulik S, Zdebik AA, Reichold M, Tobin J, Lieberer E, Sterner C, Landoure G, Arora R, Sirimanna T, Thompson D, Cross JH, Van’t Hoff W, Al Masri O, Tullus K, Yeung S, Anikster Y, Klootwijk E, Hubank M, Dillon MJ, Heitzmann D, Arcos-Burgos M, Knepper MA, Dobbie A, Gahl WA, Warth R, Sheridan E, Kleta R (2009) Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N Engl J Med 360:1960–70
Boyd-Shiwarski CR, Shiwarski DJ, Roy A, Namboodiri HN, Nkashama LJ, Xie J, McClain KL, Marciszyn A, Kleyman TR, Tan RJ, Stolz DB, Puthenveedu MA, Huang CL, Subramanya AR (2018) Potassium-regulated distal tubule WNK bodies are kidney-specific WNK1 dependent. Mol Biol Cell 29:499–509
Boyd-Shiwarski CR, Subramanya AR (2017) The renal response to potassium stress: integrating past with present. Curr Opin Nephrol Hypertens 26:411–418
Boyden LM, Choi M, Choate KA, Nelson-Williams CJ, Farhi A, Toka HR, Tikhonova IR, Bjornson R, Mane SM, Colussi G, Lebel M, Gordon RD, Semmekrot BA, Poujol A, Valimaki MJ, De Ferrari ME, Sanjad SA, Gutkin M, Karet FE, Tucci JR, Stockigt JR, Keppler-Noreuil KM, Porter CC, Anand SK, Whiteford ML, Davis ID, Dewar SB, Bettinelli A, Fadrowski JJ, Belsha CW, Hunley TE, Nelson RD, Trachtman H, Cole TR, Pinsk M, Bockenhauer D, Shenoy M, Vaidyanathan P, Foreman JW, Rasoulpour M, Thameem F, Al-Shahrouri HZ, Radhakrishnan J, Gharavi AG, Goilav B, Lifton RP (2012) Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature 482:98–102
Canonica J, Sergi C, Maillard M, Klusonova P, Odermatt A, Koesters R, Loffing-Cueni D, Loffing J, Rossier B, Frateschi S, Hummler E (2016) Adult nephron-specific MR-deficient mice develop a severe renal PHA-1 phenotype. Pflugers Arch 468:895–908
Cary R. Boyd-Shiwarski DJS, Shawn E. Griffiths, Rebecca T. Beacham, Logan Norrell, Daryl E. Morrison, Jun Wang, Jacob Mann, William Tennant, Eric N. Anderson, Jonathan Franks, Michael Calderon, Kelly A. Connolly, Claire J. Weaver, Claire C. Weckerly, Udai Bhan Pandey, Christopher J. Donnelly, Dandan Sun, Aylin R. Rodan, Arohan R. Subramanya (2022) WNK kinases sense molecular crowding and rescue cell volume via phase separation. bioRxiv 2022.01.10.475707
Castaneda-Bueno M, Ellison DH, Gamba G (2022) Molecular mechanisms for the modulation of blood pressure and potassium homeostasis by the distal convoluted tubule. EMBO Mol Med 14:e14273
Chadwick JA, Hauck JS, Lowe J, Shaw JJ, Guttridge DC, Gomez-Sanchez CE, Gomez-Sanchez EP, Rafael-Fortney JA (2015) Mineralocorticoid receptors are present in skeletal muscle and represent a potential therapeutic target. FASEB J 29:4544–4554
Chaudhary P, Wainford RD (2020) Association of urinary sodium and potassium excretion with systolic blood pressure in the Dietary Approaches to Stop Hypertension Sodium Trial. J Hum Hypertens
Chen JC, Lo YF, Lin YW, Lin SH, Huang CL, Cheng CJ (2019) WNK4 kinase is a physiological intracellular chloride sensor. Proc Natl Acad Sci U S A
Chen P, Guzman JP, Leong PK, Yang LE, Perianayagam A, Babilonia E, Ho JS, Youn JH, Wang WH, McDonough AA (2006) Modest dietary K+ restriction provokes insulin resistance of cellular K+ uptake and phosphorylation of renal outer medulla K+ channel without fall in plasma K+ concentration. Am J Physiol Cell Physiol 290:C1355–C1363
Cheng L, Poulsen SB, Wu Q, Esteva-Font C, Olesen ETB, Peng L, Olde B, Leeb-Lundberg LMF, Pisitkun T, Rieg T, Dimke H, Fenton RA (2019) Rapid aldosterone-mediated signaling in the DCT increases activity of the thiazide-sensitive NaCl cotransporter. J Am Soc Nephrol 30:1454–1470
Choi CS, Lee FN, McDonough AA, Youn JH (2002) Independent regulation of in vivo insulin action on glucose versus K(+) uptake by dietary fat and K(+) content. Diabetes 51:915–920
Choi CS, Thompson CB, Leong PK, McDonough AA, Youn JH (2001) Short-term K(+) deprivation provokes insulin resistance of cellular K(+) uptake revealed with the K(+) clamp. Am J Physiol Renal Physiol 280:F95–F102
Clase CM, Carrero JJ, Ellison DH, Grams ME, Hemmelgarn BR, Jardine MJ, Kovesdy CP, Kline GA, Lindner G, Obrador GT, Palmer BF, Cheung M, Wheeler DC, Winkelmayer WC, Pecoits-Filho R, Conference P (2019) Potassium homeostasis and management of dyskalemia in kidney diseases: conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int
Clausen T (2013) Quantification of Na+, K+ pumps and their transport rate in skeletal muscle: functional significance. J Gen Physiol 142:327–345
Cole TJ, Young MJ (2017) 30 Years of the mineralocorticoid receptor: mineralocorticoid receptor null mice: informing cell-type-specific roles. J Endocrinol 234:T83–T92
Cuevas CA, Su XT, Wang MX, Terker AS, Lin DH, McCormick JA, Yang CL, Ellison DH, Wang WH (2017) Potassium sensing by renal distal tubules requires Kir4.1. J Am Soc Nephrol 28:1814–1825
Czogalla J, Vohra T, Penton D, Kirschmann M, Craigie E, Loffing J (2016) The mineralocorticoid receptor (MR) regulates ENaC but not NCC in mice with random MR deletion. Pflugers Arch 468:849–858
de Souza AMA, West CA (2018) Adaptive remodeling of renal Na+ and K+ transport during pregnancy. Curr Opin Nephrol Hypertens 27:379–383
DiFranco M, Hakimjavadi H, Lingrel JB, Heiny JA (2015) Na, K-ATPase alpha2 activity in mammalian skeletal muscle T-tubules is acutely stimulated by extracellular K+. J Gen Physiol 146:281–294
DiFranco M, Yu C, Quinonez M, Vergara JL (2015) Inward rectifier potassium currents in mammalian skeletal muscle fibres. J Physiol 593:1213–1238
Ellison DH, Velazquez H, Wright FS (1987) Thiazide-sensitive sodium chloride cotransport in early distal tubule. Am J Physiol 253:F546–F554
Faulkner JL, Belin de Chantemele EJ (2020) Female sex, a major risk factor for salt-sensitive hypertension. Curr Hypertens Rep 22:99
Ferraro PM, Mandel EI, Curhan GC, Gambaro G, Taylor EN (2016) Dietary protein and potassium, diet-dependent net acid load, and risk of incident kidney stones. Clin J Am Soc Nephrol 11:1834–1844
Filippini T, Naska A, Kasdagli MI, Torres D, Lopes C, Carvalho C, Moreira P, Malavolti M, Orsini N, Whelton PK, Vinceti M (2020) Potassium intake and blood pressure: a dose-response meta-analysis of randomized controlled trials. J Am Heart Assoc 9:e015719
Filippini T, Violi F, D’Amico R, Vinceti M (2017) The effect of potassium supplementation on blood pressure in hypertensive subjects: a systematic review and meta-analysis. Int J Cardiol 230:127–135
Fodstad H, Gonzalez-Rodriguez E, Bron S, Gaeggeler H, Guisan B, Rossier BC, Horisberger JD (2009) Effects of mineralocorticoid and K+ concentration on K+ secretion and ROMK channel expression in a mouse cortical collecting duct cell line. Am J Physiol Renal Physiol 296:F966–F975
Frindt G, Houde V, Palmer LG (2011) Conservation of Na+ vs. K+ by the rat cortical collecting duct. Am J Physiol Renal Physiol 301:F14-20
Frindt G, Palmer LG (2010) Effects of dietary K on cell-surface expression of renal ion channels and transporters. Am J Physiol Renal Physiol 299:F890–F897
Frindt G, Yang L, Uchida S, Weinstein AM, Palmer LG (2017) Responses of distal nephron Na(+) transporters to acute volume depletion and hyperkalemia. Am J Physiol Renal Physiol 313:F62–F73
Funder J (2017) 30 Years of the mineralocorticoid receptor: mineralocorticoid receptor activation and specificity-conferring mechanisms: a brief history. J Endocrinol 234:T17–T21
Glover M, Mercier Zuber A, Figg N, O’Shaughnessy KM (2010) The activity of the thiazide-sensitive Na(+)-Cl(-) cotransporter is regulated by protein phosphatase PP4. Can J Physiol Pharmacol 88:986–995
Greenlee M, Wingo CS, McDonough AA, Youn JH, Kone BC (2009) Narrative review: evolving concepts in potassium homeostasis and hypokalemia. Ann Intern Med 150:619–625
Grimm PR, Taneja TK, Liu J, Coleman R, Chen YY, Delpire E, Wade JB, Welling PA (2012) SPAK isoforms and OSR1 regulate sodium-chloride co-transporters in a nephron-specific manner. J Biol Chem 287:37673–37690
Gruber S, Beuschlein F (2020) Hypokalemia and the prevalence of primary aldosteronism. Horm Metab Res 52:347–356
Gumz ML, Rabinowitz L, Wingo CS (2015) An integrated view of potassium homeostasis. N Engl J Med 373:60–72
Hattangady NG, Olala LO, Bollag WB, Rainey WE (2012) Acute and chronic regulation of aldosterone production. Mol Cell Endocrinol 350:151–162
Heitzmann D, Derand R, Jungbauer S, Bandulik S, Sterner C, Schweda F, El Wakil A, Lalli E, Guy N, Mengual R, Reichold M, Tegtmeier I, Bendahhou S, Gomez-Sanchez CE, Aller MI, Wisden W, Weber A, Lesage F, Warth R, Barhanin J (2008) Invalidation of TASK1 potassium channels disrupts adrenal gland zonation and mineralocorticoid homeostasis. EMBO J 27:179–187
Hene RJ, Koomans HA, Rabelink AJ, Boer P, Dorhout Mees EJ (1988) Mineralocorticoid activity and the excretion of an oral potassium load in normal man. Kidney Int 34:697–703
Hennings JC, Andrini O, Picard N, Paulais M, Huebner AK, Cayuqueo IK, Bignon Y, Keck M, Corniere N, Bohm D, Jentsch TJ, Chambrey R, Teulon J, Hubner CA, Eladari D (2017) The ClC-K2 chloride channel is critical for salt handling in the distal nephron. J Am Soc Nephrol 28:209–217
Hoorn EJ, Gritter M, Cuevas CA, Fenton RA (2020) Regulation of the renal NaCl cotransporter and its role in potassium homeostasis. Physiol Rev 100:321–356
Hoorn EJ, Zietse R (2015) Gut-kidney kaliuretic signaling: looking forward to feeding. Kidney Int 88:1230–1232
Hu R, McDonough AA, Layton AT (2020) Sex differences in solute transport along the nephrons: effects of Na(+) transport inhibition. Am J Physiol Renal Physiol 319:F487–F505
Ishizawa K, Wang Q, Li J, Yamazaki O, Tamura Y, Fujigaki Y, Uchida S, Lifton RP, Shibata S (2019) Calcineurin dephosphorylates Kelch-like 3, reversing phosphorylation by angiotensin II and regulating renal electrolyte handling. Proc Natl Acad Sci U S A 116:3155–3160
Ishizawa K, Xu N, Loffing J, Lifton RP, Fujita T, Uchida S, Shibata S (2016) Potassium depletion stimulates Na-Cl cotransporter via phosphorylation and inactivation of the ubiquitin ligase Kelch-like 3. Biochem Biophys Res Commun 480:745–751
Jensen IS, Larsen CK, Leipziger J, Sorensen MV (2016) Na(+) dependence of K(+) -induced natriuresis, kaliuresis and Na(+) /Cl(-) cotransporter dephosphorylation. Acta Physiol (Oxf) 218:49–61
Kamel KS, Schreiber M, Halperin ML (2014) Integration of the response to a dietary potassium load: a paleolithic perspective. Nephrol Dial Transplant 29:982–989
Kirabo A (2017) A new paradigm of sodium regulation in inflammation and hypertension. Am J Physiol Regul Integr Comp Physiol 313:R706–R710
Kobayashi K, Uchida S, Mizutani S, Sasaki S, Marumo F (2001) Intrarenal and cellular localization of CLC-K2 protein in the mouse kidney. J Am Soc Nephrol 12:1327–1334
Kopp C, Linz P, Wachsmuth L, Dahlmann A, Horbach T, Schofl C, Renz W, Santoro D, Niendorf T, Muller DN, Neininger M, Cavallaro A, Eckardt KU, Schmieder RE, Luft FC, Uder M, Titze J (2012) (23)Na magnetic resonance imaging of tissue sodium. Hypertension 59:167–172
Kortenoeven MLA, Cheng L, Wu Q, Fenton RA (2021) An in vivo protein landscape of the mouse DCT during high dietary K(+) or low dietary Na(+) intake. Am J Physiol Renal Physiol 320:F908–F921
Kortenoeven MLA, Esteva-Font C, Dimke H, Poulsen SB, Murali SK, Fenton RA (2021) High dietary potassium causes ubiquitin-dependent degradation of the kidney sodium-chloride cotransporter. J Biol Chem:100915
Krishna GG, Kapoor SC (1991) Potassium depletion exacerbates essential hypertension. Ann Intern Med 115:77–83
Li J, Xu S, Yang L, Yang J, Wang CJ, Weinstein AM, Palmer LG, Wang T (2019) Sex difference in kidney electrolyte transport II: impact of K(+) intake on thiazide-sensitive cation excretion in male and female mice. Am J Physiol Renal Physiol 317:F967–F977
Lindinger MI, Cairns SP (2021) Regulation of muscle potassium: exercise performance, fatigue and health implications. Eur J Appl Physiol 121:721–748
Lindinger MI, Franklin TW, Lands LC, Pedersen PK, Welsh DG (1985) Heigenhauser GJ (2000) NaHCO(3) and KHCO(3) ingestion rapidly increases renal electrolyte excretion in humans. J Appl Physiol 88:540–550
Liu W, Schreck C, Coleman RA, Wade JB, Hernandez Y, Zavilowitz B, Warth R, Kleyman TR, Satlin LM (2011) Role of NKCC in BK channel-mediated net K(+) secretion in the CCD. Am J Physiol Renal Physiol 301:F1088–F1097
Lopez-Cayuqueo KI, Chavez-Canales M, Pillot A, Houillier P, Jayat M, Baraka-Vidot J, Trepiccione F, Baudrie V, Busst C, Soukaseum C, Kumai Y, Jeunemaitre X, Hadchouel J, Eladari D, Chambrey R (2018) A mouse model of pseudohypoaldosteronism type II reveals a novel mechanism of renal tubular acidosis. Kidney Int 94:514–523
Louis-Dit-Picard H, Barc J, Trujillano D, Miserey-Lenkei S, Bouatia-Naji N, Pylypenko O, Beaurain G, Bonnefond A, Sand O, Simian C, Vidal-Petiot E, Soukaseum C, Mandet C, Broux F, Chabre O, Delahousse M, Esnault V, Fiquet B, Houillier P, Bagnis CI, Koenig J, Konrad M, Landais P, Mourani C, Niaudet P, Probst V, Thauvin C, Unwin RJ, Soroka SD, Ehret G, Ossowski S, Caulfield M, International Consortium for Blood P, Bruneval P, Estivill X, Froguel P, Hadchouel J, Schott JJ, Jeunemaitre X (2012) KLHL3 mutations cause familial hyperkalemic hypertension by impairing ion transport in the distal nephron. Nat Genet 44(456–60):S1-3
Lourdel S, Paulais M, Cluzeaud F, Bens M, Tanemoto M, Kurachi Y, Vandewalle A, Teulon J (2002) An inward rectifier K(+) channel at the basolateral membrane of the mouse distal convoluted tubule: similarities with Kir4-Kir5.1 heteromeric channels. J Physiol 538:391–404
McCormick JA, Mutig K, Nelson JH, Saritas T, Hoorn EJ, Yang CL, Rogers S, Curry J, Delpire E, Bachmann S, Ellison DH (2011) A SPAK isoform switch modulates renal salt transport and blood pressure. Cell Metab 14:352–364
McDonough AA, Veiras LC, Guevara CA, Ralph DL (2017) Cardiovascular benefits associated with higher dietary K+ vs. lower dietary Na+: evidence from population and mechanistic studies. Am J Physiol Endocrinol Metab 312:E348–E356
McDonough AA, Youn JH (2017) Potassium homeostasis: the knowns, the unknowns, and the health benefits. Physiology (Bethesda) 32:100–111
McFarlin BE, Chen Y, Priver TS, Ralph DL, Mercado A, Gamba G, Madhur MS, McDonough AA (2020) Coordinate adaptations of skeletal muscle and kidney to maintain extracellular [K(+)] during K(+)-deficient diet. Am J Physiol Cell Physiol 319:C757–C770
McKenna MJ, Gissel H, Clausen T (2003) Effects of electrical stimulation and insulin on Na+-K+-ATPase ([3H]ouabain binding) in rat skeletal muscle. J Physiol 547:567–580
Mente A, O’Donnell MJ, Rangarajan S, McQueen MJ, Poirier P, Wielgosz A, Morrison H, Li W, Wang X, Di C, Mony P, Devanath A, Rosengren A, Oguz A, Zatonska K, Yusufali AH, Lopez-Jaramillo P, Avezum A, Ismail N, Lanas F, Puoane T, Diaz R, Kelishadi R, Iqbal R, Yusuf R, Chifamba J, Khatib R, Teo K, Yusuf S, Investigators P (2014) Association of urinary sodium and potassium excretion with blood pressure. N Engl J Med 371:601–611
Morita H, Fujiki N, Miyahara T, Lee K, Tanaka K (2000) Hepatoportal bumetanide-sensitive K(+)-sensor mechanism controls urinary K(+) excretion. Am J Physiol Regul Integr Comp Physiol 278:R1134–R1139
Mukherjee A, Yang CL, McCormick JA, Martz K, Sharma A, Ellison DH (2021) Roles of WNK4 and SPAK in K(+)-mediated dephosphorylation of the NaCl cotransporter. Am J Physiol Renal Physiol 320:F719–F733
Murali SK, Little R, Poulsen SB, Ferdaus MZ, Ellison DH, McCormick JA, Fenton RA (2021) Potassium effects on NCC are attenuated during inhibition of cullin E3-ubiquitin ligases. Cells 11
Murillo-de-Ozores AR, Rodriguez-Gama A, Carbajal-Contreras H, Gamba G, Castaneda-Bueno M (2021) WNK4 kinase: from structure to physiology. Am J Physiol Renal Physiol 320:F378–F403
Ndanuko RN, Ibrahim R, Hapsari RA, Neale EP, Raubenheimer D, Charlton KE (2021) Association between the urinary sodium to potassium ratio and blood pressure in adults: a systematic review and meta-analysis. Adv Nutr
Neal B, Wu Y, Feng X, Zhang R, Zhang Y, Shi J, Zhang J, Tian M, Huang L, Li Z, Yu Y, Zhao Y, Zhou B, Sun J, Liu Y, Yin X, Hao Z, Yu J, Li KC, Zhang X, Duan P, Wang F, Ma B, Shi W, Di Tanna GL, Stepien S, Shan S, Pearson SA, Li N, Yan LL, Labarthe D, Elliott P (2021) Effect of salt substitution on cardiovascular events and death. N Engl J Med 385:1067–1077
Nesterov V, Bertog M, Korbmacher C (2022) High baseline ROMK activity in the mouse late distal convoluted and early connecting tubule probably contributes to aldosterone-independent K(+) secretion. Am J Physiol Renal Physiol 322:F42–F54
Nguyen MT, Yang LE, Fletcher NK, Lee DH, Kocinsky H, Bachmann S, Delpire E, McDonough AA (2012) Effects of K+-deficient diets with and without NaCl supplementation on Na+, K+, and H2O transporters’ abundance along the nephron. Am J Physiol Renal Physiol 303:F92-104
Nguyen TQ, Maalouf NM, Sakhaee K, Moe OW (2011) Comparison of insulin action on glucose versus potassium uptake in humans. Clin J Am Soc Nephrol 6:1533–1539
Nomura N, Shoda W, Wang Y, Mandai S, Furusho T, Takahashi D, Zeniya M, Sohara E, Rai T, Uchida S (2018) Role of ClC-K and barttin in low potassium-induced sodium chloride cotransporter activation and hypertension in mouse kidney. Biosci Rep 38
Oh KS, Oh YT, Kim SW, Kita T, Kang I, Youn JH (2011) Gut sensing of dietary K(+) intake increases renal K(+)excretion. Am J Physiol Regul Integr Comp Physiol 301:R421–R429
Oh YT, Kim J, Youn JH (2013) Role of pituitary in K+ homeostasis: impaired renal responses to altered K+ intake in hypophysectomized rats. Am J Physiol Regul Integr Comp Physiol 304:R1166–R1174
Ohta A, Schumacher FR, Mehellou Y, Johnson C, Knebel A, Macartney TJ, Wood NT, Alessi DR, Kurz T (2013) The CUL3-KLHL3 E3 ligase complex mutated in Gordon’s hypertension syndrome interacts with and ubiquitylates WNK isoforms: disease-causing mutations in KLHL3 and WNK4 disrupt interaction. Biochem J 451:111–122
Pacheco-Alvarez D, Cristobal PS, Meade P, Moreno E, Vazquez N, Munoz E, Diaz A, Juarez ME, Gimenez I, Gamba G (2006) The Na+:Cl- cotransporter is activated and phosphorylated at the amino-terminal domain upon intracellular chloride depletion. J Biol Chem 281:28755–28763
Palygin O, Pochynyuk O, Staruschenko A (2017) Role and mechanisms of regulation of the basolateral Kir 4.1/Kir 5.1K(+) channels in the distal tubules. Acta Physiol (Oxf) 219:260–273
Pei L, Solis G, Nguyen MT, Kamat N, Magenheimer L, Zhuo M, Li J, Curry J, McDonough AA, Fields TA, Welch WJ, Yu AS (2016) Paracellular epithelial sodium transport maximizes energy efficiency in the kidney. J Clin Invest 126:2509–2518
Penton D, Czogalla J, Wengi A, Himmerkus N, Loffing-Cueni D, Carrel M, Rajaram RD, Staub O, Bleich M, Schweda F, Loffing J (2016) Extracellular K(+) rapidly controls NaCl cotransporter phosphorylation in the native distal convoluted tubule by Cl(-) -dependent and independent mechanisms. J Physiol 594:6319–6331
Penton D, Moser S, Wengi A, Czogalla J, Rosenbaek LL, Rigendinger F, Faresse N, Martins JR, Fenton RA, Loffing-Cueni D, Loffing J (2019) Protein phosphatase 1 inhibitor-1 mediates the cAMP-dependent stimulation of the renal NaCl cotransporter. J Am Soc Nephrol 30:737–750
Penton D, Vohra T, Banki E, Wengi A, Weigert M, Forst AL, Bandulik S, Warth R, Loffing J (2020) Collecting system-specific deletion of Kcnj10 predisposes for thiazide- and low-potassium diet-induced hypokalemia. Kidney Int 97:1208–1218
Pham TD, Elengickal AJ, Verlander JW, Al-Qusairi L, Chen C, Abood DC, King SA, Loffing J, Welling PA, Wall SM (2022) Pendrin null mice develop severe hypokalemia following dietary K(+) restriction: role of ENaC. Am J Physiol Renal Physiol
Piala AT, Moon TM, Akella R, He H, Cobb MH, Goldsmith EJ (2014) Chloride sensing by WNK1 involves inhibition of autophosphorylation. Sci Signal 7:ra41
Picard N, Trompf K, Yang CL, Miller RL, Carrel M, Loffing-Cueni D, Fenton RA, Ellison DH, Loffing J (2014) Protein phosphatase 1 inhibitor-1 deficiency reduces phosphorylation of renal NaCl cotransporter and causes arterial hypotension. J Am Soc Nephrol 25:511–522
Polidoro JZ, Luchi WM, Seguro AC, Malnic G, Girardi ACC (2022) Paracrine and endocrine regulation of renal K(+) secretion. Am J Physiol Renal Physiol 322:F360–F377
Poulsen SB, Cheng L, Penton D, Kortenoeven MLA, Matchkov VV, Loffing J, Little R, Murali SK, Fenton RA (2021) Activation of the kidney sodium chloride cotransporter by the beta2-adrenergic receptor agonist salbutamol increases blood pressure. Kidney Int 100:321–335
Poulsen SB, Fenton RA (2019) K(+) and the renin-angiotensin-aldosterone system: new insights into their role in blood pressure control and hypertension treatment. J Physiol 597:4451–4464
Preston RA, Afshartous D, Rodco R, Alonso AB, Garg D (2015) Evidence for a gastrointestinal-renal kaliuretic signaling axis in humans. Kidney Int 88:1383–1391
Rabinowitz L (1989) Homeostatic regulation of potassium excretion. J Hypertens 7:433–442
Rabinowitz L (1996) Aldosterone and potassium homeostasis. Kidney Int 49:1738–1742
Rao R, Bhalla V, Pastor-Soler NM (2019) Intercalated cells of the kidney collecting duct in kidney physiology. Semin Nephrol 39:353–367
Rengarajan S, Lee DH, Oh YT, Delpire E, Youn JH, McDonough AA (2014) Increasing plasma [K+] by intravenous potassium infusion reduces NCC phosphorylation and drives kaliuresis and natriuresis. Am J Physiol Renal Physiol 306:F1059–F1068
Richardson C, Alessi DR (2008) The regulation of salt transport and blood pressure by the WNK-SPAK/OSR1 signalling pathway. J Cell Sci 121:3293–3304
Rieg T, Vallon V, Sausbier M, Sausbier U, Kaissling B, Ruth P, Osswald H (2007) The role of the BK channel in potassium homeostasis and flow-induced renal potassium excretion. Kidney Int 72:566–573
Rosenbaek LL, Assentoft M, Pedersen NB, MacAulay N, Fenton RA (2012) Characterization of a novel phosphorylation site in the sodium-chloride cotransporter, NCC. J Physiol 590:6121–6139
Rosenbaek LL, Rizzo F, MacAulay N, Staub O, Fenton RA (2017) Functional assessment of sodium chloride cotransporter NCC mutants in polarized mammalian epithelial cells. Am J Physiol Renal Physiol 313:F495–F504
Rossier BC, Baker ME, Studer RA (2015) Epithelial sodium transport and its control by aldosterone: the story of our internal environment revisited. Physiol Rev 95:297–340
Sacks FM, Svetkey LP, Vollmer WM, Appel LJ, Bray GA, Harsha D, Obarzanek E, Conlin PR, Miller ER 3rd, Simons-Morton DG, Karanja N, Lin PH, Group DA-SCR (2001) Effects on blood pressure of reduced dietary sodium and the Dietary Approaches to Stop Hypertension (DASH) diet. DASH-Sodium Collaborative Research Group. N Engl J Med 344:3–10
Schlingmann KP, Renigunta A, Hoorn EJ, Forst A-L, Renigunta V, Atanasov V, Mahendran S, Barakat TS, Gillion V, Godefroid N, Brooks AS, Lugtenberg D, Lake J, Debaix H, Rudin C, Knebelmann B, Tellier S, Rousset-Rouvière C, Viering D, de Baaij JHF, Weber S, Palygin O, Staruschenko A, Kleta R, Houillier P, Bockenhauer D, Devuyst O, Vargas-Poussou R, Warth R, Zdebik AA, Konrad M (2021) Defects in KCNJ16 cause a novel tubulopathy with hypokalemia, salt wasting, disturbed acid-base homeostasis, and sensorineural deafness. J Am Soc Nephrol 32:1498–1512
Scholl UI, Choi M, Liu T, Ramaekers VT, Hausler MG, Grimmer J, Tobe SW, Farhi A, Nelson-Williams C, Lifton RP (2009) Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc Natl Acad Sci U S A 106:5842–5847
Scholl UI, Lifton RP (2013) New insights into aldosterone-producing adenomas and hereditary aldosteronism: mutations in the K+ channel KCNJ5. Curr Opin Nephrol Hypertens 22:141–147
Sebastian A, Harris ST, Ottaway JH, Todd KM, Morris RC Jr (1994) Improved mineral balance and skeletal metabolism in postmenopausal women treated with potassium bicarbonate. N Engl J Med 330:1776–1781
Shalomov B, Handklo-Jamal R, Reddy HP, Theodor N, Bera AK, Dascal N (2021) A revised mechanism of action of hyperaldosteronism-linked mutations in cytosolic domains of GIRK4 (KCNJ5). J Physiol
Shibata S (2017) 30 Years of the mineralocorticoid receptor: mineralocorticoid receptor and NaCl transport mechanisms in the renal distal nephron. J Endocrinol 234:T35–T47
Shibata S, Arroyo JP, Castaneda-Bueno M, Puthumana J, Zhang J, Uchida S, Stone KL, Lam TT, Lifton RP (2014) Angiotensin II signaling via protein kinase C phosphorylates Kelch-like 3, preventing WNK4 degradation. Proc Natl Acad Sci U S A 111:15556–15561
Shoda W, Nomura N, Ando F, Mori Y, Mori T, Sohara E, Rai T, Uchida S (2017) Calcineurin inhibitors block sodium-chloride cotransporter dephosphorylation in response to high potassium intake. Kidney Int 91:402–411
Simon DB, Nelson-Williams C, Bia MJ, Ellison D, Karet FE, Molina AM, Vaara I, Iwata F, Cushner HM, Koolen M, Gainza FJ, Gitleman HJ, Lifton RP (1996) Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat Genet 12:24–30
Soleimani M, Bergman JA, Hosford MA, McKinney TD (1990) Potassium depletion increases luminal Na+/H+ exchange and basolateral Na+:CO3=:HCO3- cotransport in rat renal cortex. J Clin Invest 86:1076–1083
Sorensen MV, Grossmann S, Roesinger M, Gresko N, Todkar AP, Barmettler G, Ziegler U, Odermatt A, Loffing-Cueni D, Loffing J (2013) Rapid dephosphorylation of the renal sodium chloride cotransporter in response to oral potassium intake in mice. Kidney Int
Sorensen MV, Grossmann S, Roesinger M, Gresko N, Todkar AP, Barmettler G, Ziegler U, Odermatt A, Loffing-Cueni D, Loffing J (2013) Rapid dephosphorylation of the renal sodium chloride cotransporter in response to oral potassium intake in mice. Kidney Int 83:811–824
Sorensen MV, Saha B, Jensen IS, Wu P, Ayasse N, Gleason CE, Svendsen SL, Wang WH, Pearce D (2019) Potassium acts through mTOR to regulate its own secretion. JCI Insight 5
Su XT, Ellison DH, Wang WH (2019) Kir4.1/Kir5.1 in the DCT plays a role in the regulation of renal K(+) excretion. Am J Physiol Renal Physiol 316:F582–F586
Svendsen SL, Kornvig S, Berg P, Jensen IS, de Araujo I, Larsen CK, Leipziger J, Sorensen MV (2022) Dietary K(+) acts as a genuine diuretic. Acta Physiol (Oxf) 234:e13762
Takahashi D, Mori T, Nomura N, Khan MZ, Araki Y, Zeniya M, Sohara E, Rai T, Sasaki S, Uchida S (2014) WNK4 is the major WNK positively regulating NCC in the mouse kidney. Biosci Rep 34
Terker AS, Yarbrough B, Ferdaus MZ, Lazelle RA, Erspamer KJ, Meermeier NP, Park HJ, McCormick JA, Yang CL, Ellison DH (2016) Direct and indirect mineralocorticoid effects determine distal salt transport. J Am Soc Nephrol 27:2436–2445
Terker AS, Zhang C, McCormick JA, Lazelle RA, Zhang C, Meermeier NP, Siler DA, Park HJ, Fu Y, Cohen DM, Weinstein AM, Wang WH, Yang CL, Ellison DH (2015) Potassium modulates electrolyte balance and blood pressure through effects on distal cell voltage and chloride. Cell Metab 21:39–50
Terry EE, Zhang X, Hoffmann C, Hughes LD, Lewis SA, Li J, Wallace MJ, Riley LA, Douglas CM, Gutierrez-Monreal MA, Lahens NF, Gong MC, Andrade F, Esser KA, Hughes ME (2018) Transcriptional profiling reveals extraordinary diversity among skeletal muscle tissues. Elife 7
Thompson CB, Choi C, Youn JH, McDonough AA (1999) Temporal responses of oxidative vs. glycolytic skeletal muscles to K+ deprivation: Na+ pumps and cell cations. Am J Physiol 276:C1411–C1419
Thompson CB, McDonough AA (1996) Skeletal muscle Na, K-ATPase alpha and beta subunit protein levels respond to hypokalemic challenge with isoform and muscle type specificity. J Biol Chem 271:32653–32658
Thomson MN, Schneider W, Mutig K, Ellison DH, Kettritz R, Bachmann S (2018) Patients with hypokalemia develop WNK bodies in the distal convoluted tubule of the kidney. Am J Physiol Renal Physiol
Thomson MN, Schneider W, Mutig K, Ellison DH, Kettritz R, Bachmann S (2019) Patients with hypokalemia develop WNK bodies in the distal convoluted tubule of the kidney. Am J Physiol Renal Physiol 316:F292–F300
Titze J, Luft FC (2017) Speculations on salt and the genesis of arterial hypertension. Kidney Int 91:1324–1335
Tomilin V, Mamenko M, Zaika O, Wingo CS, Pochynyuk O (2019) TRPV4 deletion protects against hypokalemia during systemic K(+) deficiency. Am J Physiol Renal Physiol 316:F948–F956
Tomilin VN, Zaika O, Subramanya AR, Pochynyuk O (2018) Dietary K(+) and Cl(-) independently regulate basolateral conductance in principal and intercalated cells of the collecting duct. Pflugers Arch 470:339–353
Tsuchiya Y, Nakashima S, Banno Y, Suzuki Y, Morita H (2004) Effect of high-NaCl or high-KCl diet on hepatic Na+- and K+-receptor sensitivity and NKCC1 expression in rats. Am J Physiol Regul Integr Comp Physiol 286:R591–R596
Unwin R, Capasso G, Giebisch G (1994) Potassium and sodium transport along the loop of Henle: effects of altered dietary potassium intake. Kidney Int 46:1092–1099
Valinsky WC, Touyz RM, Shrier A (2018) Aldosterone, SGK1, and ion channels in the kidney. Clin Sci (Lond) 132:173–183
Veiras LC, Girardi ACC, Curry J, Pei L, Ralph DL, Tran A, Castelo-Branco RC, Pastor-Soler N, Arranz CT, Yu ASL, McDonough AA (2017) Sexual dimorphic pattern of renal transporters and electrolyte homeostasis. J Am Soc Nephrol 28:3504–3517
Veiras LC, Han J, Ralph DL, McDonough AA (2016) Potassium supplementation prevents sodium chloride cotransporter stimulation during angiotensin II hypertension. Hypertension 68:904–912
Velazquez H, Ellison DH, Wright FS (1987) Chloride-dependent potassium secretion in early and late renal distal tubules. Am J Physiol 253:F555–F562
Vitari AC, Deak M, Morrice NA, Alessi DR (2005) The WNK1 and WNK4 protein kinases that are mutated in Gordon’s hypertension syndrome phosphorylate and activate SPAK and OSR1 protein kinases. Biochem J 391:17–24
Volkl H, Lang F (1991) Electrophysiology of ammonia transport in renal straight proximal tubules. Kidney Int 40:1082–1089
Wade JB, Fang L, Coleman RA, Liu J, Grimm PR, Wang T, Welling PA (2011) Differential regulation of ROMK (Kir1.1) in distal nephron segments by dietary potassium. Am J Physiol Renal Physiol 300:F1385–F1393
Wang MX, Cuevas CA, Su XT, Wu P, Gao ZX, Lin DH, McCormick JA, Yang CL, Wang WH, Ellison DH (2018) Potassium intake modulates the thiazide-sensitive sodium-chloride cotransporter (NCC) activity via the Kir4.1 potassium channel. Kidney Int 93:893–902
Wang WH (2016) Basolateral Kir4.1 activity in the distal convoluted tubule regulates K secretion by determining NaCl cotransporter activity. Curr Opin Nephrol Hypertens 25:429–435
Wang XY, Masilamani S, Nielsen J, Kwon TH, Brooks HL, Nielsen S, Knepper MA (2001) The renal thiazide-sensitive Na-Cl cotransporter as mediator of the aldosterone-escape phenomenon. J Clin Invest 108:215–222
Webb TN, Carrisoza-Gaytan R, Montalbetti N, Rued A, Roy A, Socovich AM, Subramanya AR, Satlin LM, Kleyman TR, Carattino MD (2016) Cell-specific regulation of L-WNK1 by dietary K. Am J Physiol Renal Physiol 310:F15-26
Wei KY, Gritter M, Vogt L, de Borst MH, Rotmans JI, Hoorn EJ (2020) Dietary potassium and the kidney: lifesaving physiology. Clin Kidney J 13:952–968
Weinstein AM (1986) A mathematical model of the rat proximal tubule. Am J Physiol 250:F860–F873
Weinstein AM (2017) A mathematical model of the rat kidney: K(+)-induced natriuresis. Am J Physiol Renal Physiol 312:F925–F950
Weinstein AM (2022) A mathematical model of the rat kidney. IV. Whole kidney response to hyperkalemia. Am J Physiol Renal Physiol 322:F225–F244
Wen D, Cornelius RJ, Yuan Y, Sansom SC (2013) Regulation of BK-alpha expression in the distal nephron by aldosterone and urine pH. Am J Physiol Renal Physiol 305:F463–F476
West CA, Welling PA, West DA Jr, Coleman RA, Cheng KY, Chen C, DuBose TD Jr, Verlander JW, Baylis C, Gumz ML (2018) Renal and colonic potassium transporters in the pregnant rat. Am J Physiol Renal Physiol 314:F251–F259
Woda CB, Bragin A, Kleyman TR, Satlin LM (2001) Flow-dependent K+ secretion in the cortical collecting duct is mediated by a maxi-K channel. Am J Physiol Renal Physiol 280:F786–F793
Wright FS, Strieder N, Fowler NB, Giebisch G (1971) Potassium secretion by distal tubule after potassium adaptation. Am J Physiol 221:437–448
Wu P, Gao ZX, Zhang DD, Su XT, Wang WH, Lin DH (2019) Deletion of Kir5.1 impairs renal ability to excrete potassium during increased dietary potassium intake. J Am Soc Nephrol 30:1425–1438
Wu P, Su XT, Gao ZX, Zhang DD, Duan XP, Xiao Y, Staub O, Wang WH, Lin DH (2020) Renal tubule Nedd4-2 deficiency stimulates Kir4.1/Kir5.1 and thiazide-sensitive NaCl cotransporter in distal convoluted tubule. J Am Soc Nephrol 31:1226–1242
Xu S, Li J, Yang L, Wang CJ, Liu T, Weinstein AM, Palmer LG, Wang T (2021) Sex difference in kidney electrolyte transport III: Impact of low K intake on thiazide-sensitive cation excretion in male and female mice. Pflugers Arch 473:1749–1760
Yang L, Xu Y, Gravotta D, Frindt G, Weinstein AM, Palmer LG (2021) ENaC and ROMK channels in the connecting tubule regulate renal K+ secretion. J Gen Physiol 153
Youn JH (2013) Gut sensing of potassium intake and its role in potassium homeostasis. Semin Nephrol 33:248–256
Zhang C, Wang L, Zhang J, Su XT, Lin DH, Scholl UI, Giebisch G, Lifton RP, Wang WH (2014) KCNJ10 determines the expression of the apical Na-Cl cotransporter (NCC) in the early distal convoluted tubule (DCT1). Proc Natl Acad Sci U S A 111:11864–11869
Acknowledgements
The authors thank Dr. Jang H. Youn for the careful reading and suggestions.
Funding
DK123780 and DK132613-01 to AM. Leducq Foundation (17CVD05), the Novo Nordisk Foundation (NNF21OC0067647, NNF17OC0029724, NNF19OC0058439), and the Independent Research Fund Denmark to RAF.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
McDonough, A.A., Fenton, R.A. Potassium homeostasis: sensors, mediators, and targets. Pflugers Arch - Eur J Physiol 474, 853–867 (2022). https://doi.org/10.1007/s00424-022-02718-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-022-02718-3