
Abstract
This review integrates from the single muscle fibre to exercising human the current understanding of the role of skeletal muscle for whole-body potassium (K+) regulation, and specifically the regulation of skeletal muscle [K+]. We describe the K+ transport proteins in skeletal muscle and how they contribute to, or modulate, K+ disturbances during exercise. Muscle and plasma K+ balance are markedly altered during and after high-intensity dynamic exercise (including sports), static contractions and ischaemia, which have implications for skeletal and cardiac muscle contractile performance. Moderate elevations of plasma and interstitial [K+] during exercise have beneficial effects on multiple physiological systems. Severe reductions of the trans-sarcolemmal K+ gradient likely contributes to muscle and whole-body fatigue, i.e. impaired exercise performance. Chronic or acute changes of arterial plasma [K+] (hyperkalaemia or hypokalaemia) have dangerous health implications for cardiac function. The current mechanisms to explain how raised extracellular [K+] impairs cardiac and skeletal muscle function are discussed, along with the latest cell physiology research explaining how calcium, β-adrenergic agonists, insulin or glucose act as clinical treatments for hyperkalaemia to protect the heart and skeletal muscle in vivo. Finally, whether these agents can also modulate K+-induced muscle fatigue are evaluated.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In the year 2000, the comprehensive and excellent review on muscle potassium (K+) was published (Sejersted and Sjøgaard 2000). As in the years leading to the publication of this review, the past 2 decades has also seen the publication of numerous basic, applied and clinical research papers that span research performed using subcellular approaches, e.g. skinned fibres, patch clamp, through to single intact fibres, isolated muscle preparations in vitro, perfused muscle in situ, and the exercising human. The aim of the present review is to integrate the findings of recent in vitro research with human exercise performance and clinical research to provide a synthesis of our current understanding of the regulation of muscle K+ for exercise performance, fatigue and health.
What K+ is and why K+ is needed in the body
Potassium is a positively charged element (an electrolyte/ion, specifically a cation) ubiquitously found in the body fluid compartments of most animals, plants and other life forms (Demigné et al. 2004). It is readily obtained from dietary meat and plant products (Hamidi et al. 2011; Mangels 2014). Nonetheless, there are situations when K+ supplements are needed to maintain or achieve a proper balance of K+ within the body. Notable amongst these situations is diuretic therapy that results in elevated renal excretion of K+ (Stone et al. 2016). At other times, there is a need to eliminate excess K+, taken into the body through ingestion or injection, or through failure of K+ regulatory organs such as the kidneys and skeletal muscle.
While K+ is found in body fluids it is not uniformly distributed. There is a highly regulated distribution of K+ at multiple levels including the intestinal epithelium (regulating intestinal transport and absorption), the plasma membranes of all cells (regulating both extracellular and intracellular K+ concentrations, i.e.[K+]o and [K+]i, respectively), renal epithelial cells (regulation of whole-body balance of K+) and sweat glands (regulation to conserve K+ within the body) (McDonough and Youn 2017; Youn 2013). Potassium is also regulated within cells, notably at the surface membrane and also at mitochondrial membranes and membranes of other intracellular organelles (DiFranco et al. 2012; Lindinger 2005). This high level of regulation implies unique physical and chemical attributes of the cation, and the effects of its concentration or activity, with respect to biological functions within the body. For example, is K+ sensed by receptors (or binding sites) and if so, how is it sensed? The quick answer is that [K+] appears to be sensed indirectly (Landowne et al. 1975; Ogielska and Aldrich 1999) and possibly directly (DiFranco et al. 2015b; Hakimjavadi et al. 2018) by sensors on the surface of many types of cells. Therefore, there must be graded responses that depend on concentration and perhaps on the rate of change of [K+]. What is special about K+ with respect to its ability to being sensed? For the purposes of comparison, it is beneficial to examine two other univalent ions that are regulated together with K+, i.e. sodium (Na+) and chloride (Cl−). The molecular mass of K+ is 42 g/mol, while those for Na+ and Cl− are 23 and 35 g/mol, respectively. When in solution, these ions attract molecules including H3O+ (hydronium ion), OH− (hydroxyl ion), H+ (hydrogen ion) and H2O (water). Hence, in physiological solution, they have a hydrated radius that plays a major role in the ability of cell ion transport systems to discern between K+ and Na+, as both ions have the same charge and similar molecular mass (Conway 1981; Landowne et al. 1975). Water molecules in the hydration shell of K+ are somewhat disordered compared to those hydrating a Na+ and tend to incline their dipole moments tangentially to the hydration sphere (Mancinelli et al. 2007). Chloride, on the other hand, in addition to being negatively charged, i.e. an anion, forms hydrogen-bonded bridges with water molecules and are readily accommodated into the H-bond network of water. The hydrated radii of K+, Na+ and Cl− are 3.3 Å, 3.55 Å and 3.3 Å, respectively (Conway 1981). Membrane receptors are, therefore, able to sense and respond primarily to those ions for which they have been designed to be responsive. For example, the fact that potassium channels are sensitive not only to K+, but also to Rb+ > Cs+ > Na+ in general has been exploited by researchers studying K+ movement (Lam et al. 2014).
The [K+] in plasma at rest ranges from 3.5 to 5 mM and within cells, i.e.[K+]i, ranges from 60 to 160 mM, depending on cell type. The primary effector of the transmembrane [K+] gradient was shown to be the sarcolemmal Na+/K+ ATPase (NKA), discovered and characterised by Jens Skou over the period 1957–1962 and for which he was awarded the Nobel Prize in chemistry in 1997. Notably, Skou (1962) demonstrated the inhibition of the NKA by the glycoside ouabain. The regulation of this important membrane protein is complex and affected by catecholamines, insulin, contractile activity, membrane potential (resting EM), intracellular [Na+] ([Na+]i), and several other molecules (see Clausen 2000, 2003; Pirkmajer and Chibalin 2016).
The regulation of K+ implies that sensors exist on or in cell membranes that detect the presence of [K+]o and possibly [K+]i. An external sensor for [K+]o on the NKA (DiFranco et al. 2015b; Hakimjavadi et al. 2018), indeed regulates NKA activity. It is unknown if another sensor detects when [K+]i is adequate. Many membrane channels, including outward and inward rectifier K+ channels, are sensitive to changes in resting EM. The resting EM is largely determined by, but is typically greater (i.e. more polarised), than the equilibrium (Nernst) potential for K+, as described quantitatively by the Goldman–Hodgkin–Katz equation (Cairns and Lindinger 2008; Lindinger 2005); therefore, there appears to be a dynamic, sensitive micro-domain feedback between intracellular/extracellular [K+], resting EM and K+ channel opening. The key point is that the regulated [K+]i is 15–40 times greater than in extracellular fluids. The resultant [K+] difference across cell membranes determines the K+-equilibrium potential (EK) that is crucial for cell function. The functions of the intra- to extracellular [K+] gradient include secretion of urine (renal epithelial cells), secretion of sweat (sweat gland epithelial cells), signal transmission, and action potential generation and propagation (neurons and muscle cells, and hence brain, cardiac and skeletal muscle function), as well as a myriad of transport functions in various epithelial cells and cellular organelles throughout the body.
Overview of whole-body K+ balance
The reader is referred to the review by McDonough and Youn (2017). We use 70 kg as the mass of an average western male of normal body composition to describe K+ distribution—the values can be linearly scaled to any body mass for men and women as there are no known sex differences with respect to hydration and K+ distribution. Intracellular fluid volume (ICFV) is ~ 57% of total body water (TBW) and extracellular fluid volume (ECFV) is ~ 43% of TBW. Since the TBW is ~ 72% of body mass then the ICFV is ~ 24L and ECFV ~ 18L, with TBW of ~ 42L (ICRP 2002). The average [K+] within cells is 125 mM (Kowalchuk et al. 1988; Lindinger and Grudzien 2003; Lindinger and Heigenhauser 1987), therefore, giving about 3.0 mol of K+ within cells and 0.07 mol outside of cells. There is also K+ ‘stored’ within bone and cartilage (0.25 mol) that is slowly exchangeable with extracellular K+ but can serve as a ‘reservoir’ of K+ in times of K+ depletion (Williams and Leggett 1987). The body, therefore, has about 3.3 mol of K+—based on molecular mass and this is equal to 140 g (ICRP 2002). The majority of K+ in the body is found in skeletal muscle (68%), bone (7.5%), blood contents and spleen (3.7%), liver (3.3%), blood vessels (epithelium and smooth muscle cells—3.2%), central nervous system (2.8%), and cardiac muscle (0.6%) (Willliams and Leggett 1987).
Whole-body K+ balance at rest (Fig. 1) is a function of dietary intake, intestinal absorption and excretion and renal secretion (McDonough and Youn 2017). With increased muscular activity and sweating, then cutaneous secretion of K+ onto the skin occurs (Baker and Wolfe 2020). The level of chronic physical activity/inactivity also markedly affects the distribution of K+ in soft tissues, especially skeletal muscle (Deogenes et al. 2007; Wyckelsma et al. 2019; Zorbas et al. 2009). Losses may also occur when the intestinal system is ‘leaky’ (Agarwal et al. 1994; Priyamvada et al. 2015). The main sources of dietary K+ are meats and dairy products, and plant products including legumes, vegetables, potatoes and fruit (Hamidi et al. 2011; Mangels 2014). Dietary K+ loads can result in locally high [K+] in the gut, portal circulation and liver. There also appears to be K+-sensitive sensory mechanisms within the liver/portal circulation (Gumz et al. 2015) and or gut which feeds forward to signal K+ retention or clearance by the kidneys (Youn 2013). These mechanisms appear to be important in attenuating the hyperkalaemia and attendant risk of cardiac arrhythmias that are sometimes associated with rapid ingestion of K+ rich meals and solutions (Lindinger et al. 1999a, b; Youn 2013).

Schematic representation of the main aspects of regulation of whole-body K+ balance at rest. There is remarkably little oscillation of plasma [K+]a when ingesting many high K+ drinks and foods. There is a feed-forward system to the kidneys (and possibly skeletal muscle) to effect renal K+ clearance when high [K+] is sensed in the small intestine (Youn 2013), although unclear if this involves a central integrator. Elevated plasma [K+]a stimulates ventilation (Paterson 1997), likely through involvement of a central regulator. Elevated plasma [K+]a stimulates extraction of K+ by tissues, especially skeletal muscle
Intestinal system
With self-selected diets, the daily K+ ingestion in foods averages 2.8 g/day, of which 85% is absorbed by the intestinal tract over the wide range of intakes (Holbrook et al. 1984). Average urinary excretions of K+ averaged 77% of total intake resulting in a positive balance of 0.28 g/day (Holbrook et al. 1984). Similar values are reported by Agarwal et al. (1994): ingestion of 90 mmol/day of K+ in the diet results in absorption of 90% of intake (81 mmol) and net urinary excretion of a similar amount; thus, normal fecal K+ excretion average 9 mmol/day. Potassium is primarily absorbed by the duodenum, upon movement of digesta from the stomach to the upper gastrointestinal tract. Duodenojejunal K+ transport is dependent on the electrochemical potential difference for K+ between plasma and intestinal lumen and is associated with the osmolarity of digesta, and the absorption of Na+, water and other nutrients; peak absorption rates are estimated to be 12–15 mmol/h/cm (Agarwal et al. 1994; Lindinger et al. 1999a, b). Absorption of K+ in the human gastrointestinal tract occurs primarily by passive diffusion via both apical membrane mechanisms and paracellular route into blood via the portal circulation (Agarwal et al. 1994; Hinderling 2016). Feedforward mechanisms arising from gut K+-sensors result in increased renal K+ secretion and helps to maintain the plasma [K+] (Youn 2013; McDonough and Youn 2017). Sustained elevation in plasma [K+] is also associated with increased rates of intestinal K+ secretion, thus contributing to fecal K+ losses (Bia and Defronzo 1981).
Blood
Potassium is present in plasma and blood cells. The plasma [K+] is around 4 mM at rest in healthy individuals, even in the face of large increases or decreases in K+ intake (Hinderling 2016). Circadian variation of plasma [K+] occurs both inter-compartmentally and with renal clearance of K+ (Moore-Ede et al. 1978). Potassium absorbed from the intestinal tract may result in large elevations in plasma [K+] (Lindinger et al. 1999a, b), which in turn appears to be sensed by various tissues within the body including vascular smooth muscle, skeletal muscle, pancreas, central nervous system, peripheral nerves and kidneys. In addition to gut sensing of K+ (Youn 2013), pancreatic insulin secretion is proportional to both the glycemic and kalemic content of meals (Hiatt et al. 1972). Furthermore, erythrocytes in particular have a role in regulating plasma [K+] during periods of acute increases such as during intense exercise (Lindinger et al. 1999a, b). The associated increases in plasma [K+], adrenaline, [H+] and [lactate−] are able to stimulate a 300-fold increase in the rate of net K+ uptake by erythrocytes (Lindinger and Grudzien 2003), thus providing an acute but effective means of helping to prevent excessive increases in plasma [K+].
Renal
Elevated dietary K+ leads to increased renal K+ secretion that cannot be fully explained by the elevation of plasma [K+] (Lee et al. 2007; Lindinger et al. 2000). There is at least one feedforward system between the gut/liver/portal circulation and the kidneys whose primary function appears to proximate a balance between intake and renal excretion (Lee et al. 2007; Youn 2013; McDonough and Youn 2017). Studies in humans have shown that only 20–50% of an acute, oral K+ load is excreted by the kidneys in < 4 h (Lindinger et al. 2000), therefore, other mechanisms, i.e. extraction by skeletal muscle (Lindinger et al. 1999a, b) and liver (DeFronzo et al. 1980), are evoked to prevent life-threatening hyperkalaemia. Due to the effects of meals and circadian rhythm, the renal excretion of K+ alternates between net tubular secretion and reabsorption. The active reabsorption of K+ occurs by active apical Na+-K+-2Cl− cotransporters in the ascending limb of Henle. Tubular secretion of K+ occurs in the distal convoluted tubule and cortical collecting duct, and is mediated by the apical renal outer medullary K+ channel and the basolateral NKA (Hinderling 2016). There is net tubular reabsorption of K+ under normal conditions, perhaps balancing cutaneous losses of K+ due to passive and active sweating (Hinderling 2016).
Skin
Sweating contributes sizeable losses of K+ during prolonged exercise or exposure to heat. Ninety minutes of sweating in a cool environment leads to sweat K+ losses of 4–10 mmol, and an average sweat rate of 1L/h, independently of fluid ingestion (Maughan et al. 2005). Sweat rate and sweat gland K+ secretion both increase during exercise in hot environments and sweat [K+] may be as high as 8 mM (Baker and Wolfe 2020). Since the pool of plasma K+ is low, a total sweat loss of 2L with a mean sweat [K+] of 6 mM exceeds the plasma K+ content at rest. The net K+ loss from cells helps to defend plasma K+ content during periods of prolonged sweating. Therefore, K+-containing sports beverages should be considered for endurance-type exercise, especially in hot environments.
Skeletal muscle
Skeletal muscle has an important role in whole-body K+ balance (Lindinger et al. 1999a, b; McDonough and Youn 2017). It is well-suited for this role (see section below) because of its large mass, i.e. ~ 40% of lean body mass and its ability to ‘store’ large amounts of K+, and tolerate increased [K+]i to the point determined by the resting EM and its control of various K+ channels. Most of the ingested K+ (up to 80%) is translocated into cells (Bia and Defronzo 1981; Lindinger et al. 1999a, b), with skeletal muscle, liver and kidneys providing front lines of defense for K+ uptake against potentially life-threatening hyperkalaemia. Elevated plasma insulin stimulates the NKA in skeletal muscle and liver (Clausen and Kohn 1977), to clear K+ from plasma during and following ingestion of K+-rich foods (Hinderling 2016). Once extracellular K+ has been cleared NKA activity needs to be downregulated to avoid/minimise subsequent hypokalaemia—this is important upon cessation of intense exercise where hypokalaemia can be life-threatening (see below). Factors contributing to the downregulation of muscle NKA activity include decreased [Na+]i and increased resting EM, i.e. voltage-sensitive regulation (Bewick et al. 1999).
Skeletal muscle fibres regulate cellular and tissue K+ balance
Skeletal muscle fibres regulate the [K+] on both sides of the sarcolemma. A single transport mechanism such as the NKA, that is only capable of moving K+ into the cell, is insufficient to regulate cellular K+ balance (Pirkmajer and Chibalin 2016). Due to the importance of K+ in determining the resting EM and how it changes during muscle activity, there are requirements for other means of getting K+ across the surface and T-system membranes in both directions. Most of the K+ channels in the surface and T-system membranes are voltage-gated (termed Kv) which means that they are sensitive to the resting EM and are either in an open or closed state depending on the type of K+ channel and resting EM at that moment. Some of the Kv are termed inward rectifiers, which means that when in an ‘open’ configuration they permit a flux of K+ into the cell. Some Kv are outward-rectifiers, thus permitting efflux of K+. The main types of K+ transport proteins in skeletal muscle (Table 1), all contribute to membrane excitability, signaling and ion homeostasis. It is important to note that there is considerable variety in K+ channel terminology—in this review, we have used specific terminology consistent with identified protein structures (Gutman et al. 2005; Kubo et al. 2005; Wei et al. 2005) and focus on only the main types identified in skeletal muscle.
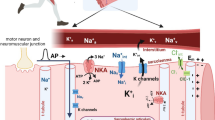
The four main types of K+ transport proteins (Fig. 2, Table 1) function in an integrated manner: (1) to maintain a normal resting EM when the muscle is quiescent (inward-rectifier K+ (Kir) channels and NKA); (2) for the repolarisation phase of the action potential (delayed and outward-rectifier K+ channel (termed KCa1.1); (3) to restore [K+]i and [Na+]i after the action potential (NKA); (4) to modulate resting EM during periods of activity (NKA, Kir, NKCC1). The NKCC1 is also involved in the regulation of muscle cell volume during periods of osmotic stress such as occurs at the onset of exercise (Kristensen and Juel 2010; Lindinger et al. 2011). In addition, the Kir 6.2 is an inward-rectifier K+ channel, sensitive to low micro-domain [ATP], that functions to link metabolic state with resting EM (Cifelli et al. 2008; Gosmanov et al. 2003). Each transport system is characterised by multiple isoforms depending on fibre-type and genetics (Jurkat-Rott et al. 2006; Kristensen and Juel 2010; Pirkmajer and Chibalin 2016). Structural defects in submits of these proteins results in myopathies (Cannon 2015). Isoforms of many of these channels are also present in the mitochondrial membrane and appear to have roles similar to those in cell surface membranes (Jurkat-Rott et al. 2006; Kristensen and Juel 2010). Skeletal muscle is a complicated tissue, capable of plasticity and adaptation to changes in its environment, responding to chronic changes by changing its phenotype. Characteristics of the phenotype are, in part, determined by the number and types of K+ transport proteins present which, in turn, are determinants of resting EM, speed of action potential repolarisation, and fatigability.

The main K+ transport systems in skeletal muscle. Top: Schematic representation of the surface or T-system membrane in a skeletal muscle cell, showing the main types of K+ transport protein and predominant direction of ion conductance. NKA = sodium–potassium ATPase, KCa1.1 = voltage-gated and calcium-sensitive delayed rectifier K+ channel, KATP = ATP-sensitive K+ channel, NKCC1 = sodium–potassium-2chloride cotransporter, [ion]O = extracellular ion concentration, [ion]I = interstitial ion concentration, [ion]i = intracellular ion concentration. Bottom: Contributions of different K+ transport systems to K+ flux into an intact, perfused, mixed muscle system. Bumetanide inhibits NKCC1. Tetracaine inhibits Kir channels. Response to ouabain or barium was nearly identical: ouabain inhibits the NKA; barium is an indiscriminate blocker of all K+ channels. Figure created using data from Lindinger et al. (2001)
Potassium transport proteins
Potassium channels, in addition to Cl− channels (Nielsen et al. 2017) are important to maintain a stable resting EM, modulate action potential duration and peak, and restore resting EM during and after an action potential (Table 1). The predominant outward-rectifier K+ channel is the “big K(+)" (KCa1.1 or BK) large conductance calcium and delayed voltage-gated K+ channel. The KCa1.1 channel is more abundant in the T-system than in the sarcolemma (Nielsen et al. 2003a; Tricarico et al. 2005) indicating the important role of the T-system in sequestering released K+ for rapid re-uptake by NKA at the end of each action potential (Lindinger 2005; Tricarico et al. 2005). The human BK channel structure was determined by Yuan et al. (2010). BK channel activation requires both membrane depolarisation and an increase in intracellular Ca2+ to micromolar levels for activation (Yang et al. 2015), therefore, this channel does not operate in normal resting muscle. With muscle activation, high concentrations of Ca2+ are attained in local micro-domains through functional coupling mechanisms with voltage-gated Ca2+ channels or channels that release Ca2+ from intracellular stores (Fakler and Adelman 2008). The KCa1.1 channel, of which there may be more than one subtype (DiFranco et al. 2012), is responsible for the down-stroke of the action potential that rapidly reestablishes the resting EM after the rapid Na+ influx causes the upstroke. KCa1.1 channels, through effects on resting EM, also function as a critical regulator of muscle contractility during activity (Bailey et al. 2019). During activity, various mechanisms work together to tailor the voltage and Ca2+ dependence of channel activation, as well as the kinetics of activation and deactivation gating, for the cell-specific function of BK channels in excitability or K+ transport (Bailey et al. 2019; Sakamoto and Kurakawa 2019).
The NKA is responsible for creating/maintaining a high [K+]i, particularly following the rapid efflux through KCa1.1 channels during the action potential and contributes about 50% of the K+ influx in resting muscle (Fig. 2). The NKA regulates plasma and whole-body [K+], with NKA activity being influenced by a combination of G-protein mediated increases in intrinsic activity as well as by adaptive modifications to the density of functional NKA units located in surface sarcolemmal and T-system membranes (Pirkmajer and Chibalin 2016). NKA activity is affected by increases in [K+]o, increases in [Na+]i, feedforward system from the gastrointestinal tract (related to dietary K+ load), and circulating hormones including the catecholamines, adrenaline and noradrenaline (anticipatory response to exercise, during all activities and especially intense exercise), insulin (to counteract the effect of Na+-mediated glucose transport into muscle), amylin and others. The NKA is tetrameric protein comprised of at least three different α-subunits and at least three different β-subunits. The composition depends on the muscle fibre-type, physical training status (Perry et al. 2016) and individual genetics. In muscles of mixed fibre-type at rest, the activity of the NKA contributes 50% to inward flux of K+ (Fig. 2). With increasing exercise intensity, there is graded increase in NKA activity towards maximal (reviewed by Clausen 2003) that usually prevents [K+]o from reaching excessive levels that cause large depolarisations of the membranes of excitable cells. Due to the membrane stabilising effects of Cl− (Cairns et al. 2004; Clausen 2011; Nielsen et al. 2017), it is now thought that the NKA need not operate at maximal capacity, even with high-intensity exercise. This is consistent with calculations presented in the work of Sejersted and coworkers, as reviewed in Sejersted and Sjøgaard (2000). Based on these data, and on the results of in vitro vastus lateralis muscle NKA activity taken prior to and immediately after the fatigue of high-intensity leg cycling exercise (Hostrup et al. 2014a; Juel et al. 2015), it is evident that moderate to high-intensity exercise can be continued even when in vivo [K+]o is substantially elevated.
The adenosine triphosphate (ATP)-dependent K+ channel (KATP) appears to be the most abundant K+ channel present in the sarcolemma (Spruce et al. 1985) and likely exceeds the number of KCa1.1 channels in the T-system (Nielsen et al. 2003a). The KATP channel is an inward-rectifier channel of the Kir6.2 sub-family having SUR1 and SUR1 sulfonylurea receptors. In resting muscle of mixed fibre-type composition in situ, the KATP channel contributes little to the inward K+ conductance but does contribute to membrane K+ leak (Lindinger et al. 2001). When micro-domain [ATP] falls below ~ 1 mM (Spruce et al. 1985), as is likely to occur in some muscle fibres during intense contractions (Söderlund and Hultman 1991), the KATP channel opening allows for a rapid and selective flux of K+ across the cell membrane. The sensitivity of the KATP channel to micro-domain [ATP] indicates a direct link to the metabolic activity of the cell (Zingman et al. 2007). When metabolic activity is high or when blood flow is inadequate cytosolic [ATP] can fall to very low levels (Tupling et al. 2001) in the vicinity of ATP-requiring proteins (NKA, calcium-ATPase). Opening of the KATP channel under these conditions allows K+ flux down its electrochemical gradient. This flux is typically outwards (facilitating some membrane polarisation) but may also be into the cell, for example when T-system [K+] is markedly elevated. In the heart, the KATP channel is involved in the prevention of catastrophic failure (Ye et al. 2018), and similar role has been proposed for skeletal muscle (Hostrup and Bangsbo 2017). The KATP channel can be modulated by changes in intracellular concentrations of H+ and inorganic phosphate, and by phosphorylation such as occurs during periods of intense contractions. Additionally, open KATP channels function to limit Ca2+ influx into fibres during repeated contractile activity (Cifelli et al. 2008).
In general, Kir channels function to stabilise the membrane both at rest and during activity (DiFranco et al. 2015b) and these Ba2+-inhibited channels collectively contribute 40–50% to the K+ influx in resting, intact muscle of mixed fibre-type (Fig. 2). Rectification refers to the electrophysiological property that the inward K+ current is larger than the outward K+ current. This occurs when EK is more negative than the resting EM, such as in quiescent muscle and is also likely in the T-system during periods of intense contractions. The Kir conductance is important in establishing and maintaining a relatively stable resting EM. In contrast to the outward-rectifier voltage-gated K+ channels (i.e. KCa1.1 and its subtypes) that remain closed at resting EM, the Kir channels are active, because the resting EM is depolarised relative to EK, thus the Kir conductance exerts a stabilising influence on resting EM through K+ efflux. The depolarisation during each action potential allows strong rectification to close Kir channels hence abolishing the Kir current during the action potential. Following each action potential, the Kv channels remain closed and Kir becomes active. During intense muscle activity, the K+ efflux through KCa1.1 channels likely elevates interstitial and T-system [K+] (Fraser et al. 2011; Shorten and Soboleva 2007). Under these conditions, the EK may become depolarised relative to resting EM which is held negative by the Cl− gradient (Cairns et al. 2004; Nielsen et al. 2017). A substantial influx of K+ may occur through Kir channels promoting the clearance of K+ from the T-system lumen together with NKA mediated K+ influx (Watanabe and Wada 2020).
The sodium–potassium–chloride cotransporter (NKCC) is the fourth main type of K+ transport protein in muscle. Only the NKCC1 isoform has been identified in skeletal muscle (Wong et al. 2001) and its activity appears to be linked to micro-domain activation of KATP (Gosmanov et al. 2003, 2004). The NKCC1 is somewhat active in resting mixed muscle and contributes up to 12% of the inward K+ conductance (Fig. 2). In resting muscle, the Cl− current is small since the Cl− equilibrium potential (ECl) approaches resting EM. At rest, under conditions that mimic those in vivo, Cl− influx via the NKCC keeps the [Cl−]i slightly higher than that predicted for passive electrochemical equilibrium, so that ECl can be about 3 mV depolarised from resting EM (van Mil et al. 1997). In consequence, there is a ‘balancing’ efflux of Cl− through ClC-1 channels that depolarises the sarcolemma. NKCC1 activity is increased with an increase in extracellular tonicity; this may be important for maintenance of cellular volume in inactive skeletal muscle during periods of intense exercise accompanied by increased plasma osmolality (Lindinger et al. 2002, 2011). In contracting muscles, the Cl− current, primarily through ClC-1 channels with a contribution from NKCC, may contribute to repolarisation during action potentials (de Paoli et al. 2013; Nielsen et al. 2017).
Now that we have introduced the main types and functions of K+ transport proteins let us examine what happens in human muscles which are of mixed fibre-type composition. This contrasts with the fibre-type predominant muscles of rodents. This is important, because much of what we know has been determined using reductionist in vitro approaches. We, therefore, need to integrate this information with what happens in exercising humans.
K+ disturbances in plasma, muscle interstitial and intracellular compartments
For many years [K+] has been measured in the plasma of exercising humans, and sometimes in the muscle intracellular compartment. The [K+]i is usually determined from muscle biopsy tissue by calculations using muscle K+ and fluid contents (Gunnarsson et al. 2013; Kowalchuk et al. 1988; Sjøgaard et al. 1985). Over the last 20 years, the [K+]o has routinely been measured in the muscle interstitium ([K+]I) using the microdialysis technique (Table 2), with a few earlier studies having used K+-sensitive microelectrodes (Hník and Vyskočil 1981; Vyskočil et al. 1983). Moreover, it has repeatedly been speculated that the [K+] in the lumen of the T-system of skeletal muscle (with a long tortuous pathway and confined space) exceeds that in the interstitium (Sejersted and Sjøgaard 2000). The T-system [K+] values have been calculated by modelling and approach 12-14 mM (Fraser et al. 2011; Shorten and Soboleva 2007), although they have yet to be directly measured. These values compare with the higher measured [K+]I values during exercise. Moreover, higher [K+]o values of up to 50 mM have been calculated for the interfibre space of superfused, isolated rat EDL muscles (Clausen 2011), that markedly exceed the interfibre [K+] values of 9-10 mM measured with K+-sensitive microelectrodes (Juel 1986) or microdialysis (Radzyukevich et al. 2009), and hence are questionable. Given this, we focus on measured [K+]I values (Table 2).
Plasma K+ concentrations
The plasma venous [K+] ([K+]v) increases rapidly with dynamic exercise as a consequence of K+ efflux from working muscle fibres as confirmed by an increased arterial-venous [K+] difference (Green et al. 2000; Kowalchuk et al. 1988; Nielsen et al. 2003b; Vøllestad et al. 1994) in concert with a reduced [K+]i (Table 2). This K+ efflux is thought to occur during each action potential (Clausen et al. 2004; Hník and Vyskočil 1981) mainly via delayed rectifier K+-channels (DiFranco et al. 2012; Duval and Léoty 1980), and with a contribution via KATP channels that open later during repeated activation (Pedersen et al. 2009). Furthermore, haemoconcentration contributes to the elevated plasma [K+] during exercise since fluid moves from plasma to both interstitial and intracellular compartments (Atanasovska et al. 2018; Juel 1988; Lindinger and Heigenhauser 1988; Lindinger et al. 1992). However, the plasma [K+], when uncorrected for fluid shifts, is the concentration what the heart and many tissue are exposed to. Following exercise cessation, the plasma [K+] falls rapidly with a half time of ~ 30 s after intense cycling (Vøllestad et al. 1994). Clearly, blood sampled 30-60 s after exercise markedly underestimates peak plasma [K+] values.
The highest [K+]v values recorded in effluent blood of leg muscles in exercising humans is 8-9 mM during treadmill running or cycling (McKenna et al. 1997; Medbø and Sejersted 1990; Vøllestad et al. 1994). When measured in the more remote arm blood vessels the plasma [K+] is lower than in leg vessels (Table 2) (Saltin et al. 1981). This difference presumably reflects some K+ uptake by non-working muscle (Lindinger et al. 1990, 1995) and erythrocytes (Lindinger et al. 1992, 1995, 1999) (Fig. 3). During repeated intense exercise, i.e. repeated 30-s sprint bouts, the rise in plasma [K+] diminishes somewhat with consecutive exercise bouts (Hargreaves et al. 1998; Lindinger et al. 1990, 1992, 1995; McKenna et al. 1993). With more prolonged running or cycling at submaximal intensities, e.g. < 75% VO2peak, there are smaller elevations of plasma [K+]. Even with intense sports such as rowing or soccer the arm measures of plasma [K+] are not extreme. Notably, physical training has often, but not always, been shown to attenuate or slow the exercise-induced rise of plasma [K+] (Christiansen 2019; Hostrup and Bangsbo 2017) so that peak plasma [K+] can be lowered by 0.3–0.5 mM during intense exercise (Harmer et al. 2000; McKenna et al. 1993, 1997). The few studies involving eccentric contractions, which likely incorporate sarcolemmal damage, have not shown the anticipated higher plasma [K+] values (Goodman et al. 2014; Piitulainen et al. 2008).

Schematic overview of regulation of whole-body K+ balance during exercise. Top: The red arrows indicate the movement of K+ released from contracting muscle. Changes in tissue [K+] during moderate to high-intensity exercise indicating regulatory roles of increased catecholamines (activator of NKA), circulating plasma (distribute high [K+] to whole body), erythrocytes (catecholamine and increased [K+]-mediated reduction of plasma [K+]), other non-contracting tissues (catecholamine and increased [K+]-mediated reduction of plasma [K+]). Bottom: Time course of change in plasma [K+] reflecting rapid increase at onset of exercise, steady-state phase when net K+ release from contracting muscles is matched by K+ extraction by other tissues, and rapid decrease upon cessation of exercise. With high-intensity exercise, plasma [K+] may remain below baseline for more than 1 h (Lindinger et al. 1992). Figure created using data from (Vøllestad et al. 1994; Lindinger et al. 1999 and Lindinger 1995)
Muscle interstitial and intracellular K+ concentrations
The simultaneous measurement of muscle [K+]I and plasma [K+]v for working knee extensor or calf muscles reveals gradients of [K+] between these compartments of up to 6 mM (Green et al. 2000; Gunnarsson et al. 2013; Nielsen et al. 2003b). The average muscle [K+]I recorded during intense exercise generally exceed that for plasma [K+]v and ranges from 9 to 14 mM (Table 2). The [K+]I also shows variability of up to 6 mM between probes inserted into the same working muscle (Juel et al. 2000). It is unknown whether those probes detecting lower [K+]I are located adjacent to quiescent fibres (within the working muscle) or adjacent to slow-twitch rather than in fast-twitch fibres given that slow-twitch fibres release less K+ per action potential than fast-twitch fibres (Clausen et al. 2004). Peak [K+]I rises with increasing intensity of dynamic exercise (Juel et al. 2000; Nielsen et al. 2003b; Gunnarsson et al. 2013), or the strength of static contractions (Vyskočil et al. 1983). This indeed is predicted since at more intense workloads there is a higher frequency of action potentials and greater motor unit recruitment. Similarly, [K+]I rises progressively with increasing stimulation frequency in animal muscle (Li et al. 2006). Moreover, after physical training the [K+]I is lowered by up to 2 mM during intense submaximal exercise (Nielsen et al. 2003b). Interestingly, when a K+-sensitive microelectrode is abutting the sarcolemma of a single frog muscle fibre the [K+]o can increase to 9–10 mM during the K+-waves associated with each action potential that summate (Hník and Vyskočil 1981). This is suggestive that a higher [K+]I exists in an apparent unstirred layer adjacent to a muscle fibre than in the bulk interstitial fluid.
Furthermore, the [K+]i falls during exercise from resting values of ~ 160 mM by varying extents but to less than 130 mM with intense exercise (Table 2). The lowest end-exercise [K+]i values for human muscle are similar to those in artificially stimulated rodent muscle (Juel 1986, 1988; Lindinger and Heigenhauser 1988). Working skeletal muscle fibres are, therefore, subjected to both raised [K+]I along with lowered [K+]i, whereas quiescent fibres (within contracting or remote muscles) are exposed only to smaller increases of [K+]o. Remarkably, the [K+]I/[K+]i value determined for human muscle at the endpoint of exercise (Cairns and Lindinger 2008; Gunnarsson et al. 2013) approaches a similar value measured for stimulated rodent muscle (Juel 1986, 1988; Lindinger and Heigenhauser 1988). Likewise, the calculated resting EM for exercising human muscle at the end-point (e.g. − 58 mV, Gunnarsson et al. 2013) coincides with the measured EM values of − 60 to − 55 mV obtained from fatigued rodent muscle (Cifelli et al. 2008; Juel 1986, 1988; Lindinger and Heigenhauser 1988). It appears that there may be a common end-point [K+]I/[K+]i value (and resting EM value) with no further rise of [K+]I or decline of [K+]i., regardless of species. This may arise from a lesser activation of the delayed rectifier K+-channels through a smaller action potential peak (DiFranco et al. 2012) and some inactivation of these channels (DiFranco et al. 2012; Duval and Léoty 1980).
Ischaemia also has a profound influence on [K+]I and resting EM in both skeletal and cardiac muscle. Prolonged tourniquet ischaemia of rabbit gastrocnemius muscle causes the mean [K+]I measured with K+-sensitive microelectrodes to increase to 12–16 mM over 2–3 h (Jennische et al. 1982). This results in the sarcolemma being depolarised to between − 60 and − 50 mV in mammalian soleus or gastrocnemius muscles (Jennische 1982; Jennische et al. 1982), which rivals the largest depolarisation measured during fatigue. These K+ disturbances with ischaemia have been attributed to impaired NKA activity (Blum et al. 1988) and possibly KATP channel opening at very low [ATP] (Tupling et al. 2001). In contrast, the [K+]I in the heart increases in a triphasic manner during myocardial ischaemia to markedly exceed that in ischaemic skeletal muscle (Watanabe and Gettes 2018; Weiss and Shine 1982; Wilde and Aksnes 1995). When global ischaemia is imposed on the isolated whole heart, [K+]I increases over the initial 5–15 min to a plateau value of 10–14 mM where it remains until climbing slowly to around 30 mM at 50–60 min (Watanabe and Gettes 2018; Weiss and Shine 1982; Wilde and Aksnes 1995). Cardiac contraction is abolished during the initial phase, when [K+]I accumulates together with a large acidosis (Watanabe and Gettes 2018; Weiss and Shine 1982). In the third phase, with excessive [K+]I, the resting force increases and irreversible damage occurs (Weiss and Shine 1982).
Physiological interactive effects with K+
Several other ionic, hormonal or metabolic changes can accentuate or blunt the effects of raised [K+]o on physiological processes. Exercise-induced changes of [Na+], [H+], [lactate−], [Cl−] or Cl− conductance, and catecholamines can all interact with K+ disturbances. Rundown of trans-sarcolemmal K+ and Na+-gradients act synergistically to reduce skeletal muscle force and M-waves (Cairns and Lindinger 2008; McKenna et al. 2008; Overgaard et al. 1999). An acidosis with lactate− accumulation appears to convey protection against hyperkalaemic effects in skeletal muscle (de Paoli et al. 2010; Nielsen et al. 2001; Pedersen et al. 2005). In contrast, the combined effects of K+ with acidosis are extremely detrimental for the heart (Fig. 4a) (Leitch and Paterson 1994a, b). Open ClC-1 channels and a normal trans-sarcolemmal Cl− gradient initially protect effects of a reduced trans-sarcolemmal K+ gradient on the resting EM (and force) in skeletal muscle (Cairns et al. 2004; Clausen 2011; Vaughan-Jones 1982) but not in cardiac muscle (Vaughan-Jones 1982). Whereas later during repeated contractions, a reduced Cl− conductance becomes protective for skeletal muscle force (Nielsen et al. 2017; Pedersen et al. 2005, 2009). Cardiac sympathetic nerve stimulation can counteract effects of hyperkalaemia on the heart (O’Neill et al. 1993), while circulating catecholamines are protective for K+-depressed contractions in both cardiac muscle (Fig. 4a, c) (Engstfeld et al. 1961; Leitch and Paterson 1994a) and skeletal muscle (Fig. 5, 7c) (Cairns et al. 1995, 2011; Clausen et al. 1993; Hansen et al. 2005; Uwera et al. 2020). Recent findings suggest that lowered ATP from inhibited glycogenolysis may, via reduced NKA activity, magnify the depressive K+ effects (Jensen et al. 2020). Hence, multiple physiological changes need to be considered together to fully appreciate the influence of K+ disturbances on cardiac and skeletal muscle performance in vivo.

Influence of raised extracellular [K+] on cardiac performance and ventricular action potentials: modulation by noradrenaline (norepinephrine) or extracellular calcium. a and b Cardiac performance in the anaesthetised rabbit in situ at 37 °C is depressed with added KCl and then improved with norepinephrine (NE) or raised extracellular Ca2+. ABP, arterial blood pressure; LVP, left ventricular pressure; dP/dt, rate of left ventricular pressure rise; pHa, arterial pH; [K+]a, arterial K+ concentration; [Ca2+]a, arterial Ca2+ concentration. c and d Action potentials in ventricular myocytes from the guinea-pig at 37 °C are depressed with 8 mM K+ Tyrode solution and restored with noradrenaline (NA) or raised extracellular Ca2+. In C, with added 0.08 μM NA, in D, with 6 mM Ca2+. a and b figures created using data from (Leitch and Paterson 1994a, b), c and d from (Paterson et al. 1993)

Raised [K+]o causes a depression of peak tetanic force by depolarising the resting EM in isolated rat skeletal muscle. Top: The peak tetanic force-[K+]o relationship in fast-twitch extensor digitorum longus muscle at 30 °C—shifted to the right (towards higher [K+]o) with added salbutamol (10 μM). Data are mean ± SEM. Bottom: The peak tetanic force-resting EM relationship determined from [K+]o effects on force and resting EM in slow-twitch soleus muscle at 30 °C. Data are mean ± SD. a figures created using data from Hansen et al. (2005), b from Cairns et al. (1995)
Integrative effects of K+ disturbances on body processes
Several physiological processes are known to be influenced by elevated interstitial or systemic K+ (Table 3) and have been reviewed previously (Clifford and Hellsten 2004; Dempsey et al. 2014; Hník and Vyskočil 1981; Juel 2007; Paterson 1996a, 1997). This section briefly highlights that K+ exerts multiple effects on different body processes and shows that moderately raised [K+]o can support exercise performance.
Muscle blood flow
A number of simultaneous neurogenic and vasodilator mechanisms act to initiate and modulate the skeletal muscle hyperemia that occurs during and following the onset of exercise (Joyner and Casey 2015). With increased muscle activity, due to the rapid (within seconds) release of K+ from skeletal muscle fibres, the raised [K+]I evokes local vasodilation and an increase in muscle blood flow (Armstrong et al. 2007; Burns et al. 2004; Clifford and Helsten 2004). Potassium infusion experiments promote hyperaemia in resting leg muscle (Juel et al. 2007; Terwoord et al. 2018), through direct effects on vascular smooth muscle (i.e. increased vascular conductance) via inward rectifying K+ channels (Burns et al. 2004; Juel et al. 2007; Terwoord et al. 2018). Never-the-less, [K+]I increases per se make a limited, albeit obligatory, contribution to the total exercise-induced hyperemia (Juel et al. 2007; Terwoord et al. 2018).
Exercise pressor reflex
The notion of a reflex originating in muscle leading to an elevation of heart rate and arterial blood pressure (McCloskey and Mitchell 1972; Saltin et al. 1981) may be instigated by several factors including K+, H+, lactate−, phosphate, adenosine, substance P, bradykinin and prostaglandins (MacLean et al. 2000). A role for K+ is supported by temporal and quantitative associations between increases of heart rate or blood pressure, and increases of either [K+]v (Fallentin et al. 1992; Saltin et al. 1981) or [K+]I (MacLean et al. 2000). Even small increases of [K+]I to just 5 mM can contribute effects (MacLean et al. 2000). However, K+ infusion experiments that increased plasma [K+] up to 6.5 mM did not increase heart rate or blood pressure (Juel et al. 2007) which questions the role of K+ per se in this reflex. Notably, high exercise heart rates cannot be accomplished via this reflex (Dempsey et al. 2014; Paterson 1996a). To mediate the exercise pressor reflex this requires stimulation of muscle afferents (Dempsey et al. 2014).
Afferent feedback
Stimulation of group III–IV muscle afferents occurs with contractions (Caron et al. 2015; Decherchi et al. 1998) or K+ administration to 5–20 mM [K+]I (Caron et al. 2015; Decherchi et al. 1998). Stimulation of this pathway to the central nervous system may, in addition to the exercise pressor reflex, contribute to elevated rating of perceived exertion and central fatigue (with reduced muscle power output) based on pharmacological blockade of these muscle afferents (Amman et al. 2009,2013; Dempsey et al. 2014). Furthermore, temporal associations between elevations of plasma [K+] and the occurrence of central fatigue during repeated static contractions indicates a possible role for K+ (Cairns et al. 2017). An elevated [K+]o may also evoke the sensation of pain via afferent feedback (Hník and Vyskočil 1981). Pain-indeed manifests with potassium chloride injection when plasma [K+] exceeds 11 mM (Durelli et al. 1982), yet ischaemic pain can be dissociated from raised [K+]I (Green et al. 2000).
Ventilation
The integrated, central regulation of ventilation at rest and during exercise subserves the simultaneous needs to provide oxygen to, and to remove carbon dioxide from, the tissues (Lindinger and Heigenhauser 2012; Keir et al. 2019). This regulation is effected by centrally integrating central and peripheral chemoreceptor, and metaboreceptor responses resulting in multi-level, local control. Potassium is well situated to act as a chemical modulator of ventilation centrally (Linton and Band 1985) and locally, because an increase in its extracellular concentration is a good indicator of increased metabolism. Large increases of plasma [K+] resulting from ingestion of high K+-containing beverages can result in elevated ventilation (Lindinger et al. 1999a, b). Raised [K+]I may contribute to hyperpnoea during exercise in two ways—by reflex drive from muscle afferents (MacLean et al. 2000; McCloskey and Mitchell 1972), and by sensitising the peripheral chemoreceptors (Linton and Band 1985; Paterson 1997). In support of the first notion, the blocking of the group III–IV afferents, which are normally activated by raised [K+]I, lowers respiratory rate and ventilation during exercise (Amann et al. 2009; Dempsey et al. 2014). Secondly, a close temporal association exists between plasma [K+]a and ventilation in exercising men (Paterson et al. 1989; 1990) which is thought to involve a K+-induced increase in the chemosensitivity of the carotid body chemoreceptors (Linton and Band 1985; Paterson 1997; Qayyum et al. 1994). This aspect becomes more important with hypoxia (Qayyum et al. 1994) and intense exercise (Paterson et al. 1989) but is only one of many factors underpinning exercise-hyperpnoea (Dempsey et al. 2014; Paterson 1997). These K+ effects on ventilation are independent of acidosis since it occurs with McArdles syndrome patients whom do not produce H+ (Paterson et al. 1990).
Neuromuscular transmission
Experimentally raising [K+]o to 8–14 mM facilitates both quantal and non-quantal acetylcholine release at the neuromuscular junction in animal models (Ceccarelli et al. 1988; da Silva et al. 2016; Vizi and Vyskočil 1979). Also, K+ causes an increased frequency of miniature endplate potentials which bolsters the endplate potential amplitude but only when [Ca2+]o is present (Ceccarelli et al. 1988; Vizi and Vyskocil 1979). Along with this, raised [K+]o increases the fusion of synaptic vesicles, (containing acetylcholine), with the presynaptic membrane of the motor-axon terminal (Ceccarelli et al. 1988). Hence, K+-induced depolarisation of presynaptic membranes augments neuromuscular transmission.
NKA activity
The early work of Skou demonstrated that raised [K+]o allosterically activates the NKA (Pirkmajer and Chibalin 2016). However, with the K+ affinity for the skeletal muscle NKA, i.e. K1/2 K, thought to be ~ 1 mM, it was regarded that the pump was virtually fully activated at the resting [K+]o of ~ 4 mM. However, recent findings show that the α2-isoform of the NKA (present in mammalian T-system membranes) is actually stimulated with up to 10–20 mM [K+]o (DiFranco et al. 2015a; Hakimjavadi et al. 2018). Indeed, the outward electrogenic NKA pump current can double between 4 and 10 mM [K+]o (DiFranco et al. 2015a) since this current has a K1/2 K, of ~ 4 mM (DiFranco et al. 2015a; Hakimjavadi et al. 2018). This enhanced NKA current and influx of K+ can provide some resistance to sarcolemmal depolarisation to maintain excitability.
Hypokalaemia and its potential dangers
Both acute hypokalaemia (plasma [K+] < 3.5 mM) and hyperkalaemia (plasma [K+] > 5.5 mM) can aggravate ventricular arrhythmias (Durfey et al. 2017; Trenor et al. 2018), particularly in the absence of the stabilising influence of plasma [Ca2+] (Durfey et al. 2017), catecholamines (Paterson et al. 1993), and for individuals with underlying heath concerns (Durfey et al. 2017; Hoppe et al. 2018). Even in healthy adults without any impairment of K+ regulation, intense exercise causes elevation of plasma [K+]a followed by abrupt hypokalaemia on exercise cessation. Indeed, the post-exercise plasma [K+] is transiently lower than resting values (Fig. 3) and can reach a nadir of 3.0–3.5 mM at 5–10 min post-exercise (Atanasovska et al. 2014, 2018; Cairns et al. 2017; Gunnarsson et al. 2013; Harmer et al. 2000; Medbø and Sejersted 1990; Vøllestad et al. 1994). The rapid and potentially large fall in plasma [K+] is consequent to a high NKA activity, resulting in a transient mismatch between K+ loss and re-uptake by muscles that were previously contracting. This post-exercise hypokalaemia, while typically short-lasting, can pose a threat to stability of the myocardium (Podrid 1990) and possibly contributes to sudden cardiac death (see below).
Hypokalaemia may also be a chronic condition associated with the use of diuretics for treatment of hypertension or chronic heart failure. In these situations, a plasma [K+] of < 3.5 mM has been associated with a higher risk of atrial fibrillation. Plasma [K+]a < 3 mM may result in Q-T interval prolongation, Torsade des pointes, ventricular fibrillation, and sudden cardiac death (Collins et al. 2017). In their study of 911,698 men and women with disease and a further 338,297 healthy controls, the prevalence of chronic hypokalaemia amongst individuals aged 55–70 year in the USA was remarkably high. Irrespective of disease, 0.2% of individuals had plasma [K+] < 3.0 mM and nearly 4% of individuals had plasma [K+] < 3.5 mM (Collins et al. 2017). Mortality rates for the hypokalaemic group were 2.5-fold greater than for those with normal plasma [K+]. In this study, there was no association between use of medications, disease state and prevalence of hypokalaemia, indicating that other factors also contribute.
A concern for individuals that have chronic hypokalaemia, whether it is known to them or not, are the effects of a further post-exercise lowering of plasma [K+]a on cardiac function. Sudden cardiac death has been reported as a result of intense sexual activities and sporting activities (Asif and Harmon 2017; Hayashi et al. 2015). While hypokalaemia has been implicated because of its potential to cause ventricular tachycardia and fibrillation, it is likely that there is initially an underlying cardiac abnormality that predisposes an individual to death via hypokalaemia (Asif and Harmon 2017; Jazayeri and Emert 2019). The occurrence of hypokalaemia appears to be rare among people participating in endurance events (Mohseni et al. 2011), likely because the submaximal intensity does not necessitate elevated NKA activities and greatly elevated concentrations of circulating catecholamines.
Hypokalaemia, particularly when acute, contributes to direct suppression of K+ channel conductances in the heart, with recent evidence indicating that indirect effects on activation of Na+ and Ca2+ channels contribute to impaired repolarisation of the cardiac sarcolemma (Weiss et al. 2017). This effect is sufficient to induce early after-depolarisations and related arrhythmias, including Torsades de pointes, polymorphic ventricular tachycardia and fibrillation. The main effect appears to be a destabilisation of cardiac Kir channels, decreasing the inward and increasing the outward K+ conductance, despite the hyperpolarisation with lowered [K+]I (Weiss et al. 2017). A secondary effect appears to be reduced NKA activity consequent to hyperpolarisation.
Compared to effects on the heart relatively little is known about hypokalaemic effects on skeletal muscle. Studies of resting EM in rodents generally show a small hyperpolarisation at 1–2 mM [K+]o, although in some fibres/conditions a depolarisation occurs at < 2 mM [K+]o (Akaike 1975; Mølgaard et al. 1980). The peak tetanic force is unchanged at 2 mM [K+]o in mouse soleus in vitro (Cairns et al. 2015) but is slightly reduced at 1.2 mM [K+]o in mouse EDL (Hayward et al. 2008). Notably, there is a slower fatigue during repeated tetanic contractions in mouse soleus muscles equilibrated at 2 mM [K+]o (Cairns et al. 2015).
Hyperkalaemia—function of cardiac and skeletal muscle
Hyperkalaemia is more common than hypokalaemia (Hoppe et al. 2018) and the chronic condition reflects nutrition, renal disease and other health concerns. Acute hyperkalaemia can result from rapid ingestion of K+-containing foods and beverages, and when coupled with intense exercise can be life-threating with destabilisation of vagal tone, and atrial and ventricular membranes (cardiac dysrhythmia) (Durfey et al. 2017). Raised [K+]I and decreased [K+]i directly impacts contraction of working skeletal muscle fibres, and elevated plasma [K+]a (which raises cardiac [K+]I) modulates cardiac contractility. When contraction occurs during the course of an ischaemic event the pronounced elevation of [K+]I likely impairs skeletal or cardiac muscle function.
It is necessary to preface the next two sections by pointing out that while hyperkalaemia accompanies intense exercise, such high-intensity exercise is typically not otherwise performed by individuals that are experiencing hyperkalaemia of non-exercise origin. Because of the attendant high NKA activities during periods of exercise-induced hyperkalaemia, sustained hyperkalaemia is short-lasting even at high work rates (Vøllestad et al. 1994).
Cardiac muscle
Large elevations of [K+]o can induce severe arrhythmias (Durfey et al. 2017), with increases to 20 mM causing cardiac arrest (Ettinger et al. 1974; Jazayeri and Emert 2019; Weiss et al. 2017). But what happens to the heart with smaller elevations of [K+]o? In the absence of underlying pathologies, the effects of hyperkalaemia on ventricular function include a shortening of the action potential due to altered gating of K+ channels. Specifically, at 8 mM [K+]o, the influx of K+ through Kir channels is increased, even in the face of a reduced driving force for K+, with decreased K+ efflux through delayed rectifier K+ channels. The net effect is an increased repolarising current late in the action potential resulting in a narrowing (Fig. 4c, d) that may compensate against any action potential prolongation and arrhythmia susceptibility (Hegyi et al. 2019). However, such protective mechanisms along with increased circulating catecholamines (Paterson et al. 1993) are not always adequate thus resulting in cardiac arrest or even sudden death (Hegyi et al. 2019).
In anesthetised mammals, the rapid administration of 8–11 mM [K+]o diminishes left ventricular pressure, contractile force, and arterial blood pressure (Fig. 4a) (Ettinger et al. 1974; Leitch and Paterson 1994a, b; Logic et al. 1968; O’Neill et al. 1993; Paterson et al. 1992; Surawicz et al. 1967). Dramatic falls in arterial blood pressure and aortic flow only occur at > 11 mM [K+]o in vivo (Paterson et al. 1992). At these [K+]o, there is also a reduced heart rate (Paterson et al. 1992) that may lower the blood pressure. In isolated Langendorff perfused rabbit hearts, rapid increases to 8–12 mM [K+]o reduce aortic blood flow and mean output pressure (index of ventricular performance) with such impairments exceeding 50% at 12 mM [K+]o (Leitch and Paterson 1994a, b; Paterson et al. 1992; Ryan and Patterson 1996). Cardiac twitch force also becomes depressed at raised [K+]o in isolated ventricular or papillary muscle. At 9–10 mM [K+]o, a 20–30% decrease occurs (Anderson et al. 2004; Robertson and Lumley1989; Ryan and Paterson 1996) followed by complete suppression around 15–18 mM [K+]o (Engstfeld et al. 1961).
Studies on intracellular action potentials in ventricular myocytes or muscle strips at 8–2 mM [K+]o show depolarisation of resting EM beyond − 65 mV (Fig. 4c, d) (Kodama et al. 1984; Paterson et al. 1992; Pool-Wilson 1984). The ventricular action potential also displays a smaller overshoot, reduced upstroke velocity, narrower width, and profound slowing of conduction (Fig. 4c, d) (Kodama et al. 1984; Paterson et al. 1992; Pool-Wilson 1984; Wan et al. 2000). Such action potential effects show regional variations in the heart for given increases of [K+]o (Wan et al. 2000). All these features can readily be explained by depolarisation-induced inactivation of voltage-gated Na+ channels and Ca2+ channels (Nobel 1979) along with greater inward-rectifier K+ currents and lesser delayed rectifier K+ currents (Hegyi et al. 2019). Increases of [K+]o to 8 mM lower the stimulation threshold, reflecting an increased sarcolemmal excitability, but 12–16 mM [K+]o reduces sarcolemmal excitability (Paterson et al. 1992). Similarly, studies using the electrocardiogram (ECG) show that raised [K+]o exerts multiple effects including a decreased or abolished P-wave, widening of the QRS complex with reduced amplitude, ST segment or QT interval depression, and an increased T-wave (Ettinger et al. 1974; Logic et al. 1968; Paterson et al. 1992; Surawicz and Lexington 1967; Weiss et al. 2017).
Raised [K+]o (8–12 mM) also exerts direct effects on sinoatrial nodal cells leading to a depolarised maximum diastolic potential, reduced slope of diastolic depolarisation, and decreased amplitude of pacemaker action potentials (Choate et al. 2001). Such [K+]o also attenuate the sympathetic drive for sinoatrial nodal pacemaking (Choate et al. 2001). These combined effects lower the heart rate, and predominates at > 10 mM [K+]o, to oppose the increased heart rate via the exercise pressor reflex which occurs at lower [K+]o (MacLean et al. 2000; Saltin et al. 1981).
Skeletal muscle
Moderately elevated [K+]o (7–10 mM) potentiates twitch and submaximal tetanic contractions in isolated non-fatigued rodent muscle (Cairns et al. 1997, 2011; Pedersen et al. 2019; Yensen et al. 2002). Notably, potassium chloride ingestion that increases plasma [K+] to 6–7 mM also augments twitches in human muscle (Grob et al. 1957). Such K+-induced force potentiation does not involve a broader action potential (Yensen et al. 2002) but appears to be mediated via a raised intracellular [Ca2+] ([Ca2+]i) (Pedersen et al. 2019; Quiñonez et al. 2010). Therefore, a moderate hyperkalaemia is likely to enhance muscle contractile performance in vivo.
The peak tetanic force-[K+]o relationship has been quantified over 7–15 mM [K+]o in isolated whole muscles of animals (Figs. 5 Top, 6a) (Ammar et al. 2015; Broch-Lips et al. 2011; Cairns et al. 1995, 1997; Uwera et al. 2020). Peak force is maintained with smaller increases of [K+]o before falling abruptly to complete suppression somewhere between 9 and 14 mM [K+]o. On the steep part of this relationship a 1 mM [K+]o increment or decrement can modulate peak tetanic force by 20–40% initial (Fig. 5 Top). Furthermore, a lowered [K+] in the solution around a mechanically skinned muscle fibre (mimicking a lowered [K+]i) that diminishes the K+ gradient across T-system membranes is sufficient to reduce peak force in single rat fast-twitch fibers (de Paoli et al. 2010; Jensen et al. 2020; Ørtenblad et al. 2003; Watanabe and Wada 2020). Moreover, raised [K+]o can reduce power or the extent of muscle/sarcomere shortening (Lucas et al. 2014; Overgaard et al. 2010; Pedersen et al. 2019). Interestingly, a reduced tolerance to raised [K+]o occurs in rat muscles with ageing, i.e. adult versus very young (Pedersen et al. 2005) or by being sedentary versus long-term physically active (Broch-Lips et al. 2011). To mimic the decline of [K+]I/[K+]i recorded during high-intensity exercise, e.g. from 4.5/125 mM to 11/110 mM (Gunnarsson et al. 2013), an appropriate test [K+]o for non-fatigued muscle in vitro, where [K+]i does not change (Cairns et al. 2015), would be 12.5 mM, = 11 mM x (125/110), rather than 11 mM. This analytical approach suggests that rundown of the trans-sarcolemmal K+-gradient of this magnitude would strikingly reduce force (Figs. 5, 6a).

Influence of raised extracellular [Ca2+] ([Ca2+]o) on K+-depressed force and action potentials in isolated mouse skeletal muscle at 25 °C. a Raised [Ca2+]o (10 mM) shifts the peak tetanic force-[K+]o relationship to the right (towards higher [K+]o) in soleus muscles. b Smaller changes of [Ca2+]o (1.3–0.5 or 2.5 mM) further depress or partially restore K+-depressed peak tetanic force (125 Hz) in soleus muscles, respectively. c Representative effect of 10 mM [K+]o and then 10 mM [K+]o plus 10 mM [Ca2+]o on intracellular action potentials in soleus fibres. Raised [K+]o causes depolarisation and a smaller action potential, then added [Ca2+]o causes a partial repolarisation and larger action potential. d Effect of 11 mM [K+]o and then 11 mM [K+]o plus 10 mM [Ca2+]o on action potential amplitude and excitability in single extensor digitorum longus fibres. Effects on action potential amplitude are similar to C. Raised [K+]o reduces sarcolemmal excitability then is partially restored with raised [Ca2+]o. Data in A,B and D are mean ± SEM. *P < 0.05. Figures created using data from Cairns et al. (2015)
Insightful early work by Vyskocil et al. (1983) furnished the measurements of both force and [K+]I during voluntary contractions of human bracioradialis muscle. They found little decline of peak force at 7-8 mM [K+]I, and then the peak MVC force fell by ~ 30% at 15 mM [K+]I (over a 1-min contraction). Thus, larger increases of [K+]I can indeed impair contraction of human muscle in vivo, although it is unlikely that this impairment is attributed solely to a K+ effect since other ionic or metabolic interactions may contribute to, or protect against, this fatigue.
Raised [K+]o is well known to depolarise the sarcolemma (Fig. 6c) (Cairns and Lindinger 2008; Sejersted and Sjøgaard 2000) hence, the relationship between peak force and resting EM, as depicted in Fig. 5 Bottom, is the key to understanding K+ effects on muscle function (Ammar et al. 2015; Cairns et al. 1995, 1997; Ørtenblad and Stephenson 2003). This relationship shows that there is a large safety margin range for depolarisation of resting EM before peak force falls markedly over a narrow EM range, i.e. − 60 to − 55 mV. Hence, force is sensitive to small changes of resting EM in this range.
K+-induced depolarisation causes a smaller action potential (Fig. 6c, d; Pedersen et al. 2005; Rich and Pinter 2003; Yensen et al. 2002) and intermittent firing of action potentials during train stimulation (Renaud and Light 1992), which together lowers tetanic [Ca2+]i (Lucas et al. 2014; Quiñonez et al. 2010). Also, some fibres lose excitability either at rest or during train stimulation (Fig. 6d; Renaud and Light 1992; Rich and Pinter, 2003) and these effects are related to an increased voltage-threshold needed to generate action potentials (Cairns et al. 1997, 2011; Pedersen et al. 2005; Rich and Pinter, 2003). All the K+-effects can be attributed to inactivation of voltage-gated Na+ channels in surface and T-system membranes (both slow and fast Na+ channel inactivation) (Rich and Pinter 2003; Ruff 1996). Notably both Na+ channel inactivation processes occur at more negative resting EM in human fast-twitch than slow-twitch fibres (Ruff 1996). In addition, depolarisation may cause inactivation of some T-system voltage-sensor proteins of excitation–contraction coupling (Ferreira-Gregorio et al. 2017; Ørtenblad and Stephenson 2003), although impairment of action potentials is likely to manifest first and hence dominate the force loss (Ørtenblad and Stephenson 2003). Together these processes reduce muscle force or cause paralysis.
Interventions to protect against hyperkalaemia
The acute management of hyperkalaemia has changed little over many years (Grob et al. 1957; Long et al. 2018), despite there now being greater understanding of the mechanisms involved with treatments. The initial aim of acute treatment is to protect the heart from arrhythmias and cardiac arrest, then prevention of skeletal muscle weakness. Clinical treatments involve three approaches: (1) counteracting the sarcolemmal processes in heart/skeletal muscle compromised by K+-induced depolarisation, (2) lowering plasma or interstitial [K+] by increased K+ uptake into cells, and/or (3) attenuating hyperkalaemia by removing K+ from the body via K+ binding agents, the kidneys and gut, or dialysis treatment which takes hours. We now discuss the first two approaches for treating hyperkalaemia and compare them with how they influence K+-induced muscle fatigue.
Calcium
Intravenous injection of a bolus of calcium (as calcium gluconate or calcium chloride) is used to rapidly treat severe hyperkalaemia, i.e. > 6.5 mM plasma [K+] (Chamberlain 1964; Durfey et al. 2017). Calcium protects via membrane stabilisation (Long et al. 2018) rather than by lowering plasma [K+] (Bosogno et al. 1994; Chamberlain 1964; Ettinger et al. 1974) with protective effects on the heart appearing within 5 min and lasting for 30–60 min (Chamberlain 1964). In vivo studies show that calcium chloride prevented the K+-induced depression of ventricular force and aortic flow (Logic et al. 1968). Similarly, raised [Ca2+]o (1.4–3.0 mM) prevented the decline of left ventricular pressure, dP/dt, and arterial blood pressure, when applied with raised [K+]o (Fig. 4b). Moreover, in the isolated perfused rabbit heart increasing [Ca2+]o stepwise from 1.8 to 10 mM, thwarted the K+-induced decrease in aortic flow (Leitch and Paterson 1994b). Interestingly, an increase to 5 mM [Ca2+]o abolished the lowering of heart rate at 12 mM [K+]o (Leitch and Paterson 1994b) which likely helped to maintain blood pressure.
Paterson et al. (1993) found that increasing [Ca2+]o (1.8–6 mM) around K+-depressed ventricular myocytes restored the action potential amplitude, upstroke velocity, and shortened action potential duration, without reversing the depolarisation (Fig. 4d). Modelling work connects these effects to an increased trans-sarcolemmal Ca2+ influx during the action potential to increase [Ca2+]i (Paterson et al. 1993). Moreover, raised [Ca2+]o reverses the adverse ECG changes in hyperkalaemic patients (Chamberlain 1964; Ettinger et al. 1974; Surawicz 1967) or in K+-depressed isolated perfused hearts (Bisogono et al. 1994). The later effects were mimicked using a Ca2+ channel ionophore, but at normal [Ca2+]o. Also, protective effects of raised [Ca2+]o on the ECG did not manifest when voltage-gated Ca2+ channel blockers are present (Bisogono et al. 1994). Hence, Ca2+ channels mediate this protective Ca2+ effect on the heart during hyperkalaemia.
Potassium-induced weakness in human skeletal muscle is treated with Ca2+ infusion (Gamstrop et al. 1957). When [Ca2+]o is raised (1.3 to 2.5–10 mM) then the peak force of K+-depressed mouse muscles is partially restored in vitro (Fig. 6a; Cairns et al. 1998, 2015; Hayward et al. 2008; Uwera et al. 2020). This Ca2+-effect occurs via repolarisation of the sarcolemma (up to 5–10 mV) (Albuquerque and Thesleff 1968; Cairns et al. 1998, 2015), which is sufficient to restore force given the steep peak tetanic force-resting EM relationship (Fig. 5). This repolarisation is intimately linked to raised [K+]i possibly due to K+ influx via the NKCC (Cairns et al. 2015). Such repolarisation promotes a restoration of action potential peak and greater percentage of excitable fibers (Fig. 6c, d) (Cairns et al. 2015; Uwera et. al. 2020). Other possible mechanisms for beneficial Ca2+ effects on action potentials include a stabilisation of voltage-gated Na+ channels (Shah et al. 2006) or screening of surface charge (Uwera et al. 2020), but such effects seem unnecessary given the measured repolarisation. Also, a Ca2+-induced recovery of depressed charge movement may occur in depolarised fibres (Ferreira-Gregorio et al. 2017).
Interestingly, studies on animal muscle in vitro have shown that raised [Ca2+]o (1.3 to 5–10 mM) increases fatigue resistance during stimulation regimes where K+ shifts are likely to occur. This includes a slower fatigue during repeated tetanic contractions in rodent fast-twitch and slow-twitch muscles (Fig. 7a, b) (Cairns et al. 1998, 2015) or during a prolonged continuous tetanus (Germinario et al. 2008; Rizvi et al. 2019). Comparable studies have not yet been performed on human muscle in vivo.

Interventions that protect against K+-induced fatigue or K+-induced paralysis in isolated rodent skeletal muscle. Raised [Ca2+]o (1.3–10 mM) increases fatigue resistance during repeated tetanic contractions (125 Hz for 500 ms, evoked every 1-s for 100 s) in mice. a fast-twitch extensor digitorum longus and b slow-twitch soleus muscles. 25 °C. Double-sigmoid functions were fitted to data points in each panel. c Salbutamol (10 μM), insulin (100 μUnits/mL), or salbutamol plus insulin (same concentrations), increase peak tetanic force (30 Hz) of rat soleus muscles suppressed with 12.5 mM [K+]o, 30 °C. Data are mean ± SEM. d Terbutaline (10 μM) increases fatigue resistance during repeated tetanic contractions (40 Hz for 300 ms, evoked every 3-s for 5 min) in mouse soleus muscles. a and b figures created from Cairns et al. (2015), c from Clausen et al. (1993), d from Juel (1988)
β-adrenergic agonists
These agents include salbutamol, terbutaline, adrenaline and noradrenaline. When administered by inhalation, nebulisation, or intravenous injection they lower resting plasma [K+] by 0.4–1.0 mM depending on dose and time (Allon and Copkney 1990; Atanasovska et al. 2018; Hostrup et al. 2014b; Long et al. 2018) and they prevent the exercise-induced rise of plasma [K+] (Wang and Clausen 1976). However, the maximum protection occurs in an hour which is much slower than that achieved with Ca2+ infusion. This lowering of plasma [K+] results from K+ uptake by skeletal muscle and other tissues via stimulation of both NKA (Clausen 2003; Pirkmajer and Chibalin 2016; Wang and Clausen 1976) and NKCC1 activity (Gosmanov et al. 2003; Wong et al. 2001). Such effects are mediated via β2-adrenergic receptors and increased cyclic adenosine monophosphate (cAMP) levels that increase protein kinase-A activity (Cairns and Borrani 2015). Such myoplasmic changes increase the affinity of NKA to raised [Na+]i (Clausen 2003; Pirkmajer and Chibilin 2016) to augment NKA activity (Cairns et al. 1995; Clausen et al. 1993; Juel 1988). Hence, β2-agonists can lessen hyperkalaemia to improve cardiac and skeletal muscle performance.
With the heart of anesthetised rabbits, exposure to noradrenaline restores left ventricular pressure, dP/dt and arterial blood pressure to counteract the depressive effects of raised [K+]o, or raised [K+]o combined with acidosis (Fig. 4A) (Leitch and Paterson 1994a, b). Similarly, noradrenaline or adrenaline, maintains aortic flow in isolated hearts depressed with 8 or 12 mM [K+]o (Leitch and Paterson 1994a, b). Adrenaline also restores force in K+-depressed papillary muscle from the frog (Engstfeld et al. 1961). Adrenaline or noradrenaline increase the action potential amplitude, upstroke velocity and duration at 8–12 mM [K+]o in isolated ventricular myocytes (Fig. 4c) (Paterson et al. 1993). These effects do not involve repolarisation of the sarcolemma even though β-agonists stimulate cardiac NKA activity (Bewick et al. 1999; Desilets and Baumgarten 1986). Instead, it likely involves enhanced trans-sarcolemmal Ca2+ influx through voltage-gated Ca2+ channels which maintains excitability (Paterson et al. 1993). Indeed, β-agonists increase both cardiac intracellular Ca2+-transients and force via greater Ca2+ influx in normal solutions (Kurihara and Konishi 1987). Moreover, some β-adrenergic enhancement of the delayed rectifier K+ current (Hegyi et al. 2019) may counter impairment of this current by hyperkalaemia.
Salbutamol inhalation attenuates the rise of plasma [K+] with exercise or potassium chloride ingestion in hyperkalaemic periodic paralysis patients to maintain force in human skeletal muscle in vivo (Wang and Clausen 1976). Indeed, a 1 mM lowering of plasma [K+] is predicted to convey a considerable protective effect on muscle force at higher [K+]o (Fig. 5 Top). Furthermore, β2-agonists restore force in K+-depressed rodent skeletal muscle by 10–60% initial in vitro (Figs. 5 Top,7c) (Cairns et al. 1995, 2011; Clausen et al. 1993; Hansen et al. 2005; Uwera et a. 2020) via stimulation of the skeletal muscle NKA (Cairns et al. 1995; Clausen et al. 1993) and likely NKCC activity (Gosmanov et al. 2003; Wong et al. 2001). Such effects are mimicked by the membrane permeable dibutyryl cAMP which supports involvement of cAMP (Clausen 2000). These processes cause a lower interfibre [K+]o in isolated human muscle preparations (Ballyani and Grafe 1988) and elevated [K+]i (Cairns et al. 1995, 2011; Clausen et al. 1993). Together these effects cause a small but variable repolarisation and a lower [Na+]i (Cairns et al. 1995; Clausen et al. 1993; Hansen et al. 2005), which together restores action potential threshold (Cairns et al. 2011) to increase the number of excitable fibres (Uwera et al. 2020), M-wave amplitude/area (Overgaard et al. 1999) and action potential peak (Uwera et al. 2020). If the muscle fibres remain, or become, excitable then β2-agonists also facilitate action potential mediated Ca2+ release from the sarcoplasmic reticulum in animal (Anderson et al. 2012; Cairns and Borrani 2015) and human muscle (Cairns and Borrani 2015; Hostrup et al. 2014a). This effect requires phosphorylation of the ryanodine receptor/Ca2+-release channel of the sarcoplasmic reticulum (Anderson et al. 2012; Cairns and Borrani 2015). However, this mechanism is unlikely to play a major protective role with β2-agonist treatment for hyperkalaemia in humans since it necessarily requires very high doses (Cairns and Borrani 2015; Hostrup et al. 2014a).
Moreover, high concentrations of β2-agonist can provide resistance to K+-induced fatigue during either repeated tetanic contractions in vitro as shown in Fig. 7d (Juel 1988), with similar effects during prolonged tetanic contractions (Cairns and Dulhunty 1994; Rizvi et al. 2018). Human studies have occasionally shown that high β2-agonist concentrations lower plasma [K+] and enhance fatigue resistance, i.e. during repeated 30-s sprints (Hostrup et al. 2014b). However, performance improvement is not consistently observed (Altarawneh et al. 2016; Hostrup et al. 2014a) possibly because the β2-agonists also modulate endogenous catecholamine levels (Hallén et al. 1996).
Insulin
Intravenous injection of insulin (usually as a bolus) lowers plasma [K+] in humans by 0.6–1.0 mM over an hour (Allon and Copkney 1990; Grob et al. 1957; Long et al. 2018). This effect manifests more rapidly than with β2-agonists and appears from 15 min (Allon and Copkney 1990). Insulin is usually administered with glucose to prevent hypoglycaemia, although this can still occur as a side-effect (Allon and Copkney 1990; Chothia et al. 2014). Insulin stimulates skeletal muscle K+ influx by increasing NKA activity, via protein kinase-C and ERK1/2 pathways (Gosmanov et al. 2003; Pirkmajer and Chibalin 2016) leading to phosphorylation of NKA (Chibalin et al. 2001), but does not also stimulate NKCC (Gosmanov et al. 2003; Wong et al. 2001). When insulin and β2-agonists are added together the fall of plasma [K+] is amplified to 1.2–1.5 mM which exceeds the effect of either agent alone (Allon and Copkney 1990). Indeed, there are post-exercise situations when both hormones are elevated in the blood (Marliss and Vranic 2002).
Acute exposure to insulin increases peak force in K+-depressed skeletal muscle in vitro by stimulating NKA activity (Cairns et al. 1995; Clausen 2000; Clausen et al. 1993), although this effect is weaker than with β2-agonists (Fig. 7C) (Clausen et al. 1993). The end result is an increased [K+]i, repolarised EM, and lowered [Na+]i, which together restores M-wave amplitude/area to restore peak force by 10–20% (Clausen 2000; Clausen et al. 2003). This insulin effect is additive to that of β-agonists through greater stimulation of NKA activity (Clausen et al. 1993). Despite these well characterised effects, insulin infusion does not also attenuate K+-induced fatigue during repeated contractions of rat muscle in situ (Karelis et al. 2003).
Glucose
When glucose/dextrose alone is administered either orally or intravenously to normal subjects or patients, it lowers resting plasma [K+] by 0.4–0.6 mM over 1–2 h (Chothia et al. 2014; Grob et al. 1957) and blunts the rise of plasma [K+] with an ingested potassium load by 0.3–0.5 mM (Allon et al. 1993). High extracellular glucose per se can phosphorylate the skeletal muscle NKA but in doing so reduces its activity (Chibalin et al. 2001). However, glucose administration triggers endogenous insulin release (Allon et al. 1993; Chothia et al. 2014) but usually to lower levels than with exogenous insulin administration (Chothia et al. 2014). Interestingly, the peptide amylin is co-secreted with insulin from pancreatic β-cells (Pirkmajer and Chibalin 2016). Amylin per se has been shown to stimulate NKA activity and is additive to the effect of insulin to restore force in K+-depressed skeletal muscle (Clausen 2000). It is tempting to speculate that effects of glucose during hyperkalaemia in vivo may involve amylin. Never-the-less, the lowering of plasma [K+] with glucose is smaller than for insulin combined with glucose (Chothia et al. 2014; Grob et al. 1957), and yields variable responses. Hence, glucose per se is not recommended as a clinical treatment (Long et al. 2018).
Interestingly, the infusion of glucose, which slightly elevates plasma glucose (5 to 7–10 mM), provides some resistance against fatigue-induced depolarisation (Karalis et al. 2005) and better maintains M-waves and force during fatiguing stimulation of rat skeletal muscle in situ (Karalis et al. 2002,2005). This may encompass acute stimulation of muscle NKA activity (Karalis et al. 2002,2005), which is independent of insulin (Karelis et al. 2003), and possible acts via greater glycolytic ATP supply for the NKA (Karalis et al. 2005). Furthermore, glucose supplementation during prolonged submaximal cycling in humans enhances maximal in vitro NKA activity (Green et al. 2007) and maintains M-waves (Stewart et al. 2007). Alternatively, the elevated glucose may act via increased lactate− production to improve function of voltage-gated Na+ channels (Rannou et al. 2012) or to lower sarcolemmal Cl− channel conductance (de Paoli et al. 2010) both of which restore M-waves and force in K+ depressed muscle (de Paoli et al. 2010).
Conclusion
K+ is a univalent cation found in all body fluids, and is important for the normal function of every cell within the body. At rest, whole-body K+ balance is influenced by dietary K+ intake, sweating, erythrocyte and skeletal muscle K+ uptake, and is regulated by the intestinal system and renal clearance in particular. Skeletal muscle fibres can regulate cellular and tissue K+ balance via four main types of K+ transport protein—NKA pumps, delayed rectifier K+ channels, inward-rectifier K+ channels including KATP channels, and NKCC1 cotransporters. Contracting skeletal muscle generates an altered K+ balance not only within itself, but also throughout the entire body. Contracting and quiescent skeletal muscle, other tissues, and cardiac muscle all encounter an altered extracellular K+ balance during and after intense exercise, with each tissue regulating its [K+]o, [K+]i, and resting EM. Contracting fibres can face exposure to 9–14 mM [K+]I during intense exercise with up to 30 mM reductions of [K+]i. In contrast, quiescent skeletal muscle fibres and cardiac muscle may be exposed to 6–10 mM [K+]o during exercise. Post-exercise hypokalaemia, with plasma [K+] < 3.5 mM can be detrimental for the heart, especially in individuals with underlying health concerns.
Moderate elevations of interstitial and plasma [K+] to 5–9 mM are favorable through stimulating muscle blood flow, the exercise pressor reflex, ventilation, neuromuscular transmission, and muscle NKA activity. Detrimental effects may occur via muscle afferents which mediate pain, perceived exertion and central fatigue. Large elevations of [K+]o i.e. > 8 mM, cause severe cardiac arrhythmias, reduce contractile performance and ultimately lead to cardiac arrest. Such effects involve depolarisation and ventricular action potential changes such as a smaller amplitude and narrowing, which are reflected in ECG changes. Similarly, large elevations of [K+]I accompanied by lowered [K+]i reduce peak tetanic force in fast-twitch and slow-twitch skeletal muscle fibres in association with depolarisation, smaller action potentials and reduced sarcolemmal excitability (intermittent firing of action potentials during trains, or completely inexcitable fibres). The treatments used for severe hyperkalaemia include an initial intravenous injection of Ca2+ salt, followed by administration of insulin (with glucose) and sometimes also with β2-agonist. Mechanisms for treatment interventions involve reducing the hyperkalaemia or protective effects on the cardiac and skeletal muscle sarcolemma to amend action potentials.
Perspectives
Future studies aimed at increasing our understanding of K+ regulation should continue to tease apart the surface and T-system membrane contributions in skeletal and cardiac muscle under different (patho) physiological conditions, with consideration for interventions for life-threatening hyperkalaemia and hypokalaemia. An accurate time course of responses of changes in the interstitial concentrations of K+, Na+ and Cl− has yet to be determined in contracting muscle, recovering muscle, and in muscle perfused with altered concentration of these ions. Interstitial methods need to employ rapid time course methods, such as electrophysiology or ion-sensitive dyes, because microdialysis provides poor time resolution. Additional studies need to target the integrated responses of ion transporting systems to address the question of what is the nature of coupling between ion transport systems operating at the microdomain (Sejersted’s “fuzzy space”) level? Are regulatory responses coordinated amongst control systems, or is each control system responding independently to the substrate/product of its reaction?
Ideally, techniques need to be developed to measure [K+] simultaneously in multiple compartments (including interstitium, T-system, sub-sarcolemmal space) during various exercise protocols or electrical stimulation regimes, and ultimately in different muscle fibre-types in humans. Hand in hand with [K+] measurements, it would be extremely valuable to measure resting EM along with an entourage of other interacting ions (Na+, Cl−, Ca2+, H+, lactate−) that constitute the exercise milieu.
More needs to be understood about K+ effects on performance of cardiac and skeletal muscle in humans. Hence: (1) determine the peak force-[K+]o relationships, and peak force–resting EM relationships for isolated human skeletal or cardiac muscle cells using close-to-in vivo models; (2) determine the peak force- and peak power-interstitial [K+] relationships in human muscles during fatiguing exercise in vivo; (3) examine the role(s) of interstitial K+ in signaling with afferent nerves to understand regulatory feedback that may contribute to elevated perception of exertion and central fatigue.
These types of studies are needed to better understand the coordinated responses of K+ transport proteins at both the surface and T-system membrane levels in vitro and in vivo. We also need to better understand how altered [K+]o and altered resting EM (by any in vivo physiological means) affects cardiac and skeletal muscle action potentials, which may lead to the development better treatments for hyperkalaemia or to combat K+-induced exercise fatigue in humans.
Abbreviations
- ATP:
-
Adenosine triphosphate
- [Ca2 +]i :
-
Intracellular calcium concentration
- cAMP:
-
Cyclic adenosine monophosphate
- [Cl−]i :
-
Intracellular chloride concentration
- ClC-1:
-
Skeletal muscle sarcolemmal chloride channel
- ECFV:
-
Extracellular fluid volume
- ECG:
-
Electrocardiogram
- ICFV:
-
Intracellular fluid volume
- KATP :
-
ATP-dependent potassium channel
- KCa1.1 :
-
Delayed rectifier potassium channel
- Kir :
-
Inward rectifier potassium channel
- [K+]a :
-
Plasma arterial potassium concentration
- [K+]i :
-
Intracellular potassium concentration
- [K+]I :
-
Interstitial potassium concentration
- [K+]o :
-
Extracellular potassium concentration
- [K+]v :
-
Plasma venous potassium concentration
- M-wave:
-
Compound extracellular muscle action potential
- [Na+]i :
-
Intracellular sodium concentration
- NKA:
-
Na+/K+ ATPase (sodium–potassium pump)
- NKCC:
-
Na+-K+-2Cl− cotransporter (sodium–potassium chloride cotransporter)
- Resting EM :
-
Resting membrane potential
- RPE:
-
Rating of perceived exertion
- TBW:
-
Total body water
- T-system:
-
Tubular system
- VO2peak:
-
Peak oxygen consumption
References
Agarwal R, Afzalpurkar R, Fordtran JS (1994) Pathophysiology of potassium absorption and secretion by the human intestine. Gastroenterology 107:548–571
Ahlborg B, Bergström J, Ekelund LG, Hultman E (1967) Muscle glycogen and muscle electrolytes during prolonged physical exercise. Acta Physiol Scand 70:129–142
Akaike N (1975) Contribution of an electrogenic sodium pump to membrane potential in mammalian skeletal muscle fibres. J Physiol 245:499–520
Albuquerque EX, Thesleff S (1968) The effect of calcium on the skeletal muscle membrane after treatment with phospholipase C. Acta Physiol Scand 72:310–321
Allon M, Copkney C (1990) Albuterol and insulin for treatment of hyperkalemia in hemodialysis patients. Kid Int 38:869–872
Allon M, Dansby L, Shanklin N (1993) Glucose modulation of the disposal of an acute potassium load in patients with end-stage renal disease. Am J Med 94:475–482
Altarawneh MM, Petersen A, Smith R, Rouffet DM, Billaut F, Perry BD, Wyckelsma WL, Tobin A, McKenna MJ (2016) Salbutamol effects on systemic potassium dynamics during and following intense continuous and intermittent exercise. Eur J Appl Physiol 116:2389–2399
Amann M, Proctor LT, Sebranek JJ, Pegelow DF, Dempsey JA (2009) Opioid-mediated muscle afferents inhibit central motor drive and limit peripheral muscle fatigue development in humans. J Physiol 587(1):271–283
Amann M, Venturelli M, Ives SJ, McDaniel J, Layec G, Rossman MJ, Richardson RS (2013) Peripheral fatigue limits endurance exercise via a sensory feedback-mediated reduction in spinal motoneuron output. J Appl Physiol 115:335–364
Ammar T, Lin W, Higgins A, Hayward LJ, Renaud JM (2015) Understanding the physiology of the asymptomatic diaphragm of the M1592V hyperkalemic periodic paralysis mouse. J Gen Physiol 146(6):509–525
Andersen JB, Gesser J, Wang T (2004) Acidosis counteracts the negative inotropic effect of K+ on ventricular muscle strips from the Toad Bufo marinus. Physiol Biochem Zoo 77(2):223–231
Andersson DC, Betzenhauser MJ, Reiken S, Umanskaya A, Shiomi T, Marks AR (2012) Stress-induced increase in skeletal muscle force requires protein kinase A phosphorylation of the ryanodine receptor. J Physiol 590:6381–6387
Armstrong ML, Dua AK, Murrant CL (2007) Potassium initiates vasodilatation induced by a single skeletal muscle contraction in hamster cremaster muscle. J Physiol 581(2):841–852. https://doi.org/10.1113/jphysiol.2007.130013 (Published online 2007 Mar 15)
Asif IM, Harmon KG (2017) Incidence and etiology of sudden cardiac death: new updates for athletic departments. Sports Health 9(3):268–279. https://doi.org/10.1177/1941738117694153
Atanasovska T, Petersen AC, Rouffet DM, Billaut F, Ng I, McKenna MJ (2014) Plasma K+ dynamics and implications during and following intense rowing exercise. J Appl Physiol 117:60–68
Atanasovska T, Smith R, Graff C, Tran CT, Melgaard J, Kanters JK, Petersen AC, Tobin A, Kjeldsen KP, McKenna MJ (2018) Protection against severe hypokalemia but impaired cardiac repolarization after intense rowing exercise in healthy humans receiving salbutamol. J Appl Physiol (1985) 125(2):624–633. https://doi.org/10.1152/japplphysiol.00680.2017
Bailey CS, Moldenhauer HJ, Park SM, Keros S, Meredith AL (2019) KCNMA1-linked channelopathy. J Gen Physiol 151(10):1173–1189. https://doi.org/10.1085/jgp.201912457
Baker LB, Wolfe AS (2020) Physiological mechanisms determining eccrine sweat composition. Eur J Appl Physiol 120(4):719–752. https://doi.org/10.1007/s00421-020-04323-7
Ballanyi K, Grafe P (1988) Changes in intracellular ion activities induced by adrenaline in human and rat skeletal muscle. Pflügers Arch 411:283–288
Bergström J, Guarnieri G, Hultman E (1971) Carbohydrate metabolism and electrolyte changes in human muscle tissue during heavy work. J Appl Physiol 30:122–125
Bewick NL, Fernandes C, Pitt AD, Rasmussen HH, Whalley DW (1999) Mechanisms of Na+-K+ pump regulation in cardiac myocytes during hyposmolar swelling. Am J Physiol Cell Physiol 276(5):C1091–C1099. https://doi.org/10.1152/ajpcell.1999.276.5.C1091
Bia MJ, DeFronzo RA (1981) Extrarenal potassium homeostasis. Am J Physiol 240(4):F257–F268. https://doi.org/10.1152/ajprenal.1981.240.4.F257
Bisogno JL, Langley A, von Dreele MM (1994) Effect of calcium to reverse the electrocardiographic effects of hyperkalaemia in the isolated rat heart: a progressive, dose-response study. Crit Care Med 22:697–704
Blum H, Schnall MD, Chance B, Buzby GP (1988) Intracellular sodium flux and high-energy phosphorus metabolites in ischemic skeletal muscle. Am J Physiol Cell Physiol 255:C377–C384
Broch-Lips M, de Paoli F, Pedersen TH, Overgaard K, Nielsen OB (2011) Effects of 8 wk of voluntary unloaded wheel running on K+ tolerance and excitability of soleus muscles in rat. J Appl Physiol 111:212–220
Burns WR, Cohen KD, Jackson WF (2004) K+-induced dilation of hamster cremastic arterioles involves both the Na+/K+-ATPase and inward-rectifier K+ channels. Microcirculation 11(3):279–293
Cairns SP, Borrani F (2015) β-Adrenergic modulation of skeletal muscle contraction: key role of excitation-contraction coupling. J Physiol 593(21):4713–4727
Cairns SP, Dulhunty AF (1994) β-Adrenoceptor activation slows high-frequency fatigue in skeletal muscle fibers of the rat. Am J Physiol Cell Physiol 266:C1204–C1209
Cairns SP, Lindinger MI (2008) Do multiple ionic interactions contribute to skeletal muscle fatigue? J Physiol 586(17):4039–4054
Cairns SP, Flatman JA, Clausen T (1995) Relation between extracellular [K+], membrane potential and contraction in rat soleus muscle: modulation by the Na+-K+ pump. Pflügers Arch 430:909–915
Cairns SP, Hing WA, Slack JR, Mills RG, Loiselle DS (1997) Different effects of raised [K+]o on membrane potential and contraction in mouse fast- and slow-twitch muscle. Am J Physiol Cell Physiol 273:C598–C611
Cairns SP, Hing WA, Slack JR, Mills RG, Loiselle DS (1998) Role of extracellular [Ca2+] in fatigue of isolated mammalian skeletal muscle. J Appl Physiol 84:1395–1406
Cairns SP, Ruzhynsky V, Renaud JM (2004) Protective role of extracellular chloride in fatigue of isolated mammalian skeletal muscle. Am J Physiol Cell Physiol 287:C762–C770
Cairns SP, Leader JP, Loiselle DS (2011) Exacerbated potassium-induced paralysis of mouse soleus muscle at 37°C vis-à-vis 25°C: implications for fatigue. Pflügers Arch 461:469–479
Cairns SP, Leader JP, Loiselle DS, Higgins A, Lin W, Renaud JM (2015) Extracellular Ca2+-induced force restoration in K+-depressed skeletal muscle of the mouse involves an elevation of [K+]i: implications for fatigue. J Appl Physiol 118:662–674
Cairns SP, Inman LAG, MacManus CP, van de Port IGL, Ruell PA, Thom JA, Thompson MW (2017) Central activation, metabolites and calcium handling during fatigue with repeated maximal isometric contractions in human muscle. Eur J Appl Physiol 117:1557–1571
Cannon SC (2015) Channelopathies of skeletal muscle excitability. Compr Physiol 5(2):761–790. https://doi.org/10.1002/cphy.c140062
Caron G, Decherchi P, Marqueste T (2015) Does metabosensitive afferent fibers activity differ from slow- and fast-twitch muscles? Exp Brain Res 233:2549–2554
Ceccarelli B, Fesce R, Grohovaz F, Haimann C (1988) The effect of potassium on exocytosis of transmitter at the frog neuromuscular junction. J Physiol 401:163–183
Chamberlain MJ (1964) Emergency treatment for hyperkalaemia. Lancet 1:464–467
Chibalin AV, Kovalenko MV, Ryder JW, Féraille E, Wallberg-Henriksson H, Zierath JR (2001) Insulin- and glucose-induced phosphorylation of the Na+, K+-adenosine triphosphatase α-subunits in rat skeletal muscle. Endocrinology 142:3474–3482
Choate JK, Nandhabalan M, Paterson DJ (2001) Raised extracellular potassium attenuates the sympathetic modulation of sino-atrial node pacemsking in the isolated guinea-pig atria. Exp Physiol 86(1):19–25
Chothia M-Y, Halperin ML, Rensburg MA, Hassan MS, Davids MR (2014) Bolus administration of intravenous glucose in the treatment of hyperkalemia: a randomized controlled trial. Nephron Physiol 126:1–8
Christiansen D (2019) Molecular stressors underlying exercise training-induced improvements in K+ regulation during exercise and Na+, K+-ATPase adaptation in human skeletal muscle. Acta Physiol 225(3):e13196
Cifelli C, Boudreault L, Gong B, Bercier JP, Renaud JM (2008) Contractile dysfunctions in ATP-dependent K+ channel-deficient mouse muscle during fatigue involve excessive depolarization and Ca2+ influx through l-type Ca2+ channels. Exp Physiol 93:1126–1138
Clausen T (2000) Effects of amylin and other peptide hormones on Na+-K+ transport and contractility in rat skeletal muscle. J Physiol 527(1):121–130
Clausen T (2003) Na+-K+ pump regulation and skeletal muscle contractility. Physiol Rev 83:1269–1324
Clausen T (2011) In isolated skeletal muscle, excitation may increase extracellular K+ 10-fold; how can contractility be maintained? Exp Physiol 96(3):356–368
Clausen T, Kohn PG (1977) The effect of insulin on the transport of sodium and potassium in rat soleus muscle. J Physiol 265(1):19–42
Clausen T, Andersen SLV, Flatman JA (1993) Na+-K+ pump stimulation elicits recovery of contractility in K+-paralysed rat muscle. J Physiol 472:521–536
Clausen T, Overgaard K, Nielsen OB (2004) Evidence that the Na+-K+ leak/pump ratio contributes to the difference in endurance between fast- and slow-twitch muscles. Acta Physiol Scand 180:209–216
Clifford PS, Hellsten Y (2004) Vasodilatory mechanisms in contracting skeletal muscle. J Appl Physiol 97:393–403
Collins AJ, Pitt B, Reaven N, Funk S, McGaughey K, Wilson D, Bushinsky DA (2017) Association of serum potassium with all-cause mortality in patients with and without heart failure, chronic kidney disease, and/or diabetes. Am J Nephrol 46(3):213–221. https://doi.org/10.1159/000479802
Conway BE (1981) Ionic hydration in chemistry and biophysics [ionenhydratation in chemie und biophysik. 12 aus: studium der physikalischen und theoretischen chemie]. Else Scie Publ Comp Amst NY. https://doi.org/10.1002/bbpc.19820860319
Costill DL, Coté R, Fink WJ, van Handel P (1981) Muscle water and electrolyte distribution during prolonged exercise. Int J Sport Med 2:130–134
da Silva AJ, Trindade MAS, Santos DOC, Lima RF (2016) Maximum-likelihood q-estimator uncovers the role of potassium at neuromuscular junctions. Biol Cybern 110:31–40
Dassau L, Conti LR, Radeke CM, Ptáček LJ, Vandenberg CA (2011) Kir2.6 regulates surface expression of Kir2.x inward rectifier potassium channels. J Biol Chem 286(11):9526–9541
de Paoli FV, Ørtenblad N, Pedersen TH, Jørgensen R, Nielsen OB (2010) Lactate per se improves the excitability of depolarized rat skeletal muscle by reduing the Cl- conductance. J Physiol 588(23):4785–4794
de Paoli FV, Broch-Lips M, Pedersen TH, Nielsen OB (2013) Relationship between membrane Cl- conductance and contractile endurance in isolated rat muscles. J Physiol 591(2):531–545
Decherchi P, Darques JL, Jammes Y (1998) Modifications of afferent activities from Tibialis anterior muscle in rat by tendon vibrations, increase of interstitial potassium or lactate concentration and electrically-induced fatigue. J Peripher Nerv Syst 3:267–276
DeFronzo RA, Felig P, Ferrannini E, Wahren J (1980) Effect of graded doses of insulin on splanchnic and peripheral potassium metabolism in man. Am J Physiol 238(5):E421-427
Demigné C, Sabboh H, Puel C, Rémésy C, Coxam V (2004) Organic anions and potassium salts in nutrition and metabolism. Nutr Res Rev 17(2):249–258. https://doi.org/10.1079/NRR200485
Dempsey JA, Blain GM, Amann M (2014) Are type III-IV muscle afferents required for a normal steady-state hyperpnoea in humans? J Physiol 592(3):464–474
Deogenes KG, Kakuris KK, Deogenov VA, Yerullis KB (2007) Electrolyte homeostasis in trained and untrained healthy subjects during prolonged hypokinesia. Clin Biochem 40(8):536–544. https://doi.org/10.1016/j.clinbiochem.2007.01.017
Désilets M, Baumgarten CM (1986) Isoproterenol directly stimulates the Na+-K+ pump in isolated cardiac myocytes. Am J Physiol Heart Circl Physiol 251:H218–H225
DiFranco M, Quinonez M, Vergara JL (2012) The delayed rectifier potassium conductance in the sarcolemma and the transverse tubular system membranes of mammalian skeletal muscle fibers. J Gen Physiol 140(2):109–137
DiFranco M, Hakimjavadi H, Lingrel JB, Heiny JA (2015) Na, K-ATPase α2 activity in mammalian skeletal muscle T-tubules is acutely stimulated by extracellular K+. J Gen Physiol 146:281–294
DiFranco M, Yu C, Quiñonez M, Vergara JL (2015) Inward rectifier potassium currents in mammalian skeletal muscle fibres. J Physiol 593(5):1213–1238. https://doi.org/10.1113/jphysiol.2014.283648
Durelli L, Mutani R, Fassio F, Delsedime M (1982) The effects of the increase of arterial potassium upon the excitability of normal and dystrophic myotonic muscles in man. J Neurol Sci 55:249–257
Durfey N, Lehnhof B, Bergeson A, Durfey SNM, Leytin V, McAteer K, Schwam E, Valiquet J (2017) Severe hyperkalemia: can the electrocardiogram risk stratify for short-term adverse events? West J Emerg Med 18(5):963–971. https://doi.org/10.5811/westjem.2017.6.33033
Duval A, Léoty C (1980) Ionic currents in slow twitch skeletal muscle of the rat. J Physiol 307:23–41
Engstfeld G, Antoni H, Fleckenstein A, Nast A, Hattingberg MV (1961) Die restitution der effegungsforteitung und kontraktionskraft des K+-gelähmten frosch- und säugetiermyokards durch adrenalin. Pflügers Arch 273:145–163
Ettinger PO, Regan TJ, Oldewurtel HA (1974) Hyperkalemia, cardiac conduction, and the electrocardiogram: a review. Am Heart J 88(3):360–371
Fakler B, Adelman JP (2008) Control of K(Ca) channels by calcium nano/microdomains. Neuron 59(6):873–881. https://doi.org/10.1016/j.neuron.2008.09.001
Fallentin N, Jensen BR, Brystrom S, Sjøgaard G (1992) Role of potassium in the reflex regulation of blood pressure during static exercise in man. J Physiol 451:643–651
Ferreira Gregorio J, Pequera G, Manno C, Ríos E, Brum G (2017) The voltage sensor of excitation-contraction coupling in mammals: inactivation and interaction with Ca2+. J Gen Physiol 149(11):1041–1058
Fialho D, Griggs RC, Matthews E (2018) Periodic paralysis. Handb Clin Neurol 148:505–520. https://doi.org/10.1016/B978-0-444-64076-5.00032-6
Fraser JA, Huang CLH, Pedersen TH (2011) Relationships between resting conductances, excitability, and t-system ionic homeostasis in skeletal muscle. J Gen Physiol 138(1):95–116
Gamstorp I, Hauge M, Helweg-Larsen HF, Mjönes H (1957) Adynamia episodica hereditaria. A disease clinically resembling familial periodic paralysis but characterized by increasing serum potassium during the paralytic attacks. Am J Med 23:385–390
Germinario E, Esposito A, Midrio M, Peron S, Palade PT, Betto R, Danieli-Betto D (2008) High-frequency fatigue of skeletal muscle: role of extracellular Ca2+. Eur J Appl Physiol 104:445–453
Goodman CA, Bennie JA, Leikis MJ, McKenna MJ (2014) Unaccustomed eccentric contractions impair plasma K+ regulation in the absence of changes in muscle Na+, K+-ATPase content. PLoS ONE 9(6):e101039
Gosmanov AR, Lindinger MI, Thomason DB (2003) Riding the tides: K+ concentration and volume regulation by muscle Na+-K+-2Cl- cotransport activity. News Physiol Sci 18:196–200. https://doi.org/10.1152/nips.01446.2003
Gosmanov AR, Fan Z, Mi X, Schneider EG, Thomason DB (2004) ATP-sensitive potassium channels mediate hyperosmotic stimulation of NKCC in slow-twitch muscle. Am J Physiol Cell Physiol 286:C586–C595
Green S, Langberg H, Skovgaard D, Bülow J, Kjær M (2000) Interstitial and arterial-venous [K+] in human calf muscle during dynamic exercise: effect of ischaemia and relation to muscle pain. J Physiol 529(3):849–861
Green HJ, Duhamel TA, Foley KP, Ouyaning J, Smith IC, Stewart RD (2007) Glucose supplements increase human muscle in vitro Na+-K+-ATPase activity during prolonged exercise. Am J Physiol Regul Integr Comp Physiol 293:R354–R362
Grob D, Liljestrand A, Johns RJ (1957) Potassium movement in normal subjects. Effect on muscle function. Am J Med 23:340–354
Gumz ML, Rabinowitz L, Wingo CS (2015) An integrated view of potassium homeostasis. N Engl J Med 373(1):60–72. https://doi.org/10.1056/NEJMra1313341
Gunnarsson TP, Christensen PM, Thomassen M, Nielsen LR, Bangsbo J (2013) Effect of intensified training on muscle ion kinetics, fatigue development, and repeated short-term performance in endurance-trained cyclists. Am J Physiol Regul Integr Comp Physiol 305:R811–R821
Gutman GA, Chandy KG, Grissmer S, Lazdunski M, McKinnon D, Pardo LA, Robertson GA, Rudy B, San-Guinetti MC, Stühmer W, Wang X (2005) International union of pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol Rev 57(4):473–508
Hakimjavadi H, Stiner CA, Radzyukevich TL, Lingrel JB, Norman N, Landero Figueroa JA, Heiny JA (2018) K+ and Rb+ affinities of the Na, K-ATPase α1 and α2 isozymes: an application of ICP-MS for quantification of Na+ pump kinetics in myofibers. Int J Mol Sci 19:2725
Hallén J, Saltin B, Sejersted OM (1996) K+ balance during exercise and role of β-adrenergic stimulation. Am J Physiol Regul Integr Comp Physiol 270:R1347–R1354
Hamidi M, Boucher BA, Cheung AM, Beyene J, Shah PS (2011) Fruit and vegetable intake and bone health in women aged 45 years and over: a systematic review. Osteoporos Int 22:1681–1693
Hansen AK, Clausen T, Nielsen OB (2005) Effects of lactic acid and catecholamines on contractility in fast-twitch muscles exposed to hyperkalemia. Am J Physiol Cell Physiol 289:C104–C112
Hargreaves M, McKenna MJ, Jenkins DG, Warmington SA, Li JL, Snow RJ, Febbraio MA (1998) Muscle metabolites and performance during high-intensity, intermittent exercise. J Appl Physiol 84(5):1687–1691
Harmer AR, McKenna MJ, Sutton JR, Snow RJ, Ruell PA, Booth J, Thompson MW, MacKay NA, Stathis CG, Crameri RM, Carey MF, Eager DE (2000) Skeletal muscle metabolic and ionic adaptations during intense exercise following sprint training in humans. J Appl Physiol 89:1793–1803
Hayashi M, Shimizu W, Albert CM (2015) The spectrum of epidemiology underlying sudden cardiac death. Circ Res 116(12):1887–1906. https://doi.org/10.1161/CIRCRESAHA.116.304521
Hayward LJ, Kim JS, Lee MY, Zhou H, Kim JW, Misra K, Salajegheh M, Wu F-F, Matsuda C, Reid V, Cros D, Hoffman EP, Renaud JM, Cannon SC, Brown RH (2008) Targeted mutation of mouse skeletal muscle sodium channel produces myotonia and potassium-sensitive weakness. J Clin Invest 118(4):1437–1449
Hegyi B, Chen-Izu Y, Izu L, Bányász T (2019) Altered K+ current profiles underlie cardiac action potential shortening in hyperkalemia and β adrenergic stimulation. Can J Physiol Pharmacol 97(8):773–780. https://doi.org/10.1139/cjpp-2019-0056
Hiatt N, Davidson MB, Bonorris G (1972) The effect of potassium chloride infusion on insulin secretion in vivo. Horm Metab Res 4:64–68
Hinderling PH (2016) The pharmacokinetics of potassium in humans is unusual. J Clin Pharmacol 56(10):1212–1220. https://doi.org/10.1002/jcph.713
Hník P, Vyskočil F (1981) Ion-selective microelectrodes: a new tool for studying ionic movements in working muscles. In the application of ion-selective microelectrodes. Elsevier/North-Holland Biomedical Press, Amsterdam, Ed Zeuthen T 157–172
Holbrook JT, Patterson KY, Bodner JE, Douglas LW, Veillon C, Kelsay JL, Mertz W, Smith JC Jr (1984) Sodium and potassium intake and balance in adults consuming self-selected diets. Am J Clin Nutr 40(4):786–793
Hoppe LK, Muhlack DC, Koenig W, Carr PR, Brenner H, Schöttker B (2018) Association of abnormal serum potassium levels with arrhythmias and cardiovascular mortality: a systematic review and meta-analysis of observational studies. Cardiovasc Drugs Ther 32(2):197–212. https://doi.org/10.1007/s10557-018-6783-0
Hostrup M, Bangsbo J (2017) Limitations in intense exercise performance of athletes: effect of speed endurance training on ion handling and fatigue development. J Physiol 595(9):2897–2913
Hostrup M, Kalsen A, Ørtenblad N, Juel C, Mørch K, Rzeppa S, Karlsson S, Backer V, Bangsbo J (2014a) β2-Adrenergic stimulation enhances Ca2+ release and contractile properties of skeletal muscles, and counteracts exercise-induced reductions in Na+-K+ATPase Vmax in trained men. J Physiol 592:5445–5459
Hostrup M, Kalsen A, Bangsbo J, Hemmersbach P, Karlsson S, Backer V (2014b) High-dose inhaled terbutaline stimulation increases muscle strength and enhances maximal sprint performance in trained men. Eur J Appl Physiol 114:2499–2508
ICRP (2002) Basic anatomical and physiological data for use in radiological protection reference values. ICRP publication 89. Ann ICRP 32:3–4
Jazayeri MA, Emert MP (2019) Sudden cardiac death: who is at risk? Med Clin North Am 103(5):913–930. https://doi.org/10.1016/j.mcna.2019.04.006
Jennische E (1982) Relation between membrane potential and lactate in gastrocnemius and soleus muscle of the cat during tourniquet ischemia and postischemic reflow. Pflügers Arch 394:329–332
Jennische E, Hagberg H, Haljamäe H (1982) Extracellular potassium concentration and membrane potential in rabbit gastrocnemius muscle during tourniquet ischemia. Pflügers Arch 392:335–339
Jensen R, Nielsen J, Ørtenblad N (2020) Inhibition of glycogenolysis prolongs action potential repriming period and impairs muscle function in rat skeletal muscle. J Physiol 598(4):789–803
Joyner MJ, Casey DP (2015) Regulation of increased blood flow (hyperemia) to muscles during exercise: a hierarchy of competing physiological needs. Physiol Rev 95(2):549–601. https://doi.org/10.1152/physrev.00035.2013
Juel C (1986) Potassium and sodium shifts during in vitro isometric muscle contraction, and the time course of the ion-gradient recovery. Pflügers Arch 406:458–463
Juel C (1988) The effect of β2-adrenoceptor activation on ion-shifts and fatigue in mouse soleus muscles stimulated in vitro. Acta Physiol Scand 134:209–216
Juel C (2007) Changes in interstitial K+ and pH during exercise: implications for blood flow regulation. Appl Physiol Nutr Metab 32(5):846–851
Juel C, Bangsbo J, Graham T, Saltin B (1990) Lactate and potassium fluxes from human skeletal muscle during and after intense, dynamic, knee extension exercise. Acta Physiol Scand 140:147–159
Juel C, Pilegaard H, Nielsen JJ, Bangsbo J (2000) Interstitial K+ in human skeletal muscle during and after dynamic graded exercise determined by microdialysis. Am J Physiol Regul Integr Comp Physiol 278:R400–R406
Juel C, Olsen S, Rentsch RL, González-Alonso J, Rosenmeier JB (2007) K+ as a vasodilator in resting human muscle: implications for exercise hyperaemia. Acta Physiol 190:311–318
Juel C, Hostrup M, Bangsbo M (2015) The effect of exercise and beta2-adrenergic stimulation on glutathioylation and function of the Na, K-ATPase in human skeletal muscle. Physiol Rep 3:e12515
Jurkat-Rott K, Fauler M, Lehmann-Horn F (2006) Ion channels and ion transporters of the transverse tubular system of skeletal muscle. J Muscle Res Cell Motil 27(5–7):275–290. https://doi.org/10.1007/s10974-006-9088-z
Karelis AD, Péronnet F, Gardiner PF (2002) Glucose infusion attenuates muscle fatigue in rat plantaris muscle during prolonged indirect stimulation in situ. Exp Physiol 87(5):585–592
Karelis AD, Péronnet F, Gardiner PF (2003) Insulin does not mediate the attenuation of fatigue associated with glucose infusion in rat plantaris muscle. J Appl Physiol 95:330–335
Karelis AD, Péronnet F, Gardiner PF (2005) Resting membrane potential of rat plantaris muscle fibers after prolonged indirect stimulation in situ: effect of glucose infusion. Can J Appl Physiol 30:105–112
Keir DA, Duffin J, Millar PJ, Floras JS (2019) Simultaneous assessment of central and peripheral chemoreflex regulation of muscle sympathetic nerve activity and ventilation in healthy young men. J Physiol 597(13):3281–3296. https://doi.org/10.1113/JP277691
Kodama I, Wilde A, Janse MJ, Durrer D, Yamada K (1984) Combined effects of hypoxia, hyperkalemia and acidosis on membrane action potential and excitability of guinea-pig ventricular muscle. J Mol Cell Cardiol 16:247–259
Kowalchuk JM, Heigenhauser GJF, Lindinger MI, Sutton JR, Jones NL (1988) Factors influencing hydrogen ion concentration in muscle after intense exercise. J Appl Physiol 65(5):2080–2089. https://doi.org/10.1152/jappl.1988.65.5.2080
Kristensen M, Juel C (2010) Potassium-transporting proteins in skeletal muscle: cellular location and fibre-type differences. Acta Physiol 198:105–123
Krustrup P, Mohr M, Nybo L, Jensen JM, Nielsen JJ, Bangsbo J (2006a) The Yo-yo IR2 test: physiological response, reliability, and application to elite soccer. Med Sci Sport Exerc 38:1666–1673
Krustrup P, Mohr M, Steenberg A, Bencke J, Kjær M, Bangsbo J (2006b) Muscle and blood metabolites during a soccer game: implications for sprint performance. Med Sci Sport Exerc 38:1165–1174
Kubo Y, Adelman JP, Clapham DE, Jan LY, Karschin A, Kurachi Y, Lazdunski M, Nichols CG, Seino S, Vandenberg CA (2005) International union of pharmacology. LIV. Nomenclature and molecular relationships of inwardly rectifying potassium channels. Pharmacol Rev 57(4):509–526
Kurihara S, Konishi M (1987) Effects of β-adrenoceptor stimulation on intracellular Ca2+ transients and tension in rat ventricular muscle. Pflügers Arch 409:427–437
Lam YL, Zeng W, Sauer DB, Jiang Y (2014) The conserved potassium channel filter can have distinct ion binding profiles: structural analysis of rubidium, cesium, and barium binding in NaK2K. J Gen Physiol 144(2):181–192. https://doi.org/10.1085/jgp.201411191
Landowne D, Potter LT, Terrar DA (1975) Structure-function relationships in excitable membranes. Annu Rev Physiol 37:485–508. https://doi.org/10.1146/annurev.ph.37.030175.002413
Lee FN, Oh G, McDonough AA, Youn JH (2007) Evidence for gut factor in K+ homeostasis. Am J Physiol Renal Physiol 293:F541-547
Leitch SP, Paterson DJ (1994a) Interactive effects of K+, acidosis, and catecholamines on isolated rabbit heart: implications for exercise. J Appl Physiol 77(3):1164–1171
Leitch SP, Paterson DJ (1994b) Role of Ca2+ in protecting the heart from hyperkalemia and acidosis in the rabbit: implications for exercise. J Appl Physiol 77(5):2391–2399
Leppik JA, Aughey RJ, Medved I, Fairweather I, Carey MF, McKenna MJ (2004) Prolonged exercise to fatigue in humans impairs skeletal muscle Na+-K+-ATPase activity, sarcoplasmic reticulum Ca2+ release, and Ca2+ uptake. J Appl Physiol 97:1414–1423
Li J, Sinoway LI, Ng Y-C (2006) Aging augments interstitial K+ concentration in active muscle of rats. J Appl Physiol 100:1158–1163
Lindinger MI (1995) Potassium regulation during exercise and recovery in humans: Implications for skeletal and cardiac muscle. J Mol Cell Cardiol 27:1011–1022
Lindinger MI (2005) Determinants of surface membrane and transverse-tubular excitability in skeletal muscle: implications for high-intensity exercise. Equine Comp Exerc Physiol 2(4):209–217
Lindinger MI, Grudzien SP (2003) Exercise-induced changes in plasma composition increase erythrocyte Na+, K+-ATPase, but not Na+-K+-2Cl- cotransporter, activity to stimulate net and unidirectional K+ transport in humans. J Physiol 553(Pt 3):987–997
Lindinger MI, Heigenhauser GJ (1987) Intracellular ion content of skeletal muscle measured by instrumental neutron activation analysis. J Appl Physiol (1985) 63(1):426–433. https://doi.org/10.1152/jappl.1987.63.1.426
Lindinger MI, Heigenhauser GJF (1988) Ion fluxes during tetanic stimulation in isolated perfused rat hindlimb. Am J Physiol Regul Integr Comp Physiol 254:R117–R126
Lindinger MI, Heigenhauser GJF (2012) Effects of gas exchange on acid-base balance. Compr Physiol 2(3):2203–2254. https://doi.org/10.1002/cphy.c100055
Lindinger MI, Heigenhauser GJF, McKelvie RS, Jones NL (1990) Role of nonworking muscle on blood metabolites and ions with intense intermittent exercise. Am J Physiol Regul Integr Comp Physiol 258:R1486–R1494
Lindinger MI, Heigenhauser GJF, McKelvie RS, Jones NL (1992) Blood ion regulation during repeated maximal exercise and recovery in humans. Am J Physiol Regul Integr Comp Physiol 262:R126–R136
Lindinger MI, McKelvie RS, Heigenhauser GJF (1995) K+ and Lac- distribution in humans during and after high-intensity exercise: role in muscle fatigue attenuation? J Appl Physiol 78(3):765–777
Lindinger MI, Franklin TW, Lands LC, Pedersen PK, Welsh DG, Heigenhauser GJF (1999) Role of skeletal muscle in plasma ion and acid-base regulation after NaHCO3 and KHCO3 loading in humans. Am J Physiol 276(1):R32-43. https://doi.org/10.1152/ajpregu.1999.276.1.R32
Lindinger MI, Horn PL, Grudzien SP (1999) Exercise-induced stimulation of K+ transport in human erythrocytes. J Appl Physiol 87:2157–2167
Lindinger MI, Franklin TW, Lands LC, Pedersen PK, Welsh DG, Heigenhauser GJF (2000) NaHCO(3) and KHCO(3) ingestion rapidly increases renal electrolyte excretion in humans. J Appl Physiol 88(2):540–550. https://doi.org/10.1152/jappl.2000.88.2.540
Lindinger MI, Hawke TJ, Vickery L, Bradford L, Lipskie SL (2001) An integrative, in situ approach to examining K+ flux in resting skeletal muscle. Can J Physiol Pharmacol 79(12):996–1006
Lindinger MI, Hawke TJ, Lipskie SL, Schaefer HD, Vickery L (2002) K+ transport and volume regulatory response by NKCC in resting rat hindlimb skeletal muscle. Cell Physiol Biochem 12(5–6):279–292. https://doi.org/10.1159/000067898
Lindinger MI, Leung M, Trajcevski KE, Hawke TJ (2011) Volume regulation in mammalian skeletal muscle: the role of sodium-potassium-chloride cotransporters during exposure to hypertonic solutions. J Physiol 589(Pt 11):2887–2899
Linton RA, Band DM (1985) The effect of potassium on carotid chemoreceptor activity and ventilation in the cat. Resp Physiol 59:65–70
Logic JR, Krotkiewski A, Koppius A, Surawicz B (1968) Negative inotropic effect of K+: its modification by Ca++ and acetylstrophanthidin in dogs. Am J Physiol 215(1):14–22
Long B, Warix JR, Koyfman A (2018) Controversies in management of hyperkalemia. J Emerg Med 55(2):192–205
Lucas B, Ammar T, Khogali S, de Jong D, Barbalinardo M, Nishi C, Hayward LJ, Renaud JM (2014) Contractile abnormalities of mouse muscles expressing hyperkalemic peiodic paralysis mutant NaV1.4 channels do not correlate with Na+ influx or channel content. Physiol Genomics 46:385–397
MacLean DA, Imadojueu VA, Sinoway LI (2000) Interstitial pH, K+, lactate, and phosphate determined with MSNA during exercise in humans. Am J Physiol Regul Integr Comp Physiol 278:R563–R571
Mancinelli R, Botti A, Bruni F, Ricci MA (2007) Soper AK (2007) Hydration of sodium, potassium, and chloride ions in solution and the concept of structure maker/breaker. J Phys Chem B 111(48):13570–13577. https://doi.org/10.1021/jp075913v
Mangels AR (2014) Bone nutrients for vegetarians. Am J Clin Nutr 100(Suppl 1):469S-S475. https://doi.org/10.3945/ajcn.113.071423
Marliss EB, Vranic M (2002) Intense exercise has unique effects on both insulin release and its roles in glucoregulation: implications for diabetes. Diabetes 51(Suppl 1):S271–S283. https://doi.org/10.2337/diabetes.51.2007.s271
Maughan RJ, Shirreffs SM, Merson SJ, Horswill CA (2005) Fluid and electrolyte balance in elite male football (soccer) players training in a cool environment. J Sports Sci 23(1):73–79
McCloskey DI, Mitchell JH (1972) Reflex cardiovascular and respiratory responses originating in exercising muscle. J Physiol 224:173–186
McDonough AA, Youn JH (2017) Potassium homeostasis: the knowns, the unknowns, and the health benefits. Physiol (Bethesda) 32(2):100–111. https://doi.org/10.1152/physiol.00022.2016
McKenna MJ, Schmidt TA, Hargreaves M, Cameron L, Skinner SL, Kjeldsen K (1993) Sprint training increases human skeletal muscle Na+-K+-ATPase concentration and improves K+ regulation. J Appl Physiol 75(1):173–180
McKenna MJ, Heigenhauser GJF, McKelvie RS, MacDougall JD, Jones NL (1997) Sprint training enhances ionic regulation during intense exercise in men. J Physiol 501(3):687–702
McKenna MJ, Medved I, Goodman CA, Brown MJ, Bjorksten AR, Murphy KT, Petersen AC, Sostaric S, Gong X (2006) N-acetycysteine attenuates the decline in muscle Na+, K+ pump activity and delays fatigue during prolonged exercise in humans. J Physiol 576(1):279–288
McKenna MJ, Bangsbo J, Renaud JM (2008) Muscle K+, Na+, and Cl- disturbances and Na+-K+ pump inactivation: implications for fatigue. J Appl Physiol 104:288–295
Medbø JI, Sejersted OM (1990) Plasma potassium changes with high intensity exercise. J Physiol 421:105–122
Mohr M, Nielsen JJ, Bangsbo J (2011) Caffeine intake improves intense intermittent exercise performance and reduces muscle interstitial potassium accumulation. J Appl Physiol 111:1372–1379
Mohseni M, Silvers S, McNeil R, Diehl N, Vadeboncoeur T, Taylor W, Shapiro S, Roth J, Mahoney S (2011) Prevalence of hyponatremia, renal dysfunction, and other electrolyte abnormalities among runners before and after completing a marathon or half marathon. Sports Health 3(2):145–151. https://doi.org/10.1177/1941738111400561
Mølgaard H, Stürup-Johansen M, Flatman JA (1980) A dichrotomy of the membrane potential response of rat soleus muscle fibers to low extracellular potassium concentrations. Pflügers Arch 383(2):181–184
Moore-Ede MC, Meguid MM, Fitzpatrick GF, Boyden CM, Ball MR (1978) Circadian variation in response to potassium infusion. Clin Pharmacol Ther 23(2):218–227. https://doi.org/10.1002/cpt1978232218
Nielsen OB, de Paoli F, Overgaard K (2001) Protective effects of lactic acid on force production in rat skeletal muscle. J Physiol 536(1):161–166
Nielsen HB, Bredmose PP, Strømstad M, Voliantis S, Quistorff B, Secher NH (2002) Bicarbonate attenuates arterial desaturation during maximal exercise in humans. J Appl Physiol 93:724–731
Nielsen JJ, Kristensen M, Hellsten Y, Bangsbo J, Juel C (2003a) Localization and function of ATP-sensitive potassium channels in human skeletal muscle. Am J Physiol Regul Integr Comp Physiol 284:R558–R563
Nielsen JJ, Mohr M, Klarskov C, Kristensen M, Krustrup P, Juel C, Bangsbo J (2003b) Effects of high-intensity intermittent training on potassium kinetics and performance in human skeletal muscle. J Physiol 554(3):857–870
Nielsen OB, de Paoli FV, Riisager A, Pedersen TH (2017) Chloride channels take center stage in acute regulation of excitability in skeletal muscle: implications for fatigue. Physiology 32:425–434
Nobel D (1979) The Initiation of the Heartbeat. The Clarendon Press, Oxford
Nordsborg N, Mohr M, Pedersen LD, Nielsen JJ, Langberg H, Bangsbo J (2003) Muscle interstitial potassium kinetics during intense exhaustive exercise: effect of previous arm exercise. Am J Physiol Regul Integr Comp Physiol 285:R143–R148
O’Neill M, Dorrington KL, Paterson DJ (1993) Cardiac sympathetic nerve stimulation enhances cardiovascular performance during hyperkalaemia in the anaesthetized pig. Exp Physiol 78(4):549–552
O’Neill M, Sears CE, Paterson DJ (1997) Interactive effects of K+, acid, norepinephrine, and ischemia on the heart: implications for exercise. J Appl Physiol 82:1046–1052
Ogielska EM, Aldrich RW (1999) Functional consequences of a decreased potassium affinity in a potassium channel pore. Ion interactions and C-type inactivation. J Gen Physiol 113(2):347–58. https://doi.org/10.1085/jgp.113.2.347
Ørtenblad N, Stephenson DG (2003) A novel signalling pathway originating in mitochondria modulates rat skeletal muscle membrane excitability. J Physiol 548(1):139–145
Overgaard K, Nielsen OB, Flatman JA, Clausen T (1999) Relations between excitability and contractility in rat soleus muscle: role of the Na+-K+ pump and Na+/K+ gradients. J Physiol 518(1):215–225
Paterson DJ (1996a) Role of potassium in the regulation of systemic physiological function during exercise. Acta Physiol Scand 156(3):287–294
Paterson DJ (1996b) Antiarrythmic mechanisms during exercise. J Appl Physiol 80(6):1853–1862
Paterson DJ (1997) Potassium and breathing in exercise. Sports Med 23(3):149–163. https://doi.org/10.2165/00007256-199723030-00002
Paterson DJ, Robbins PA, Conway J (1989) Changes in arterial plasma potassium and ventilation during exercise in man. Resp Physiol 78:323–330
Paterson DJ, Friedland JS, Bascom DA, Clement ID, Cunningham DA, Painter R, Robbins PA (1990) Changes in arterial K+ and ventilation during exercise in normal subjects and subjects with McArdles syndrome. J Physiol 429:339–348
Paterson DJ, Blake GJ, Leitch SP, Phillips SM, Brown HF (1992) Effects of catecholamines and potassium on cardiovascular performance in the rabbit. J Appl Physiol 73(4):1413–1418
Paterson DJ, Rogers J, Powell T, Brown HF (1993) Effect of catecholamines on the ventricular myocyte action potential in raised extracellular potassium. Acta Physiol Scand 148:177–186
Pedersen TH, de Paoli FV, Nielsen OB (2005) Increased excitability of acidified skeletal muscle: Role of chloride conductance. J Gen Physiol 125:237–246
Pedersen TH, de Paoli FV, Flatman JA, Nielsen OB (2009) Regulation of ClC-1 and KATP channels in action potential-firing fast-twitch muscle fibers. J Gen Physiol 134:309–322
Pedersen KK, Cheng AJ, Westerblad H, Olesen JH, Overgaard K (2019) Moderately elevated extracellular [K+] potentiates submaximal force and power in skeletal muscle via increased [Ca2+]i during contractions. Am J Physiol Cell Physiol 317:C900–C909
Perry BD, Wyckelsma VL, Murphy RM, Steward CH, Anderson M, Levinger I, Petersen AC, McKenna MJ (2016) Dissociation between short-term unloading and resistance training effects on skeletal muscle Na+, K+-ATPase, muscle function, and fatigue in humans. J Appl Physiol 121(5):1074–1086. https://doi.org/10.1152/japplphysiol.00558.2016
Petersen AC, Murphy KT, Snow RJ, Leppik JA, Aughey RJ, Garnham AP, Cameron-Smith D, McKenna MJ (2005) Depressed Na+-K+-ATPase activity in skeletal muscle at fatigue is correlated with increased Na+-K+-ATPase mRNA expression following intense exercise. Am J Physiol Regul Integr Comp Physiol 289:R288–R274
Piitulainen H, Komi P, Linnamo V, Avela J (2008) Sarcolemmal excitability as investigated with M-waves after eccentric exercise in humans. J Electromyo Kinesiol 18:672–681
Pirkmajer S, Chibalin AV (2016) Na, K-ATPase regulation in skeletal muscle. Am J Physiol Endocrinol Metab 311(1):E1–E31. https://doi.org/10.1152/ajpendo.00539.2015
Podrid PJ (1990) Potassium and ventricular arrhythmias. Am J Cardiol 65(10):33E-44E. https://doi.org/10.1016/0002-9149(90)90250-5 (discussion 52E)
Poole-Wilson PA (1984) Potassium and the heart. Clin Endocrin Metab 13(2):249–268
Priyamvada S, Gomes R, Gill RK, Saksena S, Alrefai WA, Dudeja PK (2015) Mechanisms underlying dysregulation of electrolyte absorption in inflammatory bowel disease-associated diarrhea. Inflamm Bowel Dis 21(12):2926–2935. https://doi.org/10.1097/MIB.0000000000000504
Qayyum MS, Barlow CW, O’Connor DF, Paterson DJ, Robbins PA (1994) Effect of raised potassium on ventilation in euoxia, hypoxia, and hyperoxia at rest and during light exercise in man. J Physiol 476(2):365–372
Quiñonez M, González F, Morgado-Valle C, DiFranco M (2010) Effects of membrane depolarization and changes in extracellular [K+] on the Ca2+ transients of fast skeletal muscle fibers. Implications for muscle fatigue. J Muscle Res Cell Motil 31:13–33
Radzyukevich TL, Lingrel JB, Heiny JA (2009) The cardiac glycoside binding site on the Na, K-ATPase α2 isoform plays a role in the dynamic regulation of active transport in skeletal muscle. PNAS 106:2565–2570
Rannou F, Leschiera R, Giroux-Metges MA, Pennec JP (2012) Effect of lactate on the voltage-gated sodium channels of rat skeletal muscle: modulating current opinion. J Appl Physiol 112:1454–1465
Renaud JM, Light P (1992) Effects of K+ on the twitch and tetanic contraction in the sartorius muscle of the frog, Rana pipiens. Implication for fatigue in vivo. Can J Physiol Pharmacol 70:1236–1246
Rich MM, Pinter MJ (2003) Crucial role of sodium channel fast inactivation in muscle fibre inexcitability in a rat model of critical illness myopathy. J Physiol 547(2):555–566
Rizvi MH, Abdul Azeem M, Savanur A (2019) Effects of adrenaline on contractility and endurance of isolated mammalian soleus with different calcium concentrations. J Muscle Res Cell Motil 40:373–378
Robertson MJ, Lumley P (1989) Effects of hypoxia, elevated K+ and acidosis on the potency of verapamil, diltiazem and nifedipine in the guinea-pig isolated papillary muscle. Br J Pharmacol 98:937–949
Ruff RL (1996) Sodium channel slow inactivation and the distribution of sodium channels on skeletal muscle fibres enable the performance properties of different skeletal muscle fibre types. Acta Physiol Scand 156:159–168
Ryan DM, Paterson DJ (1996) Restoration of cardiac contraction by angiotensin II during raised [K+]o in the rabbit. Acta Physiol Scand 156:419–427
Sahlin K, Alvestrand A, Brandt R, Hultman E (1978) Intracellular pH and bicarbonate concentration in human muscle during recovery from exercise. J Appl Physiol 45(3):474–480
Sakamoto K, Kurokawa J (2019) Involvement of sex hormonal regulation of K+ channels in electrophysiological and contractile functions of muscle tissues. J Pharmacol Sci 39(4):259–265. https://doi.org/10.1016/j.jphs.2019.02.009
Saltin B, Sjøgaard G, Gaffney AF, Rowell LB (1981) Potassium, lactate, and water fluxes in human quadriceps muscle during static contractions. Circl Res 48:18–24
Sejersted OM, Sjøgaard G (2000) Dynamics and consequences of potassium shifts in skeletal muscle and heart during exercise. Physiol Rev 80(4):1411–1481
Shah VN, Wingo TL, Weiss KL, Williams CK, Balser JR, Chazin WJ (2006) Calcium-dependent regulation of the voltage-gated sodium channel hH1: intrinsic and extrinsic sensors use a common molecular switch. PNAS 103(10):3592–3597
Shorten PR, Soboleva TK (2007) Anomalous ion diffusion within skeletal muscle transverse tubule networks. Theor Bio Med Model 4:18
Sjøgaard G (1983) Electrolytes in slow and fast muscle fibers of humans at rest and with dynamic exercise. Am J Physiol Regul Integr Comp Physiol 245:R25–R31
Sjøgaard G, Adams RP, Saltin B (1985) Water and ion shifts in skeletal muscle of humans with intense dynamic knee extension. Am J Physiol Regul Integr Comp Physiol 248:R190–R196
Skou JC (1962) Preparation from mammalian brain and kidney of the enzyme system involved in active transport of Na ions and K ions. Biochem Biophys Acta 58:314–325. https://doi.org/10.1016/0006-3002(62)91015-6
Söderlund K, Hultman E (1991) ATP and phosphocreatine changes in single human muscle fibers after intense electrical stimulation. Am J Physiol 261(6 Pt 1):E737–E741. https://doi.org/10.1152/ajpendo.1991.261.6.E737
Spruce AE, Standen NB, Stanfield PR (1985) Voltage-dependent ATP-sensitive potassium channels of skeletal muscle membrane. Nature 316(6030):736–738. https://doi.org/10.1038/316736a0
Stewart RD, Duhamel TA, Foley KP, Ouyang J, Smith IC, Green HJ (2007) Protection of muscle membrane excitability during prolonged cycle exercise with glucose supplementation. J Appl Physiol 103:331–339
Stone MS, Martyn L, Weaver CM (2016) Potassium intake, bioavailability, hypertension, and glucose control. Nutrients 8(7):444. https://doi.org/10.3390/nu8070444
Street D, Nielsen JJ, Bangsbo J, Juel C (2005) Metabolic alkalosis reduces exercise-induced acidosis and potassium accumulation in human skeletal muscle interstitium. J Physiol 566(2):481–489
Surawicz B, Lexington K (1967) Relationship between electrocardiogram and electrolytes. Am Heart J 73(6):814–834
Surawicz B, Chlebus H, Mazzoleni A, Lexington K (1967) Hemodynamic and electrocardiographic effects of hyperpotassemia. Differences in response to slow and rapid increases in concentration of plasma K. Am Heart J 73(5):647–664
Terwoord JD, Hearon CM, Luckasen GJ, Richards JC, Joyner MJ, Dinenno FA (2018) Elevated extracellular potassium prior to muscle contraction reduces onset and steady-state exercise hyperemia in humans. J Appl Physiol 125:615–623
Trenor B, Cardona K, Romero L, Gomez JF, Saiz J, Rajamani S, Belardinelli L, Giles W (2018) Pro-arrhythmic effects of low plasma [K+] in human ventricle: an illustrated review. Trends Cardiovasc Med 28(4):233–242. https://doi.org/10.1016/j.tcm.2017.11.002
Tricarico D, Mele A, Conte Camerino D (2005) Phenotype-dependent functional and pharmacological properties of BK channels in skeletal muscle: effects of microgravity. Neurobiol Dis 20(2):296–302. https://doi.org/10.1016/j.nbd.2005.03.011
Tupling R, Green H, Stenisterra G, Lepock J, McKee N (2001) Effects of ischemia on sarcoplasmic reticulum Ca2+ uptake and Ca2+ release in rat skeletal muscle. Am J Physiol Endocrinol Metab 281:E224–E232
Uwera F, Ammar T, McRae C, Hayward LJ, Renaud JM (2020) Lower Ca2+ enhances the K+-induced force depression in normal and hyperKPP mouse muscles. J Gen Physiol 152(7):e201912511
van Mil HG, Geukes Foppen RJ, van Siegenbeek Heukelom J (1997) The influence of bumetanide on the membrane potential of mouse skeletal muscle cells in isotonic and hypertonic media. Br J Pharmacol 120(1):39–44. https://doi.org/10.1038/sj.bjp.0700887
Vaughan-Jones RD (1982) Chloride activity and its control in skeletal and cardiac muscle. Philos Trans R Soc Lond B Biol Sci 299:537–548
Vizi ES, Vyskočil F (1979) Changes in total and quantal release of acetylcholine in the mouse diaphragm during activation and inhibition of membrane ATPase. J Physiol 286:1–14
Vøllestad NK, Hallén NJ, Sejersted OM (1994) Effect of exercise intensity on potassium balance in muscle and blood of man. J Physiol 475(2):359–368
Vyskočil F, Hník P, Rehfeldt H, Vejsada R, Ujec E (1983) The measurement of Ke+ concentration changes in human muscles during volitional contractions. Pflügers Arch 399:235–237
Wan X, Bryant SM, Hart G (2000) The effects of [K+]o on regional differences in electrical characteristics of ventricular myocytes in guinea-pig. Exp Physiol 85(6):769–774
Wang P, Clausen T (1976) Treatment of attacks in hyperkalaemic familial periodic paralysis by inhalation of salbutamol. Lancet 1:221–223
Watanabe D, Wada M (2020) Fatigue-induced change in T-system excitability and its major cause in rat fast-twitch skeletal muscle in vivo. J Physiol. https://doi.org/10.1113/JP279574 (Online ahead of print.PMID: 32833287)
Watanbe I, Gettes LS (2018) Effects of verapamil and pinacidil on extracellular K+, pH, and the incidence of ventricular fibrillation during 60 minutes of ischemia. Int Heart J 59:589–595
Wei AD, Gutman GA, Aldrich R, Chandy KG, Grissmer S, Wulff H (2005) International union of pharmacology. LII. Nomenclature and molecular relationships of calcoim-activated potassium channels. Pharmacol Rev 57(4):463–472
Weiss J, Shine KI (1982) Extracellular K+ accumulation during myocardial ischemia in isolated rabbit heart. Am J Physiol Heart Circ Physiol 242:H619–H628
Weiss JN, Qu Z, Shivkumar K (2017) The electrophysiology of hypo- and hyperkalemia. Circ Arrhythm Electrophysiol 10(3):e004667. https://doi.org/10.1161/CIRCEP.116.004667
Wilde AAM, Aksnes G (1995) Myocardial potassium loss and cell depolarisation in ischaemia and hypoxia. Cardiovasc Res 29:1–15
Williams LR, Leggett RW (1987) The distribution of intracellular alkali metals in reference man. Phys Med Biol 32(2):173–190. https://doi.org/10.1088/0031-9155/32/2/002
Wong JA, Gosmanov AR, Schneider EG, Thomason DB (2001) Insulin-independent, MAPK-dependent stimulation of NKCC activity in skeletal muscle. Am J Physiol Regul Integr Comp Physiol 281(2):R561–R571. https://doi.org/10.1152/ajpregu.2001.281.2.R561
Wyckelsma VL, Perry BD, Bangsbo J, McKenna MJ (2019) Inactivity and exercise training differentially regulate abundance of Na+-K+-ATPase in human skeletal muscle. J Appl Physiol 127(4):905–920. https://doi.org/10.1152/japplphysiol.01076.2018
Yang H, Zhang G, Cui J (2015) BK channels: multiple sensors, one activation gate. Front Physiol 6(6):29. https://doi.org/10.3389/fphys.2015.00029
Ye P, Zhu YR, Gu Y, Zhang DM, Chen SL (2018) Functional protection against cardiac diseases depends on ATP-sensitive potassium channels. J Cell Mol Med 22(12):5801–5806. https://doi.org/10.1111/jcmm.13893
Yensen C, Matar W, Renaud JM (2002) K+-induced twitch potentiation is not due to longer action potential. Am J Physiol Cell Physiol 283:C169–C177
Youn JH (2013) Gut sensing of potassium intake and its role in potassium homeostasis. Semin Nephrol 33(3):248–256. https://doi.org/10.1016/j.semnephrol.2013.04.005
Young RC, Goloman G (2013) Phasic Oscillations of Extracellular Potassium (Ko) in pregnant rat myometrium. PLoS ONE 8(5):e65110. https://doi.org/10.1371/journal.pone.0065110
Yuan P, Leonetti MD, Pico AR, Hsiung Y, MacKinnon R (2010) Structure of the human BK channel Ca2+-activation apparatus at 3.0. A resolution. Science 329(5988):182–6. https://doi.org/10.1126/science.1190414
Zingman LV, Alekseev AE, Hodgson-Zingman DM, Terzic A, A (2007) ATP-sensitive potassium channels: metabolic sensing and cardioprotection. J Appl Physiol 103(5):1888–1893. https://doi.org/10.1152/japplphysiol.00747.2007
Zorbas YG, Kakuris KK, Federenko YF, Deogenov VA (2009) Inability of healthy subjects to deposit potassium during hypokinesia and potassium supplementation. Clin Invest Med 32(1):E34-42. https://doi.org/10.25011/cim.v32i1.5085
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no competing interests.
Additional information
Communicated by Nicolas Place.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Lindinger, M.I., Cairns, S.P. Regulation of muscle potassium: exercise performance, fatigue and health implications. Eur J Appl Physiol 121, 721–748 (2021). https://doi.org/10.1007/s00421-020-04546-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00421-020-04546-8