
Abstract
By controlling urinary potassium excretion, the kidneys play a key role in maintaining whole-body potassium homeostasis. Conversely, low urinary potassium excretion (as a proxy for insufficient dietary intake) is increasingly recognized as a risk factor for the progression of kidney disease. Thus, there is a reciprocal relationship between potassium and the kidney: the kidney regulates potassium balance but potassium also affects kidney function. This review explores this relationship by discussing new insights into kidney potassium handling derived from recently characterized tubulopathies and studies on sexual dimorphism. These insights reveal a central but non-exclusive role for the distal convoluted tubule in sensing potassium and subsequently modifying the activity of the sodium-chloride cotransporter. This is another example of reciprocity: activation of the sodium-chloride cotransporter not only reduces distal sodium delivery and therefore potassium secretion but also increases salt sensitivity. This mechanism helps explain the well-known relationship between dietary potassium and blood pressure. Remarkably, in children, blood pressure is related to dietary potassium but not sodium intake. To explore how potassium deficiency can cause kidney injury, we review the mechanisms of hypokalemic nephropathy and discuss if these mechanisms may explain the association between low dietary potassium intake and adverse kidney outcomes. We discuss if potassium should be repleted in patients with kidney disease and what role dietary potassium plays in the risk of hyperkalemia. Supported by data and physiology, we reach the conclusion that we should view potassium not only as a potentially dangerous cation but also as a companion in the battle against kidney disease.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Physicians are trained to deal with disorders of plasma potassium including hypokalemia and hyperkalemia [1, 2]. Disorders of plasma potassium develop when the excretion of potassium (external potassium balance) or the exchange of potassium between the intracellular and extracellular compartments (internal potassium balance) is disturbed [3]. Because the potassium concentration in the extracellular fluid controls the resting membrane potential of excitable cells, disorders of plasma potassium can lead to muscle weakness, paralysis, and cardiac arrhythmias. Several homeostatic hormones such as insulin, catecholamines, and aldosterone maintain extracellular fluid potassium concentration by regulating potassium distribution, excretion, or both [4, 5]. Furthermore, during hypokalemia, skeletal muscle cells release potassium to the extracellular fluid to buffer the fall in potassium concentration [6]. Because potassium primarily resides inside cells, the potassium content in the extracellular fluid volume is only a fraction of total body potassium. Total body potassium is a black box that we tend to ignore clinically. Studies in which total body potassium was measured, however, offer surprising insights. Contrary to what would be expected, patients with kidney failure often have a low total body potassium content [7]. The relevance of this observation is illustrated by the association between total body potassium deficit in patients with kidney failure and increased mortality [8]. Thus, untoward changes in potassium balance can occur without notable changes in plasma potassium or with changes in the normal range. This is also relevant when considering the “evolution” of our diet in terms of electrolytes. Our diet evolved from a high potassium–low sodium diet to a low potassium–high sodium diet. Recommendations for the adequate intake of potassium (dietary reference values) vary per guideline, but generally range between 90 and 100 mmol/day (3,500–4,000 mg/day) for adults and 112 mmol/day (4,400 mg/day) for lactating women [9]. Recommendations for other age groups are 10 mmol/day (400 mg/day) for breastfed infants aged 0–4 months, 15–28 mmol/day (600–1,100 mg/day) for infants aged 4–12 months, and between 20–28 (800–1,100 mg/day) and 90–100 mmol/day (3,500–4,000 mg/day) for children and adolescents [10]. In a worldwide analysis in adults, potassium intake (estimated from urinary excretion) was 2.12 g/day and therefore ~ 40–50% lower than the recommended intake [11]. Patients with chronic kidney disease (CKD) also consume a low potassium diet either habitually or because they were instructed to reduce potassium intake through dietary counseling [12]. Population studies have clearly shown that a low potassium diet is associated with an increased risk of hypertension and cardiovascular morbidity and mortality [13, 14]. Furthermore, several studies have now also shown that a low urinary potassium excretion (as proxy for insufficient dietary intake) is associated with adverse kidney outcomes (reviewed in [15]). Although a potassium-deficient diet can result in overt hypokalemia [16], people consuming a low potassium diet will usually maintain their plasma potassium in the normal range [17]. Therefore, the impact of dietary potassium intake on health is another example of how changes in the external potassium balance can affect the internal potassium balance. The impact of low dietary potassium intake at the cellular level is becoming increasingly clear. For example, at the level of the kidney tubule, a low potassium diet increases the phosphorylation and therefore activity of the sodium-chloride cotransporter (NCC) in the distal convoluted tubule (DCT) (Fig. 1) [18]. This makes sense physiologically, because increased sodium reabsorption by NCC will reduce sodium delivery to downstream potassium-secreting nephron segments and therefore help to conserve potassium [19]. The collateral damage, however, is that NCC-mediated salt reabsorption may result in salt-sensitive hypertension, especially in the setting of a high salt diet [18]. This raises the possibility that the association between low urinary potassium excretion and negative kidney outcomes is mediated by hypertension, although blood-pressure independent effects of potassium deficiency on the kidney have also been demonstrated [20]. The focus of this review will be the reciprocal relationship between potassium and the kidney: the kidney regulates potassium balance, but potassium also affects kidney function. To address this relationship, we will first review how the kidney “senses” potassium and will do so through the insights provided by recently characterized tubulopathies. Subsequently, we will review how a low potassium diet might cause kidney damage by reviewing the clinical and pathophysiological characteristics of hypokalemic nephropathy. Although hypokalemic nephropathy is typically observed in a specific context (e.g., diarrheal diseases, eating disorders, and excessive use of diuretics or laxatives), the mechanisms of hypokalemic nephropathy may also be relevant for the effects of a low potassium diet on the kidney. Recently characterized sex differences in kidney potassium handling will be reviewed to address whether males or females are more susceptible to kaliopenic kidney injury. Next, we will address the effects of low dietary potassium intake in childhood. We will conclude this review with a perspective on current developments regarding the role of dietary potassium in patients with kidney disease.

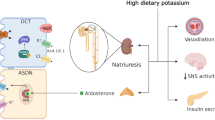
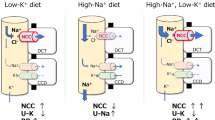
Current model of NCC activation by low K+. Low dietary potassium (K+) intake and/or a low plasma K+ concentration in peritubular capillaries (“low K+”) causes hyperpolarization of the basolateral plasma membrane with potassium chloride efflux out of cells. In the kidney, this effect has been most clearly described in the distal convoluted tubule, but probably represents a more general phenomenon [81]. Potassium chloride efflux occurs through the basolateral heteromeric potassium channel Kir4.1/5.1 (encoded by the genes KCNJ10 and KCNJ16) and the chloride channel ClC-Kb (encoded by CLCNKB). The ensuing decrease in the intracellular chloride concentration activates the kinases WNK4 and SPAK to phosphorylate and activate the sodium-chloride cotransporter (NCC, encoded by SLC12A3). The physiological effect of low K+ on NCC is increased reabsorption of sodium chloride by the DCT and a reduction in sodium delivery to downstream nephron segments. This reduces sodium reabsorption by the epithelial sodium channel (ENaC) and electrochemically coupled K+ secretion through the renal outer medullary potassium channel (ROMK) (not shown). The “collateral damage” of increased salt reabsorption through NCC is an increase in salt sensitivity
New insights from tubulopathies
The effect of low dietary potassium intake on NCC raised the question of how the DCT is capable of sensing potassium. The first clue to the answer was provided by the characterization of a tubulopathy that was coined EAST syndrome (epilepsy, ataxia, sensorineural deafness, tubulopathy) [21]. EAST syndrome is caused by mutations in KCNJ10, which encodes Kir4.1, a potassium channel that is expressed in the DCT, brain, and inner ear. EAST syndrome is characterized by a Gitelman-like tubulopathy suggesting that Kir4.1 regulates NCC activity.
This discovery spurred a flurry of research to better understand the signaling pathways involved [22, 23]. Kir4.1 is an important component of basolateral potassium conductance and enables potassium recycling required for sodium–potassium ATPase activity [24]. This has led to the current working model that a decrease in plasma potassium in peritubular capillaries causes potassium efflux through Kir4.1 leading to basolateral membrane hyperpolarization (Fig. 1). The potassium efflux is accompanied by chloride efflux through the chloride channel ClC-Kb. The subsequent reduction in intracellular chloride activates the chloride-sensitive WNK kinases [25, 26]. In turn, the WNK kinases activate SPAK to phosphorylate and therefore activate NCC. In summary, Kir4.1 could be considered a “potassium sensor” (Fig. 1). More recently, another potassium channel has been implicated in NCC regulation. We and others identified that mutations in KCNJ16 can also cause a Gitelman-like phenotype with sensorineural deafness [27]. KCNJ16 encodes for Kir5.1, which forms functional heteromers with Kir4.1 (Fig. 1). Indeed, co-expression of mutant Kir5.1 with Kir4.1 in Xenopus oocytes significantly reduced electric currents. Remarkably, KCNJ16 mutations can also cause a different phenotype that is similarly characterized not only by hypokalemia and salt wasting but also by proximal renal tubular acidosis [27]. This phenotypic variability was explained by the fact that Kir5.1 can also interact with Kir4.2 (encoded by KCNJ15). Kir4.2 is expressed in the basolateral plasma membrane of proximal tubule cells. Co-expression of these Kir5.1 mutations with Kir4.2 in oocytes also resulted in reduced currents. Depolarization of the basolateral plasma membrane in the proximal tubule could impair bicarbonate exit through the sodium-bicarbonate cotransporter NBCe1 [28]. In patients with these KCNJ16 mutations, an acid loading test demonstrated a lack of ammonia excretion despite an intact ability to acidify the urine [27]. This suggests that intracellular alkalinization inhibits ammoniagenesis. This also illustrates that basolateral potassium channels play a more general role in the nephron and also regulate acid–base balance [29]. The proposed explanation for the predominant proximal or distal phenotype is the localization of the mutations affecting the potassium channel. KCNJ16 mutations causing the predominant proximal tubule phenotype were linked to the pore-forming domain near the selectivity filter of the channel, whereas mutations causing the DCT phenotype were located in the N- or C-terminus [27]. This is reminiscent of the phenotypic variability that can be observed with mutations in the basolateral chloride channel ClC-Kb, which may cause a predominant Bartter or Gitelman phenotype [30]. The majority of patients with the KCNJ16 mutations also had hypomagnesemia. This illustrates the close interaction between potassium and magnesium handling in the kidney. The observation that mutations in SLC12A3, KCNJ10, and KCN16 inhibit NCC activity and cause hypomagnesemia illustrates that reduced NCC activity affects magnesium reabsorption. Indeed, both genetic and pharmacological NCC inhibitions reduce the activity of the transient receptor potential melastatin 6 (TRPM6) channel, which mediates transcellular magnesium reabsorption in the DCT. Furthermore, hypomagnesemia can contribute to renal potassium loss either by NCC inhibition (enhancing distal sodium delivery) or reducing the activity of the renal outer medullary potassium channel (ROMK) [31, 32]. Taken together, the regulation of NCC by Kir4.1/5.1 helps to explain NCC activation by low potassium and therefore also why low potassium predisposes to salt-sensitive hypertension (Fig. 1) [33]. However, it does not yet explain how a low potassium diet contributes to kidney injury. Here, it is also relevant to consider other tubulopathies. For example, if chronic hypokalemia would cause kidney injury, one would expect patients with Gitelman syndrome to develop CKD. Similarly, one would also expect patients with distal renal tubular acidosis (dRTA) to be at risk for CKD, especially because metabolic acidosis contributes to kidney injury [34]. Yet, CKD is an uncommon observation in patients with Gitelman syndrome and dRTA. Of note, this could also be related to a lack of longitudinal data, as a recent European survey showed that CKD is more common in patients with dRTA than in the general population, although kidney failure is still rare [35]. Similarly, Gitelman syndrome can cause glomerular proteinuria characterized by focal segmental sclerosis, thickening of the glomerular basement membrane, and podocyte effacement [36]. In contrast to patients with Gitelman syndrome or dRTA, patients with Bartter syndrome more often progress to kidney failure [30]. This discrepancy caused Walsh and colleagues to challenge the concept of “hypokalemic nephropathy” [37]. They showed that, compared to patients with Gitelman syndrome, patients with Bartter syndrome had a lower eGFR, higher plasma potassium, similar blood pressure, and higher plasma renin and aldosterone concentrations. These differences led them to speculate that hypokalemic nephropathy is not caused by chronic hypokalemia itself but rather by a direct effect of aldosterone or volume depletion.
Hypokalemic nephropathy
Hypokalemic nephropathy has been reported in patients with malnutrition, eating disorders, vomiting, surreptitious use of laxatives or diuretics, and primary aldosteronism [38]. The common denominators in these disorders are volume depletion and aldosteronism, consistent with the hypothesis of Walsh et al. [37]. Hypokalemic nephropathy often is a progressive form of CKD that can lead to kidney failure. Urinalysis is non-specific. In patients in whom a kidney biopsy is performed, chronic interstitial nephritis and fibrosis are common findings. The microscopic characteristics further include the presence of large and irregular cytoplasmic vacuoles and tubular degeneration (“vacuolar degeneration,” Fig. 2). The vacuoles may be present not only in proximal and distal tubules but also in podocytes and arterial myocytes [39]. More recently, other histological features have been identified in kidney biopsies of patients with hypokalemic nephropathy, which were called “WNK bodies” [40]. WNK bodies are punctate structures in the DCT containing the NCC-regulating WNK and SPAK kinases that localize in non-membrane-bound cytoplasmic regions. Additional characteristics of hypokalemic nephropathy have been delineated in laboratory studies using rats. Compared to rats receiving a potassium-replete diet, a potassium-deficient diet causes hypokalemia, a lower intracellular potassium concentration, lower body weight, and higher kidney weight [41]. In addition to lowering intracellular potassium, potassium deficiency also causes intracellular acidosis. In turn, this results in the production of ammonia, which may function as a local toxin and activate the alternative complement pathway [41] (Fig. 2). A possible explanation of why potassium deficiency impairs body growth but increases kidney growth is the overexpression of insulin-like growth factor-1 binding protein (IGF-1-BP). This may cause local trapping of IGF-1 and therefore low circulating levels of IFG-1 [42] (Fig. 2). Hypokalemic nephropathy was further characterized as an ischemic pattern of injury with intrarenal angiotensin II generation despite suppression of the systemic renin-angiotensin system [43]. This was accompanied by changes in vasoactive mediators, including an increase in endothelin-1 and decreases in kallikrein, nitrite/nitrate, and prostaglandin E2 (Fig. 2). Potassium depletion also causes a rapid downregulation of the water channel aquaporin-2 leading to nephrogenic diabetes insipidus [44]. This may be caused by autophagic degradation of aquaporin-2 [45] or the cellular conversion of principal cells to A-type intercalated cells triggered by suppression of Notch signaling (Fig. 2) [46]. In addition to these tubulointerstitial phenomena, potassium deficiency also impairs kidney angiogenesis. This involves the progressive loss of peritubular capillaries accompanied by macrophage infiltration and loss of vascular endothelial growth factor and endothelial nitric oxide synthase (Fig. 2) [47]. In hypokalemic nephropathy, the normalization of plasma potassium did not reverse salt sensitivity, suggesting a permanent form of kidney injury [43]. Combining the potassium-deficient diet with bicarbonate or endothelin A or B antagonists did prevent the development of kidney injury [41].

Characteristics of hypokalemic nephropathy. Hypokalemic nephropathy can cause histological or functional changes throughout the nephron. Depicted are the changes reported in previous studies either from kidney biopsies in patients (panels 1 and 3) or observations in animals placed on a potassium-deficient diet (panels 2, 4–8). Hypokalemic nephropathy is characterized by vacuolar degeneration (panel 1), which may be observed not only in proximal and distal tubules but also in podocytes and arterial myocytes [39]. More recently, so-called WNK bodies have been described occurring as punctate structures in the distal convoluted tubule (panel 3) and consisting of WNKs and SPAK, kinases that are affected by hypokalemia (Fig. 1) [40]. In vivo potassium deficiency has been shown to result in intracellular acidosis and ammoniagenesis with complement activation in cortical tubules (panel 2) [41]. The characteristic increase in kidney weight during hypokalemic nephropathy has been explained by local trapping of IGF-I by IGFBP-1, which was mainly observed in the outer medulla (panel 4) [42]. Changes in intrarenal and excreted vasoactive peptides and metabolites include an increase in angiotensin II (Ang II) and endothelin-1, and decrease in kallikrein, nitrite/nitrate, and prostaglandin E2 (panels 5 and 8) [43]. Hypokalemic nephropathy is also accompanied by loss of peritubular capillaries with macrophage infiltration and a decrease in vascular endothelial growth factor (VEGF) and endothelial nitric oxide synthase (eNOS) (panel 6) [47]. Finally, hypokalemic nephropathy causes nephrogenic diabetes insipidus possibly due to autophagy of aquaporin-2 (AQP2) and/or cellular conversion of principal cells to intercalated cells (panel 7) [45, 46]
Effects of dietary potassium in children
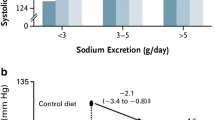
It is unknown whether low dietary potassium intake in children also leads to kidney injury (Table 1). Experimentally, this has been addressed by placing young rats on a potassium-deficient diet [48]. Potassium-depleted young rats showed a sixfold increase in plasma renin activity which was accompanied by the recruitment of renin-producing cells along the afferent arterioles. Similar to adult rats, the young rats also developed the characteristic features of hypokalemic nephropathy including tubulointerstitial injury with the proliferation of tubular epithelial cells, macrophage infiltration, and fibrosis. Potassium-depleted rats also developed a higher blood pressure which was further characterized by salt sensitivity that persisted after correction of hypokalemia. The relationship between dietary potassium intake and blood pressure has been studied more extensively in children. In a study including 233 children aged 5–17 years, urinary potassium but not sodium excretion was related to systolic blood pressure [49]. These findings were confirmed in a larger cohort with a 10-year follow-up demonstrating that higher potassium intakes were inversely associated with systolic and diastolic blood pressure, whereas sodium intake had no effect on blood pressure [50]. In a placebo-controlled study in 13-year-old children, potassium supplementation for 3 years decreased the blood pressure increase with age in girls but not in boys [51]. Another study applied a dietary approach by randomizing adolescents with prehypertension or hypertension to a “DASH” diet (Dietary Approaches to Stop Hypertension) or routine outpatient nutrition care [52]. The DASH group received more potassium and had a greater decrease in systolic blood pressure z-scores than the control group. Importantly, racial differences have been identified in urinary potassium excretion. In one metabolic balance study with fixed intakes, urinary potassium excretion was lower in Black than in White girls possibly due to lower plasma aldosterone [53]. Other proposed explanations are that Black individuals lose more potassium in stool or sweat, have genetic differences in kidney potassium handling, or have lower sodium pump activity [54, 55]. Taken together, the relationship between dietary potassium and blood pressure is already present in childhood, whereas this is less evident for the relationship between dietary sodium and blood pressure. The increasing prevalence of hypertension in children also links to the increasing prevalence of obesity [56,57,58]. Children with CKD also have a diet that is overly represented by sodium, protein, and calories, but contains too little potassium [59]. This suggests that lifestyle interventions should focus not only on caloric intake but also on sodium and potassium intake. As body weight and urinary sodium and potassium excretion show familial aggregation, such interventions should target the entire family to be successful [60, 61].
Sex differences
Because most experimental studies on hypokalemic nephropathy were performed in male animals only, it is unclear if there is sexual dimorphism in the development of hypokalemic nephropathy. It is clear, however, that there are sex differences in kidney potassium handling. Female rats and mice have a lower plasma potassium set point which coincides with higher NCC activity, depends on estrogen, and is maintained after a potassium challenge [62]. This lower plasma potassium set point may protect from hyperkalemia during pregnancy when high potassium loads are consumed [62]. Increased NCC activity in female mice is not accompanied by a larger DCT but rather a shorter DCT with higher cellular density of NCC [63]. Female mice also respond more strongly to thiazide diuretics with greater sodium and potassium excretion than males [64]. In female mice, plasma potassium decreases more after exposure to a potassium-deficient diet. This makes female mice more prone to develop nephrogenic diabetes insipidus when reducing dietary potassium [44]. In agreement with these animal data, the prevalence of hypokalemia is higher in females than in males [65]. However, no studies in human subjects have looked into sex differences in hypokalemic complications (Table 1). It is known that females with Gitelman syndrome require significantly more potassium supplementation than males [66]. This sex difference was explained by progesterone which can cause functional aldosterone antagonism with compensatory NCC upregulation. The inability to upregulate NCC in females with Gitelman syndrome could explain the more severe phenotype compared to males. This could also explain why females with Gitelman syndrome typically require even higher doses of potassium supplementation during pregnancy. In contrast, one study found a more severe phenotype in men with Gitelman syndrome [67]. Another study reported same sex siblings with differences in disease severity [68]. Therefore, the role of sex in the disease severity of Gitelman syndrome remains incompletely understood.
Perspectives for potassium
What should be the next steps to materialize the accumulating evidence for the relationships between dietary potassium intake, blood pressure, cardiovascular, and kidney outcomes? The studies reporting an association between urinary potassium excretion and kidney outcomes in patients with CKD have called for a clinical trial to address causality. The challenge of such a trial was also emphasized as it should strike a balance between investigating the beneficial effects of increasing dietary potassium intake to the dietary reference values without causing excess hyperkalemia (Table 1). In the Netherlands, a placebo-controlled, double-blind randomized clinical trial is currently ongoing to analyze the effects of potassium supplementation (40 mEq/day) for 2 years on kidney function in adult patients with progressive CKD (eGFR between 15 and 45 mL/min/1.73 m2) and hypertension [12]. This trial will also be important to analyze the safety of potassium repletion in patients with CKD in terms of hyperkalemia incidence. Here, it is important to consider the effect of dietary potassium on the plasma potassium concentration. In a recent cross-sectional analysis of non-dialysis- and dialysis-dependent patients with CKD, no associations were identified between serum and dietary potassium (assessed by 3-day food records) [69]. We previously did identify a significant but weak correlation between urinary potassium excretion and plasma potassium [12]. Most potassium supplementation studies in subjects with normal kidney function also observed a small but significant rise in plasma potassium of 0.14 mmol/L, which was independent of dose or duration of treatment [70]. The effects of potassium supplementation on plasma potassium will also depend on whether the supplementation is taken with meals and the components of this meal including glucose, sodium, and fiber content [71]. A recent study reported the results of a randomized trial on the effects of a 4-week dietary potassium intervention (40 vs. 100 mmol/day) on serum potassium levels and blood pressure in 29 patients with CKD stage G3 [72]. During the high dietary potassium intake, serum potassium was 0.21 mmol/L higher (leading to hyperkalemia in 2 patients), but failed to reduce ambulatory blood pressure. This is clearly different from potassium supplementation studies in patients with hypertension but without CKD, which show a consistent reduction in blood pressure [73]. The explanation for this discrepancy is unclear, but may relate to the fact that the relationship between urinary potassium excretion and blood pressure follows a U-shaped relationship [74]. CKD-specific factors such as relative hyperaldosteronism, metabolic acidosis, or the uremic milieu may modify or shift this U-shaped relationship. Another possibility is that potassium supplementation should be introduced earlier in the course of kidney disease progression for it to be beneficial. Ideally, a CKD stage is identified with maximal benefit and the lowest risk of hyperkalemia [75]. Longer-term intervention studies in patients with CKD with repeated measurements will be necessary to more definitively assess the effects of adequate potassium intake on plasma potassium, blood pressure, kidney function, and their interdependence. In public health, another potassium-based strategy to positively affect blood pressure is gaining ground, namely salt substitution. Salt substitution refers to the approach to replace ~ 25% of sodium chloride with potassium chloride in discretionary salt use [76]. Two large salt substitution studies in the community were recently completed. The first study (including 2,376 participants) showed that salt substitution reduced the incidence of hypertension by 51% [77]. Of note, in this study, urinary potassium excretion increased, but urinary sodium excretion was unchanged, suggesting that the effect was primarily attributable to increased potassium intake. The second study (including 20,995 participants) observed a similar reduction not only in blood pressure but also in the risk of stroke, major cardiovascular events, and death [78]. In this study, urinary potassium excretion increased and urinary sodium excretion decreased. The effect on cardiovascular outcomes could therefore be attributable to the decrease in sodium intake, the increase in potassium intake, or both. Irrespectively, salt substitution represents a relatively simple intervention that might also benefit patients with CKD. Indeed, modelling studies suggest that the prevention of cardiovascular deaths outweighs excess hyperkalemia deaths, even in patients with CKD [79]. Similar to increasing dietary potassium intake, salt substitution would also be a logical topic for a future clinical trial in patients with CKD. These studies will be necessary to further determine the contribution of dietary potassium to outcomes. In practice, this evidence should be translated to dietary recommendations which fits with the increased focus on plant-based diets in patients with kidney disease [80]. In summary, there is still a lot to learn on the role of potassium in kidney disease (Table 1). Awaiting future studies, it is becoming clear that potassium should not only be considered a potentially lethal cation but may be part of a non-pharmacological approach to improve cardiovascular and kidney outcomes in patients with kidney disease.
References
Grams ME, Hoenig MP, Hoorn EJ (2021) Evaluation of hypokalemia. JAMA 325:1216–1217
Palmer BF, Carrero JJ, Clegg DJ, Colbert GB, Emmett M, Fishbane S, Hain DJ, Lerma E, Onuigbo M, Rastogi A, Roger SD, Spinowitz BS, Weir MR (2021) Clinical management of hyperkalemia. Mayo Clin Proc 96:744–762
Gumz ML, Rabinowitz L, Wingo CS (2015) An integrated view of potassium homeostasis. N Engl J Med 373:60–72
Palmer BF (2015) Regulation of potassium homeostasis. Clin J Am Soc Nephrol 10:1050–1060
Unwin RJ, Luft FC, Shirley DG (2011) Pathophysiology and management of hypokalemia: a clinical perspective. Nat Rev Nephrol 7:75–84
McFarlin BE, Chen Y, Priver TS, Ralph DL, Mercado A, Gamba G, Madhur MS, McDonough AA (2020) Coordinate adaptations of skeletal muscle and kidney to maintain extracellular [K(+)] during K(+)-deficient diet. Am J Physiol Cell Physiol 319:C757–C770
Wouda RD, Vogt L, Hoorn EJ (2020) Personalizing potassium management in patients on haemodialysis. Nephrol Dial Transplant 36:13–18
Dolson GM, Ellis KJ, Johnson ML, Adrogué HJ (2003) Incidence and consequences of total body potassium depletion in chronic hemodialysis patients. Front Biosci 8:a126–a132
Strohm D, Ellinger S, Leschik-Bonnet E, Maretzke F, Heseker H, German Nutrition Society (DGE) (2017) Revised reference values for potassium intake. Ann Nutr Metab 71:118–124
Desloovere A, Renken-Terhaerdt J, Tuokkola J, Shaw V, Greenbaum LA, Haffner D, Anderson C, Nelms CL, Oosterveld MJS, Paglialonga F, Polderman N, Qizalbash L, Warady BA, Shroff R, VandeWalle J (2021) The dietary management of potassium in children with CKD stages 2–5 and on dialysis-clinical practice recommendations from the Pediatric Renal Nutrition Taskforce. Pediatr Nephrol 36:1331–1346
Mente A, O’Donnell MJ, Rangarajan S, McQueen MJ, Poirier P, Wielgosz A, Morrison H, Li W, Wang X, Di C, Mony P, Devanath A, Rosengren A, Oguz A, Zatonska K, Yusufali AH, Lopez-Jaramillo P, Avezum A, Ismail N, Lanas F, Puoane T, Diaz R, Kelishadi R, Iqbal R, Yusuf R, Chifamba J, Khatib R, Teo K, Yusuf S (2014) Association of urinary sodium and potassium excretion with blood pressure. N Engl J Med 371:601–611
Gritter M, Vogt L, Yeung SMH, Wouda RD, Ramakers CRB, de Borst MH, Rotmans JI, Hoorn EJ (2018) Rationale and design of a randomized placebo-controlled clinical trial assessing the renoprotective effects of potassium supplementation in chronic kidney disease. Nephron 140:48–57
O’Donnell M, Mente A, Rangarajan S, McQueen MJ, Wang X, Liu L, Yan H, Lee SF, Mony P, Devanath A, Rosengren A, Lopez-Jaramillo P, Diaz R, Avezum A, Lanas F, Yusoff K, Iqbal R, Ilow R, Mohammadifard N, Gulec S, Yusufali AH, Kruger L, Yusuf R, Chifamba J, Kabali C, Dagenais G, Lear SA, Teo K, Yusuf S (2014) Urinary sodium and potassium excretion, mortality, and cardiovascular events. N Engl J Med 371:612–623
Ma Y, He FJ, Sun Q, Yuan C, Kieneker LM, Curhan GC, MacGregor GA, Bakker SJL, Campbell NRC, Wang M, Rimm EB, Manson JE, Willett WC, Hofman A, Gansevoort RT, Cook NR, Hu FB (2021) 24-Hour urinary sodium and potassium excretion and cardiovascular risk. N Engl J Med 386:252–263
Wei KY, Gritter M, Vogt L, de Borst MH, Rotmans JI, Hoorn EJ (2020) Dietary potassium and the kidney: lifesaving physiology. Clin Kidney J 13:952–968
Black DA, Milne MD (1952) Experimental potassium depletion in man. Lancet 1:244–245
Hyatt KH, Johnson PC, Hoffler GW, Rambaut PC, Rummel JA, Hulley SB, Vogel JM, Huntoon C, Spears CP (1975) Effect of potassium depletion in normal males: an Apollo 15 simulation. Aviat Space Environ Med 46:11–15
Terker AS, Zhang C, McCormick JA, Lazelle RA, Zhang C, Meermeier NP, Siler DA, Park HJ, Fu Y, Cohen DM, Weinstein AM, Wang WH, Yang CL, Ellison DH (2015) Potassium modulates electrolyte balance and blood pressure through effects on distal cell voltage and chloride. Cell Metab 21:39–50
Hoorn EJ, Gritter M, Cuevas CA, Fenton RA (2020) Regulation of the renal NaCl cotransporter and its role in potassium homeostasis. Physiol Rev 100:321–356
Gritter M, Rotmans JI, Hoorn EJ (2019) Role of dietary K(+) in natriuresis, blood pressure reduction, cardiovascular protection, and renoprotection. Hypertension 73:15–23
Bockenhauer D, Feather S, Stanescu HC, Bandulik S, Zdebik AA, Reichold M, Tobin J, Lieberer E, Sterner C, Landoure G, Arora R, Sirimanna T, Thompson D, Cross JH, van’t Hoff W, Al Masri O, Tullus K, Yeung S, Anikster Y, Klootwijk E, Hubank M, Dillon MJ, Heitzmann D, Arcos-Burgos M, Knepper MA, Dobbie A, Gahl WA, Warth R, Sheridan E, Kleta R (2009) Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N Engl J Med 360:1960–1970
Zhang C, Wang L, Zhang J, Su XT, Lin DH, Scholl UI, Giebisch G, Lifton RP, Wang WH (2014) KCNJ10 determines the expression of the apical Na-Cl cotransporter (NCC) in the early distal convoluted tubule (DCT1). Proc Natl Acad Sci U S A 111:11864–11869
Cuevas CA, Su XT, Wang MX, Terker AS, Lin DH, McCormick JA, Yang CL, Ellison DH, Wang WH (2017) Potassium sensing by renal distal tubules requires Kir4.1. J Am Soc Nephrol 28:1814–1825
Bandulik S, Schmidt K, Bockenhauer D, Zdebik AA, Humberg E, Kleta R, Warth R, Reichold M (2011) The salt-wasting phenotype of EAST syndrome, a disease with multifaceted symptoms linked to the KCNJ10 K+ channel. Pflugers Arch 461:423–435
Bazúa-Valenti S, Chávez-Canales M, Rojas-Vega L, González-Rodríguez X, Vázquez N, Rodríguez-Gama A, Argaiz ER, Melo Z, Plata C, Ellison DH, García-Valdés J, Hadchouel J, Gamba G (2015) The effect of WNK4 on the Na+–Cl− cotransporter is modulated by intracellular chloride. J Am Soc Nephrol 26:1781–1786
Piala AT, Moon TM, Akella R, He H, Cobb MH, Goldsmith EJ (2014) Chloride sensing by WNK1 involves inhibition of autophosphorylation. Sci Signal 7:ra41
Schlingmann KP, Renigunta A, Hoorn EJ, Forst AL, Renigunta V, Atanasov V, Mahendran S, Barakat TS, Gillion V, Godefroid N, Brooks AS, Lugtenberg D, Lake J, Debaix H, Rudin C, Knebelmann B, Tellier S, Rousset-Rouvière C, Viering D, de Baaij JHF, Weber S, Palygin O, Staruschenko A, Kleta R, Houillier P, Bockenhauer D, Devuyst O, Vargas-Poussou R, Warth R, Zdebik AA, Konrad M (2021) Defects in KCNJ16 cause a novel tubulopathy with hypokalemia, salt wasting, disturbed acid-base homeostasis, and sensorineural deafness. J Am Soc Nephrol 32:1498–1512
Weiner ID, Verlander JW (2016) Recent advances in understanding renal ammonia metabolism and transport. Curr Opin Nephrol Hypertens 25:436–443
Bignon Y, Pinelli L, Frachon N, Lahuna O, Figueres L, Houillier P, Lourdel S, Teulon J, Paulais M (2020) Defective bicarbonate reabsorption in Kir4.2 potassium channel deficient mice impairs acid-base balance and ammonia excretion. Kidney Int 97:304–315
Seys E, Andrini O, Keck M, Mansour-Hendili L, Courand PY, Simian C, Deschenes G, Kwon T, Bertholet-Thomas A, Bobrie G, Borde JS, Bourdat-Michel G, Decramer S, Cailliez M, Krug P, Cozette P, Delbet JD, Dubourg L, Chaveau D, Fila M, Jourde-Chiche N, Knebelmann B, Lavocat MP, Lemoine S, Djeddi D, Llanas B, Louillet F, Merieau E, Mileva M, Mota-Vieira L, Mousson C, Nobili F, Novo R, Roussey-Kesler G, Vrillon I, Walsh SB, Teulon J, Blanchard A, Vargas-Poussou R (2017) Clinical and genetic spectrum of Bartter syndrome type 3. J Am Soc Nephrol 28:2540–2552
Ferdaus MZ, Mukherjee A, Nelson JW, Blatt PJ, Miller LN, Terker AS, Staub O, Lin DH, McCormick JA (2019) Mg(2+) restriction downregulates NCC through NEDD4-2 and prevents its activation by hypokalemia. Am J Physiol Renal Physiol 317:F825–F838
Huang CL, Kuo E (2007) Mechanism of hypokalemia in magnesium deficiency. J Am Soc Nephrol 18:2649–2652
Wang MX, Cuevas CA, Su XT, Wu P, Gao ZX, Lin DH, McCormick JA, Yang CL, Wang WH, Ellison DH (2018) Potassium intake modulates the thiazide-sensitive sodium-chloride cotransporter (NCC) activity via the Kir4.1 potassium channel. Kidney Int 93:893–902
Bugarski M, Ghazi S, Polesel M, Martins JR, Hall AM (2021) Changes in NAD and lipid metabolism drive acidosis-induced acute kidney injury. J Am Soc Nephrol 32:342–356
Lopez-Garcia SC, Emma F, Walsh SB, Fila M, Hooman N, Zaniew M, Bertholet-Thomas A, Colussi G, Burgmaier K, Levtchenko E, Sharma J, Singhal J, Soliman NA, Ariceta G, Basu B, Murer L, Tasic V, Tsygin A, Decramer S, Gil-Peña H, Koster-Kamphuis L, La Scola C, Gellermann J, Konrad M, Lilien M, Francisco T, Tramma D, Trnka P, Yüksel S, Caruso MR, Chromek M, Ekinci Z, Gambaro G, Kari JA, König J, Taroni F, Thumfart J, Trepiccione F, Winding L, Wühl E, Ağbaş A, Belkevich A, Vargas-Poussou R, Blanchard A, Conti G, Boyer O, Dursun I, Pınarbaşı AS, Melek E, Miglinas M, Novo R, Mallett A, Milosevic D, Szczepanska M, Wente S, Cheong HI, Sinha R, Gucev Z, Dufek S, Iancu D, European dRTA Consortium, Kleta R, Schaefer F, Bockenhauer D (2019) Treatment and long-term outcome in primary distal renal tubular acidosis. Nephrol Dial Transplant 34:981–991
Demoulin N, Aydin S, Cosyns JP, Dahan K, Cornet G, Auberger I, Loffing J, Devuyst O (2014) Gitelman syndrome and glomerular proteinuria a link between loss of sodium-chloride cotransporter and podocyte dysfunction? Nephrol Dial Transplant 29:iv117–iv120
Walsh SB, Unwin E, Vargas-Poussou R, Houillier P, Unwin R (2011) Does hypokalaemia cause nephropathy? An observational study of renal function in patients with Bartter or Gitelman syndrome. QJM 104:939–944
Cremer W, Bock KD (1977) Symptoms and course of chronic hypokalemic nephropathy in man. Clin Nephrol 7:112–119
Kudose S, Dounis H, D’Agati VD (2020) Multicellular vacuoles in hypokalemic nephropathy. Kidney Int 97:618
Thomson MN, Schneider W, Mutig K, Ellison DH, Kettritz R, Bachmann S (2019) Patients with hypokalemia develop WNK bodies in the distal convoluted tubule of the kidney. Am J Physiol Renal Physiol 316:F292–F300
Tolins JP, Hostetter MK, Hostetter TH (1987) Hypokalemic nephropathy in the rat. Role of ammonia in chronic tubular injury. J Clin Invest 79:1447–1458
Tsao T, Fawcett J, Fervenza FC, Hsu FW, Huie P, Sibley RK, Rabkin R (2001) Expression of insulin-like growth factor-I and transforming growth factor-β in hypokalemic nephropathy in the rat. Kidney Int 59:96–105
Suga SI, Phillips MI, Ray PE, Raleigh JA, Vio CP, Kim YG, Mazzali M, Gordon KL, Hughes J, Johnson RJ (2001) Hypokalemia induces renal injury and alterations in vasoactive mediators that favor salt sensitivity. Am J Physiol Renal Physiol 281:F620–F629
Lama A-Q, Grimm PR, Ava MZ, Paul AW (2021) Rapid development of vasopressin resistance in dietary K+ deficiency. Am J Physiol Renal Physiol 320:F748–F760
Khositseth S, Uawithya P, Somparn P, Charngkaew K, Thippamom N, Hoffert JD, Saeed F, Michael Payne D, Chen SH, Fenton RA, Pisitkun T (2015) Autophagic degradation of aquaporin-2 is an early event in hypokalemia-induced nephrogenic diabetes insipidus. Sci Rep 5:18311
Iervolino A, Prosperi F, De La Motte LR, Petrillo F, Spagnuolo M, D’Acierno M, Siccardi S, Perna AF, Christensen BM, Frische S, Capasso G, Trepiccione F (2020) Potassium depletion induces cellular conversion in the outer medullary collecting duct altering Notch signaling pathway. Sci Rep 10:5708
Reungjui S, Roncal CA, Sato W, Glushakova OY, Croker BP, Suga S, Ouyang X, Tungsanga K, Nakagawa T, Johnson RJ, Mu W (2008) Hypokalemic nephropathy is associated with impaired angiogenesis. J Am Soc Nephrol 19:125–134
Ray PE, Suga S, Liu XH, Huang X, Johnson RJ (2001) Chronic potassium depletion induces renal injury, salt sensitivity, and hypertension in young rats. Kidney Int 59:1850–1858
Geleijnse JM, Grobbee DE, Hofman A (1990) Sodium and potassium intake and blood pressure change in childhood. BMJ 300:899–902
Buendia JR, Bradlee ML, Daniels SR, Singer MR, Moore LL (2015) Longitudinal effects of dietary sodium and potassium on blood pressure in adolescent girls. JAMA Pediatr 169:560–568
Sinaiko AR, Gomez-Marin O, Prineas RJ (1993) Effect of low sodium diet or potassium supplementation on adolescent blood pressure. Hypertension 21:989–994
Couch SC, Saelens BE, Levin L, Dart K, Falciglia G, Daniels SR (2008) The efficacy of a clinic-based behavioral nutrition intervention emphasizing a DASH-type diet for adolescents with elevated blood pressure. J Pediatr 152:494–501
Palacios C, Wigertz K, Martin BR, Braun M, Pratt JH, Peacock M, Weaver CM (2009) Racial differences in potassium homeostasis in response to differences in dietary sodium in girls. Am J Clin Nutr 91:597–603
Suh A, DeJesus E, Rosner K, Lerma E, Yu W, Young JB, Rosa RM (2004) Racial differences in potassium disposal. Kidney Int 66:1076–1081
Turban S, Miller ER 3rd, Ange B, Appel LJ (2008) Racial differences in urinary potassium excretion. J Am Soc Nephrol 19:1396–1402
Sorof JM, Lai D, Turner J, Poffenbarger T, Portman RJ (2004) Overweight, ethnicity, and the prevalence of hypertension in school-aged children. Pediatrics 113:475–482
Song P, Zhang Y, Yu J, Zha M, Zhu Y, Rahimi K, Rudan I (2019) Global prevalence of hypertension in children: a systematic review and meta-analysis. JAMA Pediatr 173:1154–1163
Wühl E (2019) Hypertension in childhood obesity. Acta Paediatr 108:37–43
Chen W, Ducharme-Smith K, Davis L, Hui WF, Warady BA, Furth SL, Abraham AG, Betoko A (2017) Dietary sources of energy and nutrient intake among children and adolescents with chronic kidney disease. Pediatr Nephrol 32:1233–1241
Connor SL, Connor WE, Henry H, Sexton G, Keenan EJ (1984) The effects of familial relationships, age, body weight, and diet on blood pressure and the 24 hour urinary excretion of sodium, potassium, and creatinine in men, women, and children of randomly selected families. Circulation 70:76–85
Li Y, Sun Y, Li X, Dong L, Cheng F, Luo R, Wang C, Song J, He FJ, MacGregor GA, Zhang P (2021) Sodium and potassium excretion of schoolchildren and relationship with their family excretion in China. Nutrients 13:2864
Veiras LC, Girardi ACC, Curry J, Pei L, Ralph DL, Tran A, Castelo-Branco RC, Pastor-Soler N, Arranz CT, Yu ASL, McDonough AA (2017) Sexual dimorphic pattern of renal transporters and electrolyte homeostasis. J Am Soc Nephrol 28:3504–3517
Tahaei E, Coleman R, Saritas T, Ellison DH, Welling PA (2020) Distal convoluted tubule sexual dimorphism revealed by advanced 3D imaging. Am J Physiol Renal Physiol 319:F754–F764
Jing L, Ryo H, Shuhua X, Laxiang W, Lei Y, Alan MW, Lawrence P, Tong W (2017) Gender difference in kidney electrolyte transport. I. Role of AT1a receptor in thiazide-sensitive Na+-Cl− cotransporter activity and expression in male and female mice. Am J Physiol Renal Physiol 313:F505–F513
Wysowski DK, Kornegay C, Nourjah P, Trontell A (2003) Sex and age differences in serum potassium in the United States. Clin Chem 49:190–192
Berry MR, Robinson C, Karet Frankl FE (2013) Unexpected clinical sequelae of Gitelman syndrome: hypertension in adulthood is common and females have higher potassium requirements. Nephrol Dial Transplant 28:1533–1542
Lin SH, Cheng NL, Hsu YJ, Halperin ML (2004) Intrafamilial phenotype variability in patients with Gitelman syndrome having the same mutations in their thiazide-sensitive sodium/chloride cotransporter. Am J Kidney Dis 43:304–312
Turman MA (1998) Concomitant occurrence of Gitelman and Bartter syndromes in the same family? Pediatr Nephrol 12:23–25
Ramos CI, González-Ortiz A, Espinosa-Cuevas A, Avesani CM, Carrero JJ, Cuppari L (2021) Does dietary potassium intake associate with hyperkalemia in patients with chronic kidney disease? Nephrol Dial Transplant 36:2049–2057
Cappuccio FP, Buchanan LA, Ji C, Siani A, Miller MA (2016) Systematic review and meta-analysis of randomised controlled trials on the effects of potassium supplements on serum potassium and creatinine. BMJ Open 6:e011716
Preston RA, Afshartous D, Rodco R, Alonso AB, Garg D (2015) Evidence for a gastrointestinal-renal kaliuretic signaling axis in humans. Kidney Int 88:1383–1391
Turban S, Juraschek SP, Miller ER, Anderson CAM, White K, Charleston J, Appel LJ (2021) Randomized trial on the effects of dietary potassium on blood pressure and serum potassium levels in adults with chronic kidney disease. Nutrients 13:2678
Aburto NJ, Hanson S, Gutierrez H, Hooper L, Elliott P, Cappuccio FP (2013) Effect of increased potassium intake on cardiovascular risk factors and disease: systematic review and meta-analyses. BMJ 346:f1378
Filippini T, Naska A, Kasdagli MI, Torres D, Lopes C, Carvalho C, Moreira P, Malavolti M, Orsini N, Whelton PK, Vinceti M (2020) Potassium intake and blood pressure: a dose-response meta-analysis of randomized controlled trials. J Am Heart Assoc 9:e015719
Burnier M (2020) Increasing potassium intake to prevent kidney damage: a new population strategy? Kidney Int 98:59–61
Greer RC, Marklund M, Anderson CAM, Cobb LK, Dalcin AT, Henry M, Appel LJ (2020) Potassium-enriched salt substitutes as a means to lower blood pressure: benefits and risks. Hypertension 75:266–274
Bernabe-Ortiz A, Sal YRVG, Ponce-Lucero V, Cardenas MK, Carrillo-Larco RM, Diez-Canseco F, Pesantes MA, Sacksteder KA, Gilman RH, Miranda JJ (2020) Effect of salt substitution on community-wide blood pressure and hypertension incidence. Nat Med 26:374–378
Neal B, Wu Y, Feng X, Zhang R, Zhang Y, Shi J, Zhang J, Tian M, Huang L, Li Z, Yu Y, Zhao Y, Zhou B, Sun J, Liu Y, Yin X, Hao Z, Yu J, Li KC, Zhang X, Duan P, Wang F, Ma B, Shi W, Di Tanna GL, Stepien S, Shan S, Pearson SA, Li N, Yan LL, Labarthe D, Elliott P (2021) Effect of salt substitution on cardiovascular events and death. N Engl J Med 385:1067–1077
Marklund M, Singh G, Greer R, Cudhea F, Matsushita K, Micha R, Brady T, Zhao D, Huang L, Tian M, Cobb L, Neal B, Appel LJ, Mozaffarian D, Wu JHY (2020) Estimated population wide benefits and risks in China of lowering sodium through potassium enriched salt substitution: modelling study. BMJ 369:m824
Carrero JJ, Gonzalez-Ortiz A, Avesani CM, Bakker SJL, Bellizzi V, Chauveau P, Clase CM, Cupisti A, Espinosa-Cuevas A, Molina P, Moreau K, Piccoli GB, Post A, Sezer S, Fouque D (2020) Plant-based diets to manage the risks and complications of chronic kidney disease. Nat Rev Nephrol 16:525–542
Boyd-Shiwarski CR, Subramanya AR (2017) The renal response to potassium stress: integrating past with present. Curr Opin Nephrol Hypertens 26:411–418
Acknowledgements
We thank Manon Zuurmond for the illustrations.
Funding
This work was funded by the Dutch Kidney Foundation (grants CP16.01 and 21OK + 13).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wieërs, M.L.A.J., Mulder, J., Rotmans, J.I. et al. Potassium and the kidney: a reciprocal relationship with clinical relevance. Pediatr Nephrol 37, 2245–2254 (2022). https://doi.org/10.1007/s00467-022-05494-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-022-05494-5