
Abstract
In smooth muscle cells (SMCs), the intracellular chloride ion (Cl−) concentration is high due to accumulation by Cl−/HCO3 − exchange and Na+–K+–Cl− cotransportation. The equilibrium potential for Cl− (E Cl) is more positive than physiological membrane potentials (E m), with Cl− efflux inducing membrane depolarization. Early studies used electrophysiology and nonspecific antagonists to study the physiological relevance of Cl− channels in SMCs. More recent reports have incorporated molecular biological approaches to identify and determine the functional significance of several different Cl− channels. Both “classic” and cGMP-dependent calcium (Ca2+)-activated (ClCa) channels and volume-sensitive Cl− channels are present, with TMEM16A/ANO1, bestrophins, and ClC-3, respectively, proposed as molecular candidates for these channels. The cystic fibrosis transmembrane conductance regulator (CFTR) has also been described in SMCs. This review will focus on discussing recent progress made in identifying each of these Cl− channels in SMCs, their physiological functions, and contribution to diseases that modify contraction, apoptosis, and cell proliferation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Chloride (Cl−) is the predominant extracellular and intracellular anion with intracellular concentration [Cl−]i varying widely between different cell types. In many cells, such as frog skeletal muscle, [Cl−]i is similar to that predicted by passive distribution determined by the Donnan equilibrium [47]. In contrast, in vascular smooth muscle cells (SMCs), [Cl−]i is much higher than would be expected [14]. [Cl−]i ranging from ~30 to ~50 mM has been recorded in SMCs using a variety of techniques, including radioisotopes, fluorescent dyes, and ion-selective electrodes (see [57]). High [Cl−]i is maintained by active accumulation through Cl−/HCO3 − anion exchange and Na+–K+–Cl− cotransportation [1, 90]. The estimated equilibrium potential for Cl− (E Cl −) is between −30 and −20 mV in SMCs [57, 66]. Physiological membrane potential (E m) in vascular and nonvascular SMCs ranges between ~−60 and ~−40 mV [6, 44, 83, 85, 86, 118]. Cl− channel activation would result in Cl− efflux, leading to membrane depolarization, voltage-dependent calcium (Ca2+) channel activation, an elevation in [Ca2+]i, and contraction [19, 46, 65]. In addition to modulation of membrane potential and contractility, intracellular Cl− has also been proposed to regulate intracellular pH and cell volume in SMCs [14].
Cl− channels are subdivided into five families: transmembrane protein 16 (TMEM16)/anoctamin (ANO), bestrophins, voltage-gated Cl− channels (CLCs), cystic fibrosis (CF) transmembrane conductance regulator (CFTR), and ligand-gated Cl− channels, including glycine and γ-aminobutyric acid (GABA) receptors [30]. This review will summarize knowledge of TMEM16A/ANO, bestrophins, CLCs, and CFTR due to limited evidence for other Cl− channel members in SMCs. The predicted membrane topologies for each of these Cl− channels are illustrated in Fig. 1. Ligand-gated Cl− channels have been described in airway SMCs, where both GABAA and GlyR1 channels are expressed and functional [81, 143]. A distinct type of Cl− current (I Cl,acid) activated by acidic extracellular pH has also been reported in aortic SMCs that may be generated by CLC-3 [71, 76].

Functional significance of SMC Cl− currents
Several early studies demonstrated Cl− flux in a variety of different vascular SMC types [11, 108, 124, 137]. Noradrenaline (NE) stimulated 36Cl− efflux in rat aorta, portal vein, and rabbit pulmonary arteries [11, 108, 124]. Subsequent findings showed that NE-induced depolarization of rat anococcygeus muscle cells was Cl− current-dependent; endothelin (ET) activated Cl− currents in porcine coronary artery, human mesenteric artery SMCs, and cultured aortic SMCs; and histamine activated Cl− currents in rabbit pulmonary artery SMCs [59, 121, 123].
Research using a variety of nonselective Cl− channel antagonists further supported the concept that Cl− flux contributes to vasoconstriction. 4,4′-Diisothiocyanatostilbene-2,2′-disulfonic acid (DIDS) and indaryloxyacetic acid (IAA-94), but not niflumic acid (NFA), hyperpolarized and relaxed pressurized rat cerebral arteries [84]. NFA reduced NE- but not K+-induced contractions in rat aorta and mesenteric arteries [16, 17, 62]. Histamine-induced depolarization and contraction were also attenuated by NFA in rabbit middle cerebral and basilar arteries, respectively [37, 120]. IAA-94 inhibited ET-induced vasoconstriction in cultured vascular SMCs [114]. Anion replacement has also been utilized to strengthen functional evidence obtained using nonspecific Cl− channel inhibitors. Substitution of extracellular Cl− with methanesulfonate potentiated NE-, serotonin-, endothelin-1-, and histamine-induced, but not K+-induced contractions in rabbit basilar arteries and rat aorta [19, 20, 62]. Lowering extracellular Cl− potentiated pressure-induced constriction and inhibited histamine-induced contraction in rat cerebral arteries [84, 120]. Substitution with Br− and NO3 −, which are more permeant anions than Cl−, increased contraction to NE in rat portal vein [125].
In addition to modulating SMC contractility, both volume-sensitive Cl− channels and Ca2+-activated Cl− channels (ClCa) channels have been proposed to control SMC proliferation [12, 138, 142]. DIDS, but not IAA-94 or 5-nitro-2,2″-dicarboxylic acid (NPPB), another nonselective Cl− channel blocker, suppressed ET-1-induced proliferation in cultured aortic SMCs [138]. In contrast, NPPB and IAA-94, but not DIDS, inhibited insulin-like growth factor (IGF)-induced proliferation in porcine coronary artery SMCs [12]. Under chronic hypoxic conditions, NFA and IAA-94 also inhibited proliferation of rat pulmonary artery SMCs [142].
In summary, studies measuring ion flux and those using nonselective Cl− channel blockers and extracellular anion replacement suggested that Cl− currents regulate SMC function. More recent studies have identified some of the proteins that generate and regulate these Cl− currents and investigated their physiological functions and pathological alterations.
Molecular identification of Cl− channels in SMCs
Classic Ca2+-activated Cl− (ClCa) channels
ClCa currents have been described in a variety of SMC types, including those from human mesenteric, rabbit ear, pulmonary and coronary arteries, rat portal vein and cultured cells from rat pulmonary, and cultured pig aorta [2, 26, 58, 64, 91, 130, 145]. Nonspecific Cl− channel blockers previously shown to modulate SMC functions were demonstrated to inhibit whole-cell ClCa currents, supporting relevance [64]. Cl− channel blockers also inhibited spontaneous transient inward currents (STICs) in rabbit portal vein SMCs [48]. STICS occur due to the simultaneous activation of multiple Cl− channels by a Ca2+ spark, a local intracellular Ca2+ transient that occurs due to ryanodine (RyR)-mediated sarcoplasmic reticulum Ca2+ release [53]. In some SMC types, including those from airways, Ca2+ sparks activate both STICs and spontaneous transient outward currents (STOCs), which occur due to the simultaneous activation of multiple large conductance Ca2+-activated potassium (BKCa) channels. A single Ca2+ spark can activate both ClCa and BKCa channel, eliciting a STOC followed by a STIC [151]. STICs induce depolarization, whereas STOCs hyperpolarize the membrane potential. Thus, bimodal regulation of ClCa and BKCa channels by Ca2+ sparks permits fine tuning of membrane potential [150].
ClCa currents exhibit a distinct phenotype. The IV relationship is outwardly rectifying at low intracellular Ca2+ concentrations ([Ca2+]i) [45]. Elevating [Ca2+]i linearizes the ClCa IV relationship [65]. The relative permeability of SMC ClCa currents is SCN− > I− > Br− > Cl− > aspartate [41]. IP3R- or RyR-mediated SR Ca2+ release, Ca2+ entry through voltage-dependent Ca2+ channels (VDCC), and local Ca2+ influx through transient receptor potential (TRP) channels have all been demonstrated to activate ClCa currents in SMCs [9, 64–66]. Some of these regulatory mechanisms appear to be cell type-specific, as blockers of nonselective cation channels but not VDCCs inhibited ClCa currents in cerebral artery SMCs [9]. In contrast, Ca2+ entry through VDCCs activated ClCa currents in rat portal vein and rabbit coronary artery SMCs [64, 92]. Extracellular Ca2+ removal had no immediate effect on ClCa currents in pig aorta and rabbit ear artery and portal vein SMCs, suggesting that external Ca2+ was not a primary direct source for activation [2, 26, 129].
Studies illustrating that Ca2+ sparks activate STICs in rabbit portal vein, rat coronary artery, and tracheal SMCs provide direct evidence that intracellular Ca2+ release can activate ClCa channels, at least in some SMC types [40, 51, 128, 151]. However, STICs do not occur in many SMC types, including those that generate Ca2+ sparks and express ClCa channels. These findings indicate that some SMC types locate ClCa channels in close proximity to sites of intracellular Ca2+ release and, more specifically, nearby RyR channels that generate Ca2+ sparks [53]. Such organization permits local control of ClCa channel activity. In contrast, other SMC types appear to position ClCa channels away from Ca2+ spark sites, eliminating this regulatory mechanism.
Bestrophins, CLCs, CLCAs, and a tweety-3 homolog have been proposed to generate ClCa currents[66]. Tweety appeared to be an unlikely candidate due to its relatively high conductance [113]. Similarly, recombinant CLCA channels generate currents that were kinetically distinct from ClCa currents in SMCs [66]. The voltage dependence of recombinant bestrophins or CLCs was also dissimilar to those of SMC ClCa currents [45, 87, 111]. Recently discovered TMEM16A/ANO1 channels displayed properties similar to native ClCa channels [10, 105, 141]. TMEM16A/ANO1 channel message and protein have been described in rat cerebral, pulmonary and carotid artery, murine portal vein, and cultured rat pulmonary artery SMCs [21, 72, 117]. Evidence supporting the contribution of TMEM16A/ANO1 channels to ClCa currents includes the fact that recombinant channels and native SMC ClCa currents exhibit similar Ca2+ dependence and IV linearization by an elevation in [Ca2+]i (Fig. 2) [10, 72, 82, 106, 117]. TMEM16A/ANO1 knockdown reduced ClCa current density in rat cerebral artery and cultured pulmonary artery SMCs [72, 117]. Cell swelling and membrane stretch activated TMEM16A/ANO1 currents in cerebral artery SMCs [9]. Selective TMEM16A/ANO1 knockdown attenuated intravascular pressure-induced cerebral artery depolarization and vasoconstriction [9]. T16Ainh-A01, a TMEM16A/ANO1 inhibitor, relaxed methoxamine-contracted murine and human blood vessels, suggesting that agonists can activate these ion channels to induce contraction [22]. These studies provide strong evidence that TMEM16A/ANO1 channels generate classic ClCa currents in SMCs.

Original electrophysiological recordings of recombinant TMEM16A/ANO1 and SMC ClCa currents. Whole-cell currents of TMEM16A-expressing HEK-293 cells in different free [Ca2+]i [106]. Reproduced with permission from [106]. © the Biochemical Society. Whole-cell recordings of Cl− currents in cerebral artery SMCs with 200 nM and 1 μM free [Ca2+]i (adapted from ref. [117])
TMEM16A/ANO1 channels also appear to generate functional ClCa currents in nonvascular SMCs. TMEM16A/ANO1 is expressed in sheep, rat, and mice urethral SMCs [103]. Electronic field stimulation (EFS)- and NE-induced uterine contractions were inhibited by NFA and exposure to Cl−-free Krebs solution [103]. The authors suggested that TMEM16A/ANO1 regulates the development and maintenance of excitatory contractile responses in urethral SMCs [103]. TMEM16A/ANO1 is expressed in airway SMCs, and activation contributes to methacholine-induced contraction [146]. Benzbromarone, a TMEM16A/ANO1 blocker, inhibited methacholine-induced contraction of mouse and human airway SMCs [50]. TMEM16A/ANO1 is also expressed in interstitial cells of Cajal (ICC), which control SMC contraction and induce rhythmic slow waves in the gastrointestinal tract [38, 49, 52, 104]. In TMEM16A knockout mice, rhythmic contractions are reduced or absent in gastric and small intestine SMCs [49, 52].
Recent studies suggest that alterations in TMEM16A/ANO1 function contribute to cardiovascular pathology. ClCa currents were elevated in pulmonary artery SMCs of rats exposed to hypoxia for 7 days [70]. TMEM16A/ANO1 mRNA/protein and ClCa currents were elevated in pulmonary artery SMCs of rats with chronic hypoxic pulmonary hypertension (CHPH) [112]. ClCa currents and TMEM16A/ANO1 expression were also increased in conduit and intralobar pulmonary artery SMCs from monocrotaline (MCT)-treated rats, another pulmonary hypertension model [32]. NFA and T16Ainh-A01 both attenuated an elevation in serotonin-induced vasocontraction in pulmonary arteries from both CHPH and MCT rats [32, 112]. In contrast, TMEM16A/ANO1 protein and ClCa currents were both lower in basilar artery SMCs isolated from 2-kidney, 2-clip renohypertensive (2k2c)-rats [133]. The authors concluded that TMEM16A/ANO1 is a negative regulator of cell proliferation and may be important in hypertension-induced cerebrovascular remodeling.
In an ovalbumin (OVA)-sensitized mouse model of chronic asthma, TMEM16A/ANO1 expression was higher, suggesting contribution to airway hyperresponsiveness [146]. NFA and benzbromarone prevented airway hyperresponsiveness and augmented airway SMC contraction. Agonist-mediated contraction was also attenuated in airway SMCs of TMEM16A/ANO1−/− mice [146]. An increase in TMEM16A protein expression and ClCa channel activity was observed in asthmatic mouse models and human asthmatic patients, although this increase in protein was primarily observed in epithelial, not smooth muscle, cells [50].
In summary, studies suggest that TMEM16A/ANO1 channels generate ClCa currents, and activation leads to membrane depolarization and constriction in both vascular and nonvascular SMCs. Diseases are associated with altered TMEM16A/ANO1 expression and functionality, with differential changes described that may depend on multiple factors, including the pathology involved.
cGMP-dependent ClCa channels
A ClCa current distinct from classic ClCa that requires cGMP for Ca2+ activation was initially discovered in rat mesenteric artery SMCs [93]. Subsequently, this current has been described in multiple vascular and colonic SMCs [55, 73, 74]. cGMP-dependent ClCa currents are voltage-independent and require lower [Ca2+]i for activation than classic ClCa currents [74, 94]. Halide permeability is also different to classic ClCa currents, at Br− > I− > Cl− [74, 94]. cGMP-dependent ClCa currents are highly sensitive to Zn2+ and relatively insensitive to both NFA and DIDS, effective classic ClCa blockers [73]. cGMP-dependent and classic ClCa current densities are approximately equal in SMCs from many vascular beds, although deviations from this stereotype have been described [74].
cGMP-dependent ClCa currents should induce membrane depolarization and vasoconstriction. Such an effect is counterintuitive to the recognized actions of cGMP-mediated PKG activation, which activates several K+ channels, including BKCa, leading to membrane hyperpolarization and relaxation [73, 116]. Conceivably, cGMP-dependent ClCa currents act as a break to oppose the cGMP-mediated vasodilation, permitting an additional level of fine tuning of membrane potential and contractility.
The molecular identity of cGMP-dependent Cl− channels is unclear, but bestrophins, a family of four proteins (1 through 4), can control this current. Cl− currents generated by recombinant bestrophins are Ca2+-activated, but do not resemble those of classical ClCa (Fig. 3) [4, 13, 97, 111]. Bestrophin-3 mRNA and protein are present in rat mesenteric arteries, rat aorta, and cultured A7r5 cells [75]. In contrast, bestrophin-1 and bestrophin-2 are weakly expressed in these tissues [75]. In line with these observations, studies have focused primarily on identifying physiological functions of bestrophin-3 in SMCs [8]. Bestrophin-3 is found in rabbit, but not rat, pulmonary arteries suggesting species-specific expression [66]. The presence of bestrophin protein has been described to match that of cGMP-dependent ClCa currents in SMCs. Bestrophin-3 knockdown reduced cGMP-dependent ClCa currents in cultured A7r5 cells and rat mesenteric artery SMCs, but did not alter classic ClCa currents [75]. Vasomotion in rat mesenteric arteries was reported to have a strong Cl− dependency that required cGMP [5, 93]. Replacement of extracellular Cl− with less permeable aspartate inhibited vasomotion in rat mesenteric arteries [5]. Consistent with a role for bestrophins, bestrophin-3 knockdown reduced synchronized vasomotion, but not tonic contractility, in rat mesenteric arteries [8]. ClCa current has not been uniformly observed after bestrophin-3 expression in heterologous expression systems; therefore, it is unclear whether the protein forms a prototypical ion channel or is an accessory subunit [88, 96].

Recombinant bestrophin-3 and SMC cGMP-dependent ClCa currents. Whole-cell mBest3 currents expressed in COS-7 cells at a [Ca2+]i of 500 nM [88]. Adapted with permission from ref. [88]. © the American Physiological Society (APS). Whole-cell niflumic acid (NFA)-insensitive cGMP-dependent ClCa current recorded in a mesenteric artery SMC [5]. Adapted with kind permission from Springer Science+Business Media: (ref. [5], Fig. 7)
In addition to regulating vasomotion, bestrophin-3 has been demonstrated to inhibit H2O2-induced apoptosis in basilar artery SMCs [55]. Bestrophin-3 knockdown reduced cell viability, whereas bestrophin-3 overexpression prevented apoptosis. Supporting a protective role, bestrophin-3 overexpression reduced ER stress-induced cell death in cultured renal epithelial cells [67].
In summary, both cGMP-dependent and cGMP-independent ClCa currents have been observed in vascular SMCs [74]. Data indicate that two distinct ClCa channels generate these currents, including that bestrophin-3 tissue distribution closely matches that of cGMP-dependent ClCa currents [75]. The majority of research on bestrophins in SMCs has been in mesenteric arteries. Future studies should investigate bestrophin functions in other vascular beds and whether bestrophins form a prototypical ion channel or an accessory subunit to another ion channel protein. Although bestrophin-3 locates near the cell surface in mesenteric artery SMCs, other bestrophin family members (bestrophin-1 and bestrophin-2) are intracellular proteins when expressed in heterologous expression systems [8, 61, 97]. Conceivably, in SMCs of different vascular beds, other bestrophin proteins may be expressed and perform additional physiological functions.
Volume-sensitive Cl− channels
In many cell types, cell swelling stimulates compensatory K+, Cl−, and H2O efflux as a mechanism to reestablish cell volume [31]. Volume-sensitive Cl− channels are expressed in many cell types, including vascular SMCs, and appear to contribute to this process [42]. Although controversy exists as to whether Cl− channel-3 (ClC-3), a member of the ClCn gene family, operates as a prototypical ion channel, this protein has been proposed to act as a volume-sensitive Cl− channel (Fig. 4) [54]. Currently, ClC-3 is the only molecular candidate for a volume-sensitive Cl− channel in SMCs. Therefore, evidence supporting ClC-3 will be summarized in this section.

Recombinant ClC-3 and SMC volume-regulated Cl− currents. Osmotic regulation of whole-cell currents recorded from gpClC3-transfected NIH/3 T3 cells under isotonic, hypotonic, and hypertonic conditions [28]. Adapted by permission from Macmillan Publishers Ltd: ref. [28]. Volume regulation of whole-cell currents recorded from A10 vascular SMCs under similar conditions [149]. Reproduced with permission from ref. [149]. © 2008 The American Society for Biochemistry and Molecular Biology. All rights reserved
ClC-3 message was detected in canine pulmonary and renal artery SMCs [140]. Hypotonic solution activated an outwardly rectifying Cl− conductance with a similar phenotype to cardiac myocyte ClC-3, including anion permeability and inhibition by DIDS and extracellular ATP [28, 140]. Similar data were obtained when studying cultured human aortic and coronary artery vascular SMCs and isolated canine pulmonary artery and colonic SMCs [25, 29, 63]. ClC-3 overexpression elevates volume-regulated Cl− currents in aortic SMCs [76]. Intracellular dialysis of ClC-3 antibodies abolished volume-activated Cl− currents in canine pulmonary artery SMCs [127]. ClC-3 knockdown inhibited volume-sensitive Cl− currents in A10 vascular SMCs [126, 149]. PKC activators differentially regulate swelling-activated Cl− currents in rabbit portal vein versus canine pulmonary artery SMCs and cardiac myocytes, an effect that may be attributed to differences in intracellular signaling pathways involved [148]. ClC-3 expression and volume-sensitive Cl− currents were larger in femoral artery than vein SMCs, perhaps due to differences in venous and arterial blood pressures to which these vessels are exposed [56].
Other evidence questions whether ClC-3 acts as a volume-sensitive Cl− channel in SMCs. ClC-3 expression in Xenopus oocytes and HEK-293 cells did not produce volume-sensitive Cl− currents, suggesting that results may be cell type-dependent [33, 76, 109]. When expressed in immortalized cell lines, ClC-3 was an intracellular channel that was not volume-regulated [69, 89, 135]. There is also variability in the contribution of ClC-3 to ClCa currents in different cell types. For example, ClC-3 knockout reduced ClCa currents in aortic SMCs, but had no effect in parotid acinar cells [3, 36]. Cell-specific differences may arise due to variability in CaMKII activation, as ClC-3 regulation is CaMKII-dependent in aortic SMCs [36]. Further uncertainty derives from data indicating that volume-sensitive Cl− currents in pulmonary artery SMCs and other cell types, including cardiac myocytes, are unaltered in ClC-3 knockout (Clcn3−/−) mice [3, 39, 110, 132, 139]. One explanation for this finding may be that ClC-3 knockout leads to compensatory upregulation of other volume-regulated ion channels [139]. Consistent with this concept, mRNA for ClC-1 and ClC-2, but not ClC-4 or ClC-5, is elevated in Clcn3−/− mice atrial myocytes [139].
Volume-regulated Cl− channels may depend on an association between ClC-3 and NADPH oxidase(Nox)-dependent reactive oxygen species (ROS) signaling in SMCs [76]. ClC-3 locates to membrane of organelles, including endosomes, where it regulates Nox1-mediated ROS generation [43, 80]. ClC-3 acts as a Cl−/H+ exchanger that neutralizes electron flow generated by Nox1 [80]. SMCs from ClC-3−/− mice did not generate endosomal ROS or activate transcription factor nuclear factor (NF)-κB in response to tumor necrosis factor (TNF)-α and interleukin (IL)-1β [80]. As a result, volume-regulated Cl− current was not activated by TNF-α and IL-1β in ClC-3−/− mice [76].
Evidence has been provided that ClC channels control SMC function. In pig artery SMCs, ClC-2 knockdown suppressed IGF-1-induced proliferation [12]. ClC-3 knockdown inhibited endothelin-1 (ET-1)-induced aortic SMC proliferation by arresting the cell cycle [115, 131]. Aortic SMCs from Clcn3−/− mice proliferated more slowly than those from wild-type controls [80]. TNF-α and carotid artery injury both stimulated ClC-3 expression with injury-induced carotid artery neointima formation reduced in Clcn3−/− mice [15]. ClC-3 overexpression inhibited apoptosis in pulmonary artery SMCs [18].
ClC-3 is associated with changes in SMC function during disease. A hypotonicity-induced decrease in [Cl−]i and an increase in rat basilar artery SMC size correlated with hypertension in 2k2c rats, suggesting that volume-sensitive Cl− channels are more active and may be involved in vascular remodeling [107]. ClC-3 mRNA and protein were both elevated in pulmonary artery SMCs of rats with experimentally induced pulmonary hypertension [18]. Static pressure stimulated ClC-3 expression, volume-sensitive Cl− currents, and proliferation in aortic SMCs, and these changes were attenuated by Cl− channel blockers and ClC-3 knockdown [95]. Ca2+-independent Cl− currents, but not ClCa currents, were larger in proliferating pulmonary artery SMCs from rats exposed to hypoxia, suggesting that antagonists of this current may be useful in the treatment of pulmonary hypertension [70]. Volume-sensitive Cl− currents increased as femoral artery SMCs switched from a contractile to a proliferative state during vascular remodeling [56]. ClC-3 mRNA and protein were higher in aortic SMCs of diabetic rats than controls, suggesting that the channel may be associated with pathology [34]. Although the contribution of ClC-3 to volume-regulated Cl− currents is controversial and requires additional study, ClC-3 may represent a therapeutic target in SMC-associated diseases, including during proliferative vascular disease.
In summary, whether ClC-3 generates volume-sensitive Cl− channels in vascular SMCs is controversial. It is unclear whether ClC-3 is located primarily intracellular or in the plasma membrane. This uncertainty arises, in part, due to the presence of swelling-activated Cl− currents in the cells of Clcn3−/− mice [3, 76]. However, ClC-3 is expressed in SMCs and both knockdown and knockout result in physiological changes [140]. ClC-3 expression levels are also altered in disease states. Further studies are required to determine SMC CLC-3 cellular localization and whether ClC-3 is a Cl− channel or an accessory protein.
CFTR
The cystic fibrosis transmembrane conductance regulator (CFTR), a cAMP-activated ATP-gated anion channel, has primarily been studied in epithelial cells, where it was originally identified [98]. CFTR channels have subsequently been found in a number of other cell types, including neurons, cardiac myocytes, and endothelial cells [35, 119, 136]. CFTR functions in SMCs were initially proposed from experiments using highly nonspecific pharmacological modulators [23, 68, 147]. Subsequent studies using immunofluorescence and Western blotting demonstrated CFTR expression in rat thoracic aorta and intrapulmonary artery [101, 102]. cAMP pathway and CFTR activators both activated iodide efflux in cultured vascular SMCs and relaxed precontracted, depolarized endothelium-denuded aortic and intrapulmonary artery rings via a mechanism sensitive to CFTRinh-172, a more selective CFTR blocker [100, 102]. cAMP pathway agonists and pharmacological CFTR activators stimulated iodide efflux in depolarized cultured aortic SMCs of wild-type mice, but not in cells of CFTR−/− mice [100]. Vasoconstrictors also contracted aortic rings from CFTR−/− mice more than those from CFTR+/+ mice [100]. These studies suggested that stimulation of the cAMP pathway and CFTR activation were functional when the SMC membrane potential was more positive than the E Cl. Under this condition, CFTR channel activation appears to oppose vasoconstriction. A study demonstrating that myogenic tone is enhanced in both CFTR−/− cerebral and mesenteric arteries supports the concept that CFTR activation hyperpolarizes membrane potential [77].
CFTR is also expressed in nonvascular SMCs [78, 122]. cAMP pathway agonists and CFTR activators stimulated iodide efflux and induced CFTRinh-172-sensitive relaxation of tracheal SMCs [122]. CFTR knockdown attenuated histamine-induced intracellular Ca2+ release in airway SMCs [78]. CFTR−/− mice also exhibit ileal SMC phenotypes that vary when studied on different mouse strains [99]. Furthermore, CFTR channel knockout results in small intestine circular smooth muscle dysfunction 7 days postnatal in mice [24].
SMC dysfunction, including bronchoconstriction, airway hyperresponsiveness, gastric dysmotility, and intestinal obstruction, may contribute to the cystic fibrosis disease phenotype [78]. Thus, CFTR modulators may have therapeutic benefit by acting on airway SMCs. Conceivably, CFTR activators may also have antihypertensive actions, although many questions still remain regarding function in SMCs. CFTR knockout may induce many different compensatory mechanisms that could modify contractility. Conceivably, CFTR may regulate other Cl− channels in vascular SMCs. CFTR expression inhibits both volume-sensitive Cl− and ClCa current in bovine pulmonary artery endothelial cells, and upregulation of its expression results in a corresponding downregulation in both channels in recombinant cells [60, 123, 134]. Whether similar regulating mechanisms exist in SMCs is unclear, but possible.
Importantly, CFTR channels have not been directly measured in SMCs using electrophysiological techniques, including patch-clamp electrophysiology. Similarly, SMC-specific inducible CFTR−/− knockout mice should be studied and systemic blood pressure measurements performed. Such data would provide stronger support for physiological functions of vascular SMC CFTR.
Conclusions
Research has focused primarily on discovering the molecular identity, physiological functions, and pathological significance of cation channels expressed in SMCs. In contrast, little is known of anion channels, specifically Cl− channels that are expressed in SMCs. This knowledge gap has arisen, in part, due to a lack of specific Cl− channel modulators and uncertain molecular identity of the proteins present. Recent discoveries of TMEM16A/ANO1, bestrophin, ClC-3, and CFTR expression in SMCs have provided new insights (Fig. 5). Identification of these proteins has permitted the use of molecular biology techniques to inhibit Cl− channel expression and study the effects on SMC function. Evidence suggests that multiple Cl− channel types are expressed in SMCs. These channels can control physiological functions, including contractility and proliferation, and can contribute to SMC pathologies.

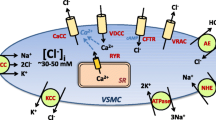
Cl− channels present in vascular SMCs. Cl− accumulates in SMCs due to the Na+–K+–Cl− cotransporter (NKCC1) and the Cl−-HO3 − exchanger-2 (AE2). cGMP-dependent and independent ClCa channels, a volume-sensitive Cl− channel, and the cystic fibrosis transmembrane conductance regulator (CFTR) have been identified. The molecular identity of the first three channels has been proposed to be bestrophin, TMEM16A/Ano1, and ClC-3, respectively. Numerous mechanisms of Ca2+ activation of ClCa channels in vascular SMCs have been suggested, including IP3R- or RyR-mediated SR Ca2+ release, Ca2+ entry through voltage-dependent Ca2+ channels (VDCC), and local Ca2+ influx through nonselective cation channels (NSCC). Activation of these channels leads to Cl− efflux and subsequent depolarization of the cell membrane that activates voltage-dependent Ca2+ channels (VDCC). ClC-3 channels have been proposed to be activated by membrane swelling. ClC-3 is present in the plasma membrane and in intracellular compartments, including endosomes. Endosomal ClC-3 channels may regulate volume-regulated Cl− channels via ROS production. CaMKII inhibits TMEM16A and activates ClC-3 channels. CFTR is a cAMP-activated ATP-gated anion channel that appears to be functional when the SMC membrane potential becomes more positive than the Cl− equilibrium potential. Under this condition, CFTR channel activation would lead to Cl− influx and oppose vasoconstriction
Future directions
Future studies should aim to identify intracellular signaling pathways that regulate different Cl− channels in SMCs and downstream functional effects of such modulation. Many ion channels have one or more auxiliary and regulatory subunits, and these proteins can, in some cases, exhibit SMC-specific expression (e.g., KCa channel β1 subunits [7]). It is possible that Cl− channels have auxiliary subunits, although this remains to be determined. Similarly, whether some proteins identified are pore-forming Cl− channels or accessory subunits is unclear, including some bestrophins and ClC proteins. Similarly, different Cl− channels may interact and regulate each other directly, for example through heteromultimer formation, and indirectly, via signaling networks. Many of these research directions will benefit from the discovery of specific Cl− channel modulators and animals with inducible, SMC-specific genetic alterations of the proteins under investigation. The next decade should see a significant increase in knowledge of Cl− channel signaling, physiology, and pathology in SMCs.
References
Aickin CC, Vermue NA (1983) Microelectrode measurement of intracellular chloride activity in smooth muscle cells of guinea-pig ureter. Pflugers Arch 397:25–28
Amedee T, Large WA, Wang Q (1990) Characteristics of chloride currents activated by noradrenaline in rabbit ear artery cells. J Physiol 428:501–516
Arreola J, Begenisich T, Nehrke K, Nguyen HV, Park K, Richardson L, Yang B, Schutte BC, Lamb FS, Melvin JE (2002) Secretion and cell volume regulation by salivary acinar cells from mice lacking expression of the Clcn3 Cl − channel gene. J Physiol 545:207–216
Barro SR, Spitzner M, Schreiber R, Kunzelmann K (2009) Bestrophin-1 enables Ca2 + -activated Cl − conductance in epithelia. J Biol Chem 284:29405–29412
Boedtkjer DM, Matchkov VV, Boedtkjer E, Nilsson H, Aalkjaer C (2008) Vasomotion has chloride-dependency in rat mesenteric small arteries. Pflugers Arch 457:389–404
Brayden JE, Nelson MT (1992) Regulation of arterial tone by activation of calcium-dependent potassium channels. Science 256:532–535
Brenner R, Perez GJ, Bonev AD, Eckman DM, Kosek JC, Wiler SW, Patterson AJ, Nelson MT, Aldrich RW (2000) Vasoregulation by the beta1 subunit of the calcium-activated potassium channel. Nature 407:870–876
Broegger T, Jacobsen JC, Secher DV, Boedtkjer DM, Kold-Petersen H, Pedersen FS, Aalkjaer C, Matchkov VV (2011) Bestrophin is important for the rhythmic but not the tonic contraction in rat mesenteric small arteries. Cardiovasc Res 91:685–693
Bulley S, Neeb ZP, Burris SK, Bannister JP, Thomas-Gatewood CM, Jangsangthong W, Jaggar JH (2012) TMEM16A/ANO1 channels contribute to the myogenic response in cerebral arteries. Circ Res 111:1027–1036
Caputo A, Caci E, Ferrera L, Pedemonte N, Barsanti C, Sondo E, Pfeffer U, Ravazzolo R, Zegarra-Moran O, Galietta LJ (2008) TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science 322:590–594
Casteels R, Kitamura K, Kuriyama H, Suzuki H (1977) The membrane properties of the smooth muscle cells of the rabbit main pulmonary artery. J Physiol 271:41–61
Cheng G, Kim MJ, Jia G, Agrawal DK (2007) Involvement of chloride channels in IGF-I-induced proliferation of porcine arterial smooth muscle cells. Cardiovasc Res 73:198–207
Chien LT, Zhang ZR, Hartzell HC (2006) Single Cl − channels activated by Ca2+ in Drosophila S2 cells are mediated by bestrophins. J Gen Physiol 128:247–259
Chipperfield AR, Harper AA (2000) Chloride in smooth muscle. Prog Biophys Mol Biol 74:175–221
Chu X, Filali M, Stanic B, Takapoo M, Sheehan A, Bhalla R, Lamb FS, Miller FJ Jr (2011) A critical role for chloride channel-3 (CIC-3) in smooth muscle cell activation and neointima formation. Arterioscler Thromb Vasc Biol 31:345–351
Criddle DN, de Moura RS, Greenwood IA, Large WA (1996) Effect of niflumic acid on noradrenaline-induced contractions of the rat aorta. Br J Pharmacol 118:1065–1071
Criddle DN, de Moura RS, Greenwood IA, Large WA (1997) Inhibitory action of niflumic acid on noradrenaline- and 5-hydroxytryptamine-induced pressor responses in the isolated mesenteric vascular bed of the rat. Br J Pharmacol 120:813–818
Dai YP, Bongalon S, Hatton WJ, Hume JR, Yamboliev IA (2005) ClC-3 chloride channel is upregulated by hypertrophy and inflammation in rat and canine pulmonary artery. Br J Pharmacol 145:5–14
Dai Y, Zhang JH (2001) Role of Cl − current in endothelin-1-induced contraction in rabbit basilar artery. Am J Physiol Heart Circ Physiol 281:H2159–H2167
Dai Y, Zhang JH (2002) Manipulation of chloride flux affects histamine-induced contraction in rabbit basilar artery. Am J Physiol Heart Circ Physiol 282:H1427–H1436
Davis AJ, Forrest AS, Jepps TA, Valencik ML, Wiwchar M, Singer CA, Sones WR, Greenwood IA, Leblanc N (2010) Expression profile and protein translation of TMEM16A in murine smooth muscle. Am J Physiol Cell Physiol 299:C948–C959
Davis AJ, Shi J, Pritchard HA, Chadha PS, Leblanc N, Vasilikostas G, Yao Z, Verkman AS, Albert AP, Greenwood IA (2013) Potent vasorelaxant activity of the TMEM16A inhibitor T16A(inh) -A01. Br J Pharmacol 168:773–784
Day RM, Agyeman AS, Segel MJ, Chevere RD, Angelosanto JM, Suzuki YJ, Fanburg BL (2006) Serotonin induces pulmonary artery smooth muscle cell migration. Biochem Pharmacol 71:386–397
De Lisle RC, Meldi L, Mueller R (2012) Intestinal smooth muscle dysfunction develops postnatally in cystic fibrosis mice. J Pediatr Gastroenterol Nutr 55:689–694
Dick GM, Bradley KK, Horowitz B, Hume JR, Sanders KM (1998) Functional and molecular identification of a novel chloride conductance in canine colonic smooth muscle. Am J Physiol 275:C940–C950
Droogmans G, Callewaert G, Declerck I, Casteels R (1991) ATP-induced Ca2+ release and Cl − current in cultured smooth muscle cells from pig aorta. J Physiol 440:623–634
Duan DD (2011) The ClC-3 chloride channels in cardiovascular disease. Acta Pharmacol Sin 32:675–684
Duan D, Winter C, Cowley S, Hume JR, Horowitz B (1997) Molecular identification of a volume-regulated chloride channel. Nature 390:417–421
Duan D, Zhong J, Hermoso M, Satterwhite CM, Rossow CF, Hatton WJ, Yamboliev I, Horowitz B, Hume JR (2001) Functional inhibition of native volume-sensitive outwardly rectifying anion channels in muscle cells and Xenopus oocytes by anti-ClC-3 antibody. J Physiol 531:437–444
Duran C, Thompson CH, Xiao Q, Hartzell HC (2010) Chloride channels: often enigmatic, rarely predictable. Annu Rev Physiol 72:95–121
Eggermont J, Trouet D, Carton I, Nilius B (2001) Cellular function and control of volume-regulated anion channels. Cell Biochem Biophys 35:263–274
Forrest AS, Joyce TC, Huebner ML, Ayon RJ, Wiwchar M, Joyce J, Freitas N, Davis AJ, Ye L, Duan DD, Singer CA, Valencik ML, Greenwood IA, Leblanc N (2012) Increased TMEM16A-encoded calcium-activated chloride channel activity is associated with pulmonary hypertension. Am J Physiol Cell Physiol 303:C1229–C1243
Friedrich T, Breiderhoff T, Jentsch TJ (1999) Mutational analysis demonstrates that ClC-4 and ClC-5 directly mediate plasma membrane currents. J Biol Chem 274:896–902
Fu J, Fu J, Gao B, Wang L, Zhang N, Li X, Ji Q (2010) Expression patterns of ClC-3 mRNA and protein in aortic smooth muscle, kidney and brain in diabetic rats. Clin Invest Med 33:E146–E154
Gadsby DC, Nairn AC (1999) Regulation of CFTR Cl − ion channels by phosphorylation and dephosphorylation. Adv Second Messenger Phosphoprotein Res 33:79–106
Ganapathi SB, Wei SG, Zaremba A, Lamb FS, Shears SB (2013) Functional regulation of ClC-3 in the migration of vascular smooth muscle cells. Hypertension 61:174–179
Gokina NI, Bevan JA (2000) Histamine-induced depolarization: ionic mechanisms and role in sustained contraction of rabbit cerebral arteries. Am J Physiol Heart Circ Physiol 278:H2094–H2104
Gomez-Pinilla PJ, Gibbons SJ, Bardsley MR, Lorincz A, Pozo MJ, Pasricha PJ, Van de Rijn M, West RB, Sarr MG, Kendrick ML, Cima RR, Dozois EJ, Larson DW, Ordog T, Farrugia G (2009) Ano1 is a selective marker of interstitial cells of Cajal in the human and mouse gastrointestinal tract. Am J Physiol Gastrointest Liver Physiol 296:G1370–G1381
Gong W, Xu H, Shimizu T, Morishima S, Tanabe S, Tachibe T, Uchida S, Sasaki S, Okada Y (2004) ClC-3-independent, PKC-dependent activity of volume-sensitive Cl channel in mouse ventricular cardiomyocytes. Cell Physiol Biochem 14:213–224
Greenwood IA, Helliwell RM, Large WA (1997) Modulation of Ca(2+)-activated Cl − currents in rabbit portal vein smooth muscle by an inhibitor of mitochondrial Ca2+ uptake. J Physiol 505(Pt 1):53–64
Greenwood IA, Large WA (1999) Modulation of the decay of Ca2 + -activated Cl − currents in rabbit portal vein smooth muscle cells by external anions. J Physiol 516(Pt 2):365–376
Guan YY, Wang GL, Zhou JG (2006) The ClC-3 Cl − channel in cell volume regulation, proliferation and apoptosis in vascular smooth muscle cells. Trends Pharmacol Sci 27:290–296
Hara-Chikuma M, Yang B, Sonawane ND, Sasaki S, Uchida S, Verkman AS (2005) ClC-3 chloride channels facilitate endosomal acidification and chloride accumulation. J Biol Chem 280:1241–1247
Harder DR (1984) Pressure-dependent membrane depolarization in cat middle cerebral artery. Circ Res 55:197–202
Hartzell C, Putzier I, Arreola J (2005) Calcium-activated chloride channels. Annu Rev Physiol 67:719–758
Hill MA, Zou H, Potocnik SJ, Meininger GA, Davis MJ (2001) Invited review: arteriolar smooth muscle mechanotransduction: Ca(2+) signaling pathways underlying myogenic reactivity. J Appl Physiol 91:973–983
Hodgkin AL, Horowicz P (1959) The influence of potassium and chloride ions on the membrane potential of single muscle fibres. J Physiol 148:127–160
Hogg RC, Wang Q, Large WA (1994) Effects of Cl channel blockers on Ca-activated chloride and potassium currents in smooth muscle cells from rabbit portal vein. Br J Pharmacol 111:1333–1341
Huang F, Rock JR, Harfe BD, Cheng T, Huang X, Jan YN, Jan LY (2009) Studies on expression and function of the TMEM16A calcium-activated chloride channel. Proc Natl Acad Sci U S A 106:21413–21418
Huang F, Zhang H, Wu M, Yang H, Kudo M, Peters CJ, Woodruff PG, Solberg OD, Donne ML, Huang X, Sheppard D, Fahy JV, Wolters PJ, Hogan BL, Finkbeiner WE, Li M, Jan YN, Jan LY, Rock JR (2012) Calcium-activated chloride channel TMEM16A modulates mucin secretion and airway smooth muscle contraction. Proc Natl Acad Sci U S A 109:16354–16359
Hume JR, Leblanc N (1989) Macroscopic K + currents in single smooth muscle cells of the rabbit portal vein. J Physiol 413:49–73
Hwang SJ, Blair PJ, Britton FC, O'Driscoll KE, Hennig G, Bayguinov YR, Rock JR, Harfe BD, Sanders KM, Ward SM (2009) Expression of anoctamin 1/TMEM16A by interstitial cells of Cajal is fundamental for slow wave activity in gastrointestinal muscles. J Physiol 587:4887–4904
Jaggar JH, Porter VA, Lederer WJ, Nelson MT (2000) Calcium sparks in smooth muscle. Am J Physiol Cell Physiol 278:C235–C256
Jentsch TJ, Friedrich T, Schriever A, Yamada H (1999) The CLC chloride channel family. Pflugers Arch 437:783–795
Jiang L, Liu Y, Ma MM, Tang YB, Zhou JG, Guan YY (2013) Mitochondria dependent pathway is involved in the protective effect of bestrophin-3 on hydrogen peroxide-induced apoptosis in basilar artery smooth muscle cells. Apoptosis 18:556–565
Kang XL, Zhang M, Liu J, Lv XF, Tang YB, Guan YY (2012) Differences between femoral artery and vein smooth muscle cells in volume-regulated chloride channels. Can J Physiol Pharmacol 90:1516–1526
Kitamura K, Yamazaki J (2001) Chloride channels and their functional roles in smooth muscle tone in the vasculature. Jpn J Pharmacol 85:351–357
Klockner U (1993) Intracellular calcium ions activate a low-conductance chloride channel in smooth-muscle cells isolated from human mesenteric artery. Pflugers Arch 424:231–237
Klockner U, Isenberg G (1991) Endothelin depolarizes myocytes from porcine coronary and human mesenteric arteries through a Ca-activated chloride current. Pflugers Arch 418:168–175
Kunzelmann K, Allert N, Kubitz R, Breuer WV, Cabantchik ZI, Normann C, Schumann S, Leipziger J, Greger R (1994) Forskolin and PMA pretreatment of HT29 cells alters their chloride conductance induced by cAMP, Ca2+ and hypotonic cell swelling. Pflugers Arch 428:76–83
Kunzelmann K, Milenkovic VM, Spitzner M, Soria RB, Schreiber R (2007) Calcium-dependent chloride conductance in epithelia: is there a contribution by Bestrophin? Pflugers Arch 454:879–889
Lamb FS, Barna TJ (1998) Chloride ion currents contribute functionally to norepinephrine-induced vascular contraction. Am J Physiol 275:H151–H160
Lamb FS, Clayton GH, Liu BX, Smith RL, Barna TJ, Schutte BC (1999) Expression of CLCN voltage-gated chloride channel genes in human blood vessels. J Mol Cell Cardiol 31:657–666
Lamb FS, Volk KA, Shibata EF (1994) Calcium-activated chloride current in rabbit coronary artery myocytes. Circ Res 75:742–750
Large WA, Wang Q (1996) Characteristics and physiological role of the Ca(2+)-activated Cl − conductance in smooth muscle. Am J Physiol 271:C435–C454
Leblanc N, Ledoux J, Saleh S, Sanguinetti A, Angermann J, O'Driscoll K, Britton F, Perrino BA, Greenwood IA (2005) Regulation of calcium-activated chloride channels in smooth muscle cells: a complex picture is emerging. Can J Physiol Pharmacol 83:541–556
Lee WK, Chakraborty PK, Roussa E, Wolff NA, Thevenod F (2012) ERK1/2-dependent bestrophin-3 expression prevents ER-stress-induced cell death in renal epithelial cells by reducing CHOP. Biochim Biophys Acta 1823:1864–1876
Lee SY, Lee CO (2005) Inhibition of Na + –K + pump and L-type Ca2+ channel by glibenclamide in guinea pig ventricular myocytes. J Pharmacol Exp Ther 312:61–68
Li X, Shimada K, Showalter LA, Weinman SA (2000) Biophysical properties of ClC-3 differentiate it from swelling-activated chloride channels in Chinese hamster ovary-K1 cells. J Biol Chem 275:35994–35998
Liang W, Ray JB, He JZ, Backx PH, Ward ME (2009) Regulation of proliferation and membrane potential by chloride currents in rat pulmonary artery smooth muscle cells. Hypertension 54:286–293
Ma Z, Qi J, Fu Z, Ling M, Li L, Zhang Y (2013) Protective role of acidic pH-activated chloride channel in severe acidosis-induced contraction from the aorta of spontaneously hypertensive rats. PLoS One 8:e61018
Manoury B, Tamuleviciute A, Tammaro P (2010) TMEM16A/anoctamin 1 protein mediates calcium-activated chloride currents in pulmonary arterial smooth muscle cells. J Physiol 588:2305–2314
Matchkov VV, Aalkjaer C, Nilsson H (2004) A cyclic GMP-dependent calcium-activated chloride current in smooth-muscle cells from rat mesenteric resistance arteries. J Gen Physiol 123:121–134
Matchkov VV, Aalkjaer C, Nilsson H (2005) Distribution of cGMP-dependent and cGMP-independent Ca(2+)-activated Cl(−) conductances in smooth muscle cells from different vascular beds and colon. Pflugers Arch 451:371–379
Matchkov VV, Larsen P, Bouzinova EV, Rojek A, Boedtkjer DM, Golubinskaya V, Pedersen FS, Aalkjaer C, Nilsson H (2008) Bestrophin-3 (vitelliform macular dystrophy 2-like 3 protein) is essential for the cGMP-dependent calcium-activated chloride conductance in vascular smooth muscle cells. Circ Res 103:864–872
Matsuda JJ, Filali MS, Moreland JG, Miller FJ, Lamb FS (2010) Activation of swelling-activated chloride current by tumor necrosis factor-alpha requires ClC-3-dependent endosomal reactive oxygen production. J Biol Chem 285:22864–22873
Meissner A, Yang J, Kroetsch JT, Sauve M, Dax H, Momen A, Noyan-Ashraf MH, Heximer S, Husain M, Lidington D, Bolz SS (2012) Tumor necrosis factor-alpha-mediated downregulation of the cystic fibrosis transmembrane conductance regulator drives pathological sphingosine-1-phosphate signaling in a mouse model of heart failure. Circulation 125:2739–2750
Michoud MC, Robert R, Hassan M, Moynihan B, Haston C, Govindaraju V, Ferraro P, Hanrahan JW, Martin JG (2009) Role of the cystic fibrosis transmembrane conductance channel in human airway smooth muscle. Am J Respir Cell Mol Biol 40:217–222
Milenkovic VM, Langmann T, Schreiber R, Kunzelmann K, Weber BH (2008) Molecular evolution and functional divergence of the bestrophin protein family. BMC Evol Biol 8:72
Miller FJ Jr, Filali M, Huss GJ, Stanic B, Chamseddine A, Barna TJ, Lamb FS (2007) Cytokine activation of nuclear factor kappa B in vascular smooth muscle cells requires signaling endosomes containing Nox1 and ClC-3. Circ Res 101:663–671
Mizuta K, Xu D, Pan Y, Comas G, Sonett JR, Zhang Y, Panettieri RA Jr, Yang J, Emala CW Sr (2008) GABAA receptors are expressed and facilitate relaxation in airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 294:L1206–L1216
Namkung W, Yao Z, Finkbeiner WE, Verkman AS (2011) Small-molecule activators of TMEM16A, a calcium-activated chloride channel, stimulate epithelial chloride secretion and intestinal contraction. FASEB J 25:4048–4062
Neild TO, Keef K (1985) Measurements of the membrane potential of arterial smooth muscle in anesthetized animals and its relationship to changes in artery diameter. Microvasc Res 30:19–28
Nelson MT, Conway MA, Knot HJ, Brayden JE (1997) Chloride channel blockers inhibit myogenic tone in rat cerebral arteries. J Physiol 502(Pt 2):259–264
Nelson MT, Patlak JB, Worley JF, Standen NB (1990) Calcium channels, potassium channels, and voltage dependence of arterial smooth muscle tone. Am J Physiol 259:C3–C18
Nelson MT, Quayle JM (1995) Physiological roles and properties of potassium channels in arterial smooth muscle. Am J Physiol 268:C799–C822
Nilius B, Prenen J, Szucs G, Wei L, Tanzi F, Voets T, Droogmans G (1997) Calcium-activated chloride channels in bovine pulmonary artery endothelial cells. J Physiol 498(Pt 2):381–396
O'Driscoll KE, Hatton WJ, Burkin HR, Leblanc N, Britton FC (2008) Expression, localization, and functional properties of Bestrophin 3 channel isolated from mouse heart. Am J Physiol Cell Physiol 295:C1610–C1624
Ogura T, Furukawa T, Toyozaki T, Yamada K, Zheng YJ, Katayama Y, Nakaya H, Inagaki N (2002) ClC-3B, a novel ClC-3 splicing variant that interacts with EBP50 and facilitates expression of CFTR-regulated ORCC. FASEB J 16:863–865
Owen NE (1984) Regulation of Na/K/Cl cotransport in vascular smooth muscle cells. Biochem Biophys Res Commun 125:500–508
Pacaud P, Loirand G, Gregoire G, Mironneau C, Mironneau J (1992) Calcium-dependence of the calcium-activated chloride current in smooth muscle cells of rat portal vein. Pflugers Arch 421:125–130
Pacaud P, Loirand G, Mironneau C, Mironneau J (1989) Noradrenaline activates a calcium-activated chloride conductance and increases the voltage-dependent calcium current in cultured single cells of rat portal vein. Br J Pharmacol 97:139–146
Peng H, Matchkov V, Ivarsen A, Aalkjaer C, Nilsson H (2001) Hypothesis for the initiation of vasomotion. Circ Res 88:810–815
Piper AS, Large WA (2004) Single cGMP-activated Ca(+)-dependent Cl(−) channels in rat mesenteric artery smooth muscle cells. J Physiol 555:397–408
Qian JS, Pang RP, Zhu KS, Liu DY, Li ZR, Deng CY, Wang SM (2009) Static pressure promotes rat aortic smooth muscle cell proliferation via upregulation of volume-regulated chloride channel. Cell Physiol Biochem 24:461–470
Qu Z, Cui Y, Hartzell C (2006) A short motif in the C-terminus of mouse bestrophin 3 [corrected] inhibits its activation as a Cl channel. FEBS Lett 580:2141–2146
Qu Z, Wei RW, Mann W, Hartzell HC (2003) Two bestrophins cloned from Xenopus laevis oocytes express Ca(2+)-activated Cl(−) currents. J Biol Chem 278:49563–49572
Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL (1989) Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245:1066–1073
Risse PA, Kachmar L, Matusovsky OS, Novali M, Gil FR, Javeshghani S, Keary R, Haston CK, Michoud MC, Martin JG, Lauzon AM (2012) Ileal smooth muscle dysfunction and remodeling in cystic fibrosis. Am J Physiol Gastrointest Liver Physiol 303:G1–G8
Robert R, Norez C, Becq F (2005) Disruption of CFTR chloride channel alters mechanical properties and cAMP-dependent Cl − transport of mouse aortic smooth muscle cells. J Physiol 568:483–495
Robert R, Savineau JP, Norez C, Becq F, Guibert C (2007) Expression and function of cystic fibrosis transmembrane conductance regulator in rat intrapulmonary arteries. Eur Respir J 30:857–864
Robert R, Thoreau V, Norez C, Cantereau A, Kitzis A, Mettey Y, Rogier C, Becq F (2004) Regulation of the cystic fibrosis transmembrane conductance regulator channel by beta-adrenergic agonists and vasoactive intestinal peptide in rat smooth muscle cells and its role in vasorelaxation. J Biol Chem 279:21160–21168
Sancho M, Garcia-Pascual A, Triguero D (2012) Presence of the Ca2 + -activated chloride channel anoctamin 1 in the urethra and its role in excitatory neurotransmission. Am J Physiol Ren Physiol 302:F390–F400
Sanders KM, Koh SD, Ward SM (2006) Interstitial cells of cajal as pacemakers in the gastrointestinal tract. Annu Rev Physiol 68:307–343
Schroeder BC, Cheng T, Jan YN, Jan LY (2008) Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell 134:1019–1029
Scudieri P, Sondo E, Caci E, Ravazzolo R, Galietta LJ (2013) TMEM16A–TMEM16B chimaeras to investigate the structure-function relationship of calcium-activated chloride channels. Biochem J 452:443–455
Shi XL, Wang GL, Zhang Z, Liu YJ, Chen JH, Zhou JG, Qiu QY, Guan YY (2007) Alteration of volume-regulated chloride movement in rat cerebrovascular smooth muscle cells during hypertension. Hypertension 49:1371–1377
Smith JM, Jones AW (1985) Calcium-dependent fluxes of potassium-42 and chloride-36 during norepinephrine activation of rat aorta. Circ Res 56:507–516
Steinmeyer K, Schwappach B, Bens M, Vandewalle A, Jentsch TJ (1995) Cloning and functional expression of rat CLC-5, a chloride channel related to kidney disease. J Biol Chem 270:31172–31177
Stobrawa SM, Breiderhoff T, Takamori S, Engel D, Schweizer M, Zdebik AA, Bosl MR, Ruether K, Jahn H, Draguhn A, Jahn R, Jentsch TJ (2001) Disruption of ClC-3, a chloride channel expressed on synaptic vesicles, leads to a loss of the hippocampus. Neuron 29:185–196
Sun H, Tsunenari T, Yau KW, Nathans J (2002) The vitelliform macular dystrophy protein defines a new family of chloride channels. Proc Natl Acad Sci U S A 99:4008–4013
Sun H, Xia Y, Paudel O, Yang XR, Sham JS (2012) Chronic hypoxia-induced upregulation of Ca2 + -activated Cl − channel in pulmonary arterial myocytes: a mechanism contributing to enhanced vasoreactivity. J Physiol 590:3507–3521
Suzuki M, Mizuno A (2004) A novel human Cl(-) channel family related to Drosophila flightless locus. J Biol Chem 279:22461–22468
Takenaka T, Epstein M, Forster H, Landry DW, Iijima K, Goligorsky MS (1992) Attenuation of endothelin effects by a chloride channel inhibitor, indanyloxyacetic acid. Am J Physiol 262:F799–F806
Tang YB, Liu YJ, Zhou JG, Wang GL, Qiu QY, Guan YY (2008) Silence of ClC-3 chloride channel inhibits cell proliferation and the cell cycle via G/S phase arrest in rat basilar arterial smooth muscle cells. Cell Prolif 41:775–785
Taniguchi J, Furukawa KI, Shigekawa M (1993) Maxi K + channels are stimulated by cyclic guanosine monophosphate-dependent protein kinase in canine coronary artery smooth muscle cells. Pflugers Arch 423:167–172
Thomas-Gatewood C, Neeb ZP, Bulley S, Adebiyi A, Bannister JP, Leo MD, Jaggar JH (2011) TMEM16A channels generate Ca(2)(+)-activated Cl(−) currents in cerebral artery smooth muscle cells. Am J Physiol Heart Circ Physiol 301:H1819–H1827
Thorneloe KS, Nelson MT (2004) Properties of a tonically active, sodium-permeable current in mouse urinary bladder smooth muscle. Am J Physiol Cell Physiol 286:C1246–C1257
Tousson A, Van Tine BA, Naren AP, Shaw GM, Schwiebert LM (1998) Characterization of CFTR expression and chloride channel activity in human endothelia. Am J Physiol 275:C1555–C1564
Uchida R, Nishijima H, Yamazaki J, Shimamura K, Kitamura K (1990) Effects of Cl channel blockers on the contractions recorded in the rabbit basilar artery. Jpn J Pharmacol 79(Suppl I):210
Van RC, Lazdunski M (1993) Endothelin and vasopressin activate low conductance chloride channels in aortic smooth muscle cells. Pflugers Arch 425:156–163
Vandebrouck C, Melin P, Norez C, Robert R, Guibert C, Mettey Y, Becq F (2006) Evidence that CFTR is expressed in rat tracheal smooth muscle cells and contributes to bronchodilation. Respir Res 7:113
Vennekens R, Trouet D, Vankeerberghen A, Voets T, Cuppens H, Eggermont J, Cassiman JJ, Droogmans G, Nilius B (1999) Inhibition of volume-regulated anion channels by expression of the cystic fibrosis transmembrane conductance regulator. J Physiol 515(Pt 1):75–85
Wahlstrom BA (1973) A study on the action of noradrenaline on ionic content and sodium, potassium and chloride effluxes in the rat portal vein. Acta Physiol Scand 89:522–530
Wahlstrom BA, Svennerholm B (1974) Potentiation and inhibition of noradrenaline induced contractions of the rat portal vein in anion substituted solutions. Acta Physiol Scand 92:404–411
Wang L, Chen L, Jacob TJ (2000) The role of ClC-3 in volume-activated chloride currents and volume regulation in bovine epithelial cells demonstrated by antisense inhibition. J Physiol 524(Pt 1):63–75
Wang GX, Hatton WJ, Wang GL, Zhong J, Yamboliev I, Duan D, Hume JR (2003) Functional effects of novel anti-ClC-3 antibodies on native volume-sensitive osmolyte and anion channels in cardiac and smooth muscle cells. Am J Physiol Heart Circ Physiol 285:H1453–H1463
Wang Q, Hogg RC, Large WA (1992) Properties of spontaneous inward currents recorded in smooth muscle cells isolated from the rabbit portal vein. J Physiol 451:525–537
Wang Q, Large WA (1991) Modulation of noradrenaline-induced membrane currents by papaverine in rabbit vascular smooth muscle cells. J Physiol 439:501–512
Wang Q, Large WA (1993) Action of histamine on single smooth muscle cells dispersed from the rabbit pulmonary artery. J Physiol 468:125–139
Wang GL, Wang XR, Lin MJ, He H, Lan XJ, Guan YY (2002) Deficiency in ClC-3 chloride channels prevents rat aortic smooth muscle cell proliferation. Circ Res 91:E28–E32
Wang J, Xu H, Morishima S, Tanabe S, Jishage K, Uchida S, Sasaki S, Okada Y (2007) Single-channel properties of volume-sensitive Cl − channel in ClC-3 deficient cardiomyocytes. Jpn J Physiol 55:379–383
Wang M, Yang H, Zheng LY, Zhang Z, Tang YB, Wang GL, Du YH, Lv XF, Liu J, Zhou JG, Guan YY (2012) Downregulation of TMEM16A calcium-activated chloride channel contributes to cerebrovascular remodeling during hypertension by promoting basilar smooth muscle cell proliferation. Circulation 125:697–707
Wei L, Vankeerberghen A, Cuppens H, Eggermont J, Cassiman JJ, Droogmans G, Nilius B (1999) Interaction between calcium-activated chloride channels and the cystic fibrosis transmembrane conductance regulator. Pflugers Arch 438:635–641
Weylandt KH, Valverde MA, Nobles M, Raguz S, Amey JS, Diaz M, Nastrucci C, Higgins CF, Sardini A (2001) Human ClC-3 is not the swelling-activated chloride channel involved in cell volume regulation. J Biol Chem 276:17461–17467
Weyler RT, Yurko-Mauro KA, Rubenstein R, Kollen WJ, Reenstra W, Altschuler SM, Egan M, Mulberg AE (1999) CFTR is functionally active in GnRH-expressing GT1-7 hypothalamic neurons. Am J Physiol 277:C563–C571
White CR, Elton TS, Shoemaker RL, Brock TA (1995) Calcium-sensitive chloride channels in vascular smooth muscle cells. Proc Soc Exp Biol Med 208:255–262
Xiao GN, Guan YY, He H (2002) Effects of Cl − channel blockers on endothelin-1-induced proliferation of rat vascular smooth muscle cells. Life Sci 70:2233–2241
Yamamoto-Mizuma S, Wang GX, Liu LL, Schegg K, Hatton WJ, Duan D, Horowitz TL, Lamb FS, Hume JR (2004) Altered properties of volume-sensitive osmolyte and anion channels (VSOACs) and membrane protein expression in cardiac and smooth muscle myocytes from Clcn3−/− mice. J Physiol 557:439–456
Yamazaki J, Duan D, Janiak R, Kuenzli K, Horowitz B, Hume JR (1998) Functional and molecular expression of volume-regulated chloride channels in canine vascular smooth muscle cells. J Physiol 507(Pt 3):729–736
Yang YD, Cho H, Koo JY, Tak MH, Cho Y, Shim WS, Park SP, Lee J, Lee B, Kim BM, Raouf R, Shin YK, Oh U (2008) TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature 455:1210–1215
Yang Z, Zhang Z, Xu Y, Wang T, Ma D, Ye T (2008) Effects of calcium-activated chloride channels on proliferation of pulmonary artery smooth muscle cells in rats under chronic hypoxic condition. J Nanjing Med Univ 22:39–43
Yim PD, Gallos G, Xu D, Zhang Y, Emala CW (2011) Novel expression of a functional glycine receptor chloride channel that attenuates contraction in airway smooth muscle. FASEB J 25:1706–1717
Yu K, Duran C, Qu Z, Cui YY, Hartzell HC (2012) Explaining calcium-dependent gating of anoctamin-1 chloride channels requires a revised topology. Circ Res 110:990–999
Yuan XJ (1997) Role of calcium-activated chloride current in regulating pulmonary vasomotor tone. Am J Physiol 272:L959–L968
Zhang CH, Li Y, Zhao W, Lifshitz LM, Li H, Harfe BD, Zhu MS, ZhuGe R (2013) The transmembrane protein 16A Ca(2+)-activated Cl − channel in airway smooth muscle contributes to airway hyperresponsiveness. Am J Respir Crit Care Med 187:374–381
Zhao YJ, Wang J, Rubin LJ, Yuan XJ (1998) Roles of K + and Cl − channels in cAMP-induced pulmonary vasodilation. Exp Lung Res 24:71–83
Zhong J, Wang GX, Hatton WJ, Yamboliev IA, Walsh MP, Hume JR (2002) Regulation of volume-sensitive outwardly rectifying anion channels in pulmonary arterial smooth muscle cells by PKC. Am J Physiol Cell Physiol 283:C1627–C1636
Zhou JG, Ren JL, Qiu QY, He H, Guan YY (2005) Regulation of intracellular Cl − concentration through volume-regulated ClC-3 chloride channels in A10 vascular smooth muscle cells. J Biol Chem 280:7301–7308
ZhuGe R, Bao R, Fogarty KE, Lifshitz LM (2010) Ca2+ sparks act as potent regulators of excitation–contraction coupling in airway smooth muscle. J Biol Chem 285:2203–2210
ZhuGe R, Sims SM, Tuft RA, Fogarty KE, Walsh JV Jr (1998) Ca2+ sparks activate K + and Cl − channels, resulting in spontaneous transient currents in guinea-pig tracheal myocytes. J Physiol 513(Pt 3):711–718
Zielenski J (2000) Genotype and phenotype in cystic fibrosis. Respiration 67:117–133
Acknowledgments
We thank M. Dennis Leo and Sarah K. Burris for reading the manuscript. This work was supported by National Institutes of Health Heart, Lung and Blood grants (HL67061, HL094378, and HL110347) awarded to Jonathan H. Jaggar.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bulley, S., Jaggar, J.H. Cl− channels in smooth muscle cells. Pflugers Arch - Eur J Physiol 466, 861–872 (2014). https://doi.org/10.1007/s00424-013-1357-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-013-1357-2