
Abstract
Recent studies suggest that primary changes in vascular resistance can cause sustained changes in arterial blood pressure. In this review, we summarize current knowledge about Cl− homeostasis in vascular smooth muscle cells. Within vascular smooth muscle cells, Cl− is accumulated above the electrochemical equilibrium, causing Cl− efflux, membrane depolarization, and increased contractile force when Cl− channels are opened. At least two different transport mechanisms contribute to raise [Cl−] i in vascular smooth muscle cells, anion exchange, and cation-chloride cotransport. Recent work suggests that TMEM16A-associated Ca2+-activated Cl− currents mediate Cl− efflux in vascular smooth muscle cells leading to vasoconstriction. Additional proteins associated with Cl− flux in vascular smooth muscle are bestrophins, which modulate vasomotion, the volume-activated LRRC8, and the cystic fibrosis transmembrane conductance regulator (CFTR). Cl− transporters and Cl− channels in vascular smooth muscle cells (VSMCs) significantly contribute to the physiological regulation of vascular tone and arterial blood pressure.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
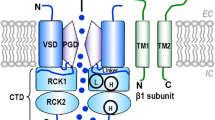
Adequate blood pressure is essential for proper organ function. While too low blood pressure (hypotension) can cause organ failure, elevated blood pressure (hypertension) is a major risk factor for cardiovascular disease, stroke, and chronic renal failure, which affects more than 25 % of the adult population worldwide [58]. Arterial blood pressure is determined by the volume of blood pumped by the heart (cardiac output) and the peripheral resistance of the vasculature. By Starling’s Law, an increase in ventricular end-diastolic volume, which critically depends on the extracellular fluid volume, will increase stroke volume and hence cardiac output. A major hypothesis originally developed by Arthur Guyton in the early 1970s proposes that the kidneys control the level of long-term blood pressure by regulating extracellular fluid volume via the pressure natriuresis mechanism [43, 44]. According to this concept, any impaired capacity of the kidneys to excrete Na+ will cause fluid retention, an increased cardiac output, and ultimately an increased blood pressure. The importance of renal Na+ handling for blood pressure regulation and the pathogenesis of hypertension is strongly supported by genetic, experimental, and clinical evidence [22, 61, 69]. However, several more recent observations have challenged an exclusive role of the kidney for long-term blood pressure homeostasis [68, 76]. For example, selective genetic inactivation of the mineralocorticoid receptor in vascular smooth muscle cells (VSMCs) reduced vascular myogenic tone, diminished the aging-related increase of blood pressure, and prevented arterial stiffening induced by aldosterone and high Na+ intake in mice [35, 66]. These effects occurred without measurable changes in renal Na+ handling, supporting that vascular mechanisms also impact on long-term blood pressure control. As the resistance to flow is inversely proportional to the fourth power of the radius of the blood vessel, small arteries and the arteriolar network are the main determinants of peripheral resistance. Their diameter is determined by contraction of the contractile cells within the wall, the VSMCs. In these cells, the cytosolic Cl− concentration ([Cl−] i ) is normally above the electrochemical equilibrium because of active Cl− accumulation [20]. Hence, opening of Cl− channels in the plasma membrane of VSMCs causes a depolarizing Cl− efflux, which can activate voltage-dependent Ca2+ channels (Fig. 1). The ensuing rise in the cytosolic Ca2+ concentration ([Ca2+] i ) enhances force development by activating myosin light chain kinase via an increased formation of Ca2+-calmodulin. Accordingly, alterations of [Cl−] i in VSMCs should also modulate vascular tone. In this review, we give an update about the role of Cl− for the regulation of peripheral resistance and arterial blood pressure.

Chloride homeostasis in vascular smooth muscle cells. NKCC and AE (in concert with NHE) accumulate chloride into vascular smooth muscle cells (VSMCs), while KCC cotransporters, which lower the intracellular Cl− concentration ([Cl −] i ), obviously play a minor role for steady state [Cl−] i . As a consequence, [Cl−] i in VSMCs is normally above the electrochemical equilibrium. Hence, opening of plasma membrane Cl− channels causes a depolarizing Cl− efflux. Cl− channels identified in VSMCs include Ca2+-activated Cl− channels (CaCC), volume-regulated Cl− channels (VRAC), and the cAMP-regulated cystic fibrosis transmembrane conductance regulator (CFTR). Depolarization results in the activation of voltage-dependent Ca2+ channels (VDCC). The ensuing rise in the cytosolic Ca2+ concentration ([Ca 2+] i ) enhances force development by activating myosin light chain kinase via an increased formation of Ca2+-calmodulin. CaCC, which can be activated through Ca2+ release via ryanodine receptors (RYR) in the sarcoplasmic reticulum (SR) or Ca2+ release via IP3 receptors activated by agonist stimulation of G protein-coupled receptors, boost responses to vasoconstrictive signaling. Activation of CFTR appears to occur predominantly at membrane potentials positive to the equilibrium potential for Cl−, hence causing Cl− influx and hyperpolarization. These mechanisms do not take place in all types of VSMCs, because the set of transport proteins expressed in VSMCs varies considerably along the vascular tree.
Chloride transporters in vascular smooth muscle cells
In contrast to ions like Na+ and K+, in many cell types, the electrochemical gradient of Cl− across the plasma membrane is not very far from the electrochemical equilibrium, which is expected from a passive distribution across the plasma membrane. Depending on the expression and activity of the various proteins that mediate Cl− transport, [Cl−] i can be either below or above the electrochemical equilibrium. In smooth muscle cells including VSMCs, the Cl− equilibrium potential is normally above the resting membrane potential (Em) (Table 1). At least two different mechanisms contribute to raise [Cl−] i in VSMCs [20]: Cl−/HCO3 − exchange and Na+-K+-2Cl− cotransport. There is some evidence for a third mechanism, which has been referred to as “pump III” [20], but its molecular identity and driving force have not been identified so far. The Cl−/H+ antiporter ClC-3 is also expressed in VSMCs (like in other mammalian tissues) [55] and has been proposed to be involved in myogenic tone regulation [32]; however, ClC-3 mainly localizes intracellularly to endosomes [95].
Anion exchanger
Cl− accumulation via anion exchange was initially shown for guinea pig ureter and vas deferens smooth muscle cells [2, 3]. In these cells, HCO3 − is not at equilibrium due to the action of Na+/H+ exchange, which holds the intracellular pH at a more alkaline level than it would be if H+ ions were in equilibrium with Em [101]. The outward gradient for HCO3 − thus provides the driving force for Cl− accumulation via anion exchange. This mechanism has also been confirmed for smooth muscle cells cultured from embryonic rat aorta [54]. Of the three known Na+-independent anion exchangers of the SLC4A family of bicarbonate transporters, only AE2 and AE3 were shown to be expressed in native arteries and in VSMCs [13]. AE2 was among seven genes, for which considerable association with increased blood pressure has been reported, but this association could not be confirmed in replication samples [94]. Conversely, a loss of AE2 or AE3 transport activity may cause a decrease of [Cl−] i in VSMC leading to a diminished vascular tone and low blood pressure. Unfortunately, in vivo studies on AE2 knockout mice have been hampered because of the severe phenotype with growth retardation and early lethality around the time of weaning [38]. By contrast, AE3-knockout mice develop normally and show normal survival, although they are more susceptible to seizures [46] and suffer from impaired vision [4]. Cardiac performance and mean arterial pressure were not altered in anesthetized AE3-knockout mice [80].
Cation-chloride cotransporter
The first compelling evidence for Na+-K+-2Cl− cotransport in VSMCs was the finding of an ouabain-insensitive K+ influx, which is sensitive to the NKCC1 inhibitor bumetanide and the presence of both extracellular Na+ and Cl− [75]. Inhibition of Na+-K+-2Cl− cotransport with bumetanide decreased [Cl−] i , hyperpolarized VSMCs, and attenuated the activation of l-type Ca2+ channels [5, 30]. Bumetanide also suppressed the constriction of mouse mesenteric arteries and rat afferent arterioles in response to angiotensin II [107].
Two different molecules mediate Na+-K+-2Cl− cotransport, NKCC1 and NKCC2, which belong to the family of cation-chloride cotransporters [36]. While the expression of NKCC2, which is mutated in Bartter syndrome type 1 [93], is restricted to epithelial cells of the thick ascending limb and the macula densa [36], where it mediates apical NaCl uptake, NKCC1 is expressed broadly including both epithelial and non-epithelial cells including VSMCs [47, 49]. Supporting a functional role for vascular tone of NKCC1 expression in VSMCs, the inhibitory action of bumetanide on contractions of mesenteric arteries was completely absent in NKCC1-knockout mice [52]. The role of NKCC1 for vascular contractility may differ along the vascular tree: When stimulated with phenylephrine, aortic smooth muscle rings from NKCC1-knockout mice exhibited no significant differences in maximum contractility and only moderate dose-response shifts, while a sharp reduction in mechanical force was noted in the portal vein [70]. Consistent with a role of NKCC1 in setting baseline vascular tone, bumetanide caused a decrease in blood pressure only in wild type but not in NKCC1-knockout mice [37]. Measurements of baseline blood pressure in NKCC1-knockout mice, however, yielded conflicting results: While one study demonstrated a lower systolic blood pressure [70], another study found even higher blood pressures in NKCC1-knockout mice compared with their wild-type controls, when they were fed a high salt diet [50]. The reason for this discrepancy remains unclear and may be related to the different blood pressure recording techniques used (tail cuff vs. telemetry), genetic backgrounds, and housing conditions. Furthermore, disruption of NKCC1 causes an increased expression of renal Na+ transporters and reduced plasma ANP concentration [106] and a disinhibition of basal renin release through a direct effect on juxtaglomerular cells [19]. Thus, different levels of compensation may add additional variation between different studies and further complicate interpretation of the results.
Evidence for an elevated Na+-K+-2Cl− cotransport in VSMCs has also been obtained in several experimental models of hypertension in the rat [74]. A co-segregation of elevated Na+-K+-2Cl− cotransport in erythrocytes and blood pressure was observed for F2 hybrids of Milan normotensive and hypertensive rats suggesting a genetic association of anomalous NKCC1 function and hypertension [10]. More recently, also an epigenetic upregulation of NKCC1 activity has been detected in the aorta from spontaneously hypertensive rats [21]. These findings support the notion that activation of NKCC1 is a general phenomenon of hypertension.
While NKCCs use the Na+ gradient to accumulate Cl− into the cell, K+-Cl− cotransporters (KCCs) use the K+ gradient to lower [Cl−] i . There are some reports that K+-Cl− cotransport takes place in smooth muscle cells [1, 88]. Whether it is active under baseline conditions is so far unclear. KCC1 and KCC3, but not the neuronal specific KCC2, are expressed in VSMCs [87, 96]. KCC3-knockout mice, which are a model for the rare human genetic disorder Andermann syndrome [84], are hypertensive [11]. As expected from a KCC knockout, [Cl−] i in VSMCs of the saphenous artery is increased [87], but the response of isolated saphenous arteries to changes in intravascular pressure, stimulation of alpha1-adrenoceptors, exogenous nitric oxide, or pharmacological blockade of Ca2+-activated Cl− channels with niflumic acid is unchanged [87]. These results argue against a major vascular intrinsic component of the increased arterial blood pressure in KCC3-knockout mice. Obviously, the local control of vascular smooth muscle tone does not require KCC3. Normalization of blood pressure by pharmacological autonomic blockade and increased urinary excretion rates of epinephrine and norepinephrine in KCC3-knockout mice rather point to a neurogenic origin of this phenotype [87].
Chloride channels in vascular smooth muscle cells
A contribution of Cl− efflux to membrane depolarization induced by norepinephrine in VSMCs was detected already in the 1970s in the rat portal vein [104, 105] and rabbit pulmonary artery [18] by radioactive tracer flux experiments. Ca2+-activated Cl− currents in VSMCs were first reported in cells isolated from the pulmonary artery [15] and portal vein [16]. Since a rise of the cytosolic Ca2+ concentration is an early event after agonist stimulation of VSMCs, it was proposed that the opening of Ca2+-activated Cl− channels may be a key step in the electromechanical coupling in VSMCs [57] (Fig. 1). Studies using pharmacological blockers and Cl−-selective electrodes suggested also important roles of Cl− currents for vasomotion [77], the myogenic response [31, 73, 110], and in VSMC proliferation [59]. However, for several reasons, the precise nature of the Cl− transport proteins involved in these functions remained elusive until recently. Cl− channels display largely similar low anion selectivities [33]; their expression varies greatly between different vascular beds [56, 63], different Cl− channels coexist in the same VSMC [63], and classical Cl− channel blockers like niflumic acid, DIDS, and flufenamic acid are unselective and have undefined modes of action [41, 102]. The molecular identification of Cl− transport proteins is further complicated by the fact that all cellular expression systems express endogenous Cl− channels.
Ca2+-activated Cl− channels
More than 25 years after the first characterization of Ca2+-activated Cl− currents in salamander photoreceptors [6] and Xenopus oocytes [7, 71], TMEM16A (anoctamin 1 (ANO1)) was independently identified by three groups as a component of Ca2+-activated Cl− channels [17, 90, 111]. It is a member of the TMEM16 family of transmembrane proteins, which has ten members in humans also known as anoctamins. Heterologous expression of TMEM16A in HEK293 cells and salamander oocytes induced Cl− currents that recapitulated the biophysical and pharmacological properties of native Ca2+-activated Cl− channels [17, 90, 111]. Similar results were found for TMEM16B [90]. Treatment with silencing small interfering TMEM16A RNAs reduced Ca2+-activated Cl− channels in pancreatic and bronchial epithelial cell lines [17] and suppressed salivary production, which depends on functional Ca2+-activated Cl− channels in wild-type mice [111]. The reconstitution of purified human TMEM16A protein in proteoliposomes produced Ca2+- and voltage-gated Cl− currents with a submicromolar Ca2+ sensitivity, thus directly demonstrating that TMEM16A forms the pore of Ca2+-activated Cl− channels [99].
Expression of TMEM16A was identified in various VSMC types from rodent and human arteries [25, 27, 28, 62, 100, 108], while expression of TMEM16B appeared to be very low [27]. Inhibition of TMEM16A by small interfering RNAs or TMEM16A-specific antibodies [14, 25, 27, 28, 62, 100, 108] suppressed Ca2+-activated Cl− currents in VSMCs from mouse aorta, rat cerebral artery, and rat and rabbit pulmonary arteries. A systematic analysis of TMEM16A expression in mice by combined Western blotting and immunohistochemistry revealed high levels of expression in the large conduit arteries as well as in the second order and smaller arterioles in the brain, skeletal muscle, and retina, whereas TMEM16A expression in primary arterioles and small arteries was small or absent [45]. Throughout the arterial tree, expression densities of Ca2+-activated Cl− currents paralleled those of TMEM16A (Fig. 2), and targeted disruption of TMEM16A in VSMCs entirely eliminated Ca2+-activated Cl− currents as well as TMEM16A immunoreactivity in all vascular segments investigated, suggesting that in the mouse, Ca2+-activated Cl− currents in arterial VSMCs are largely generated by TMEM16A. In contrast to the mouse, TMEM16A was detected in rat and human small arteries [25, 27, 28, 62, 100, 108]. Thus some species differences appear to exist. Moreover, T16Ainh-A01 [28], an aminophenylthiazole that had been identified as a low μM inhibitor of TMEM16A-mediated currents by a cell-based high throughput screen [72], induced only a partial block of Ca2+-activated Cl− currents in rabbit pulmonary artery VSMCs even when administered at high concentrations [28].

Expression of TMEM16A in murine small blood vessels of various tissues. Double immunostaining for TMEM16A (left), smooth muscle actin (SMA), or endothelial marker CD31 (middle), and overlay (right) of various whole mount preparations. The first row is retina. Whereas first order arterioles do not show strong TMEM16A staining (arrow), smaller arterioles are more intensely stained. Scale bar indicates 30 μm. The second row is small intestine. While no arterioles are present in the projection images of the confocal stack and the small arteries do not express TMEM16A, an intense staining is found in the so-called intestinal cells of Cajal (ICC) [40], where TMEM16A is required for pacemaker activity [48]. Scale bar indicates 50 μm. The third row is a cerebellar slice. Significant TMEM16A expression is only found at the branching points of the largest vessel (three arrows), whereas smaller arterioles show stronger expression. Scale bar indicates 50 μm. The fourth row is a skeletal muscle slice. A small artery running from the top to the bottom (arrow) shows increasing TMEM16A expression with decreasing diameter caused by multiple branching events. Scale bar indicates 30 μm.
Downregulation of TMEM16A-associated Ca2+-activated Cl− currents through siRNA significantly diminished the vasoconstrictor response to noradrenaline and vasopressin in rat mesenteric arteries [25, 27, 62, 100, 108] and to intravascular pressure elevation in rat cerebral arteries [14]. Mouse mesenteric arteries and human visceral adipose tissue arteries pre-constricted with the thromboxane mimetic U46619 were also relaxed by pharmacological inhibition of TMEM16A with T16Ainh-A01 [28]. In mice with an inducible VSMC-specific knockout of TMEM16A, the vasoconstrictor effects of U46619 were attenuated in retinal arterioles, but not in small mesenteric arteries, which have a very weak expression of TMEM16A [45]. These mice displayed reduced response to U46619 in isolated perfused hind limbs, were hypotensive, had smaller arterial pressure amplitudes, and developed less severe hypertension in response to chronic infusion of angiotensin II [45]. Collectively, these findings indicate that TMEM16A has a role for the Windkessel function of large conduit arteries and participates in the physiological regulation of basal vascular tone and vascular reactivity of resistance vessels to agonist-induced vasoconstriction in vivo.
Gating of TMEM16A is both voltage- and Ca2+ dependent. At [Ca2+] i in the nM range, the current displays outward rectification which is lost when [Ca2+] i is increased. The activation velocity is also strongly Ca2+ dependent: while the channel opens slowly at nM [Ca2+] i , activation is accelerated with increasing [Ca2+] i . These biophysical properties are well known from earlier studies on Ca2+-activated Cl− currents [57] and may serve presently unknown functions in VSMC. Recent observations suggest that Ca2+ exerts its modulatory influence on TMEM16A by directly binding to the channel protein. The Ca2+-activated Cl− current produced by reconstituted TMEM16A was not affected by calmodulin [99], and calmodulin did not alter activation or anion permeability of heterologously expressed mouse TMEM16A [112, 113]. In addition to [Ca2+] i and membrane voltage, PI(4,5)P2 has been shown to act as a very potent inhibitor of TMEM16A at concentrations as low as 1 μM [81]. Since cytosolic PI(4,5)P2 has been estimated to be in a range of 10–210 μM, relieve from tonic PI(4,5)P2 inhibition may constitute a gating mechanism.
Activation of TMEM16A by localized increase in [Ca2+] i also likely underlies the elusive stretch-regulated Cl− current in the myogenic response [14]. The myogenic response helps to maintain blood flow constant in the face of fluctuations of perfusion pressure and depends on the intrinsic capacity of VSMCs in resistance vessels to increase their tone in response to increases in transmural pressure. Key events contributing to the myogenic response are a membrane depolarization and opening of l-type Ca2+ channels leading to an elevation of [Ca2+] i [51]. Several mechanisms have been proposed to mediate the membrane depolarization in the myogenic response, including opening of mechanosensitive ion channels, activation of sphingosine-1-phosphate signaling, and activation of angiotensin AT1 receptors [89, 109, 114, 115]. The idea that increases in transmural pressure also activates Cl− channels to depolarize and constrict cerebral resistance vessels originated from the observation that Cl− channel blockers induced hyperpolarization and dilatation and that reductions in the extracellular Cl− concentration augmented the myogenic tone [73]. Furthermore, direct measurements of Cl− flux using a Cl−-selective electrode demonstrated a pressure-dependent increase in Cl− efflux in intact cerebral resistance arteries [31]. Cell swelling induced by hypotonicity activated Cl− currents with a comparable pharmacology also in pulmonary and renal arteries [110]. Initially, volume-regulated anion channels (VRAC) have been suggested to underlie this current [73]. However, specific inhibition by small interfering RNA demonstrated that a knockdown of TMEM16A causes a reduction of the myogenic response in cerebral resistance arteries by ∼50 % [14]. Moreover, the Cl− current evoked by cell swelling could be blocked by TMEM16A downregulation using short interfering RNAs, TMEM16A antibodies, or the fast Ca2+ chelating agent BAPTA and displayed similar biophysical properties as TMEM16A. Further studies will be required to assess whether activation of TMEM16A by localized increases in [Ca2+] i also underlies other volume-regulated Cl− currents in the cardiovascular system. In addition, it will be important to evaluate possible functions of the LRRC8 gene family, which recently has been shown to encode components of the volume-regulated anion channel VRAC [82, 103] that has been suggested to be important for VSMC proliferation [92].
Several pathologies are associated with an altered expression of TMEM16A. Pulmonary arterial VSMC expresses a prominent TMEM16A current [62]. Chronic hypoxia for 3–4 weeks in rats strongly upregulated TMEM16A protein expression and Ca2+-activated Cl− currents, which likely enhanced pulmonary reactivity to serotonin, a well-established mediator of chronic hypoxic pulmonary hypertension [97]. Very similar observations were made in rats treated with monocrotaline, an experimental drug for the induction of pulmonary hypertension [34]. In 2-kidney, 2-clip hypertensive rats, TMEM16A expression and Ca2+-activated Cl− currents were decreased in the basilar artery in an inverse relation to blood pressure [108]. Interestingly, knockdown of TMEM16A expression in cultured basilar VSMCs promoted angiotensin II effects on cell cycle, whereas TMEM16A overexpression suppressed the action of angiotensin II.
Besides the Ca2+-activated Cl− current encoded by TMEM16A, a cGMP-dependent Ca2+-activated Cl− current is co-expressed in many VSMC types. This current differs in several biophysical aspects from the classic Ca2+-activated Cl− current; in particular, it is voltage independent over the entire Ca2+ range. Downregulation of bestrophin-3 gene expression with small interfering RNAs suppressed the cGMP-dependent Ca2+-activated Cl− current in isolated cells as well as in vivo [64]. The amplitude of vasomotion was significantly dampened after downregulation of bestrophin-3, whereas vascular contractility was unaffected [12]. This suggests that bestrophins are involved in the regulation of tissue perfusion by synchronizing VSMC function. Bestrophin-3 belongs to the bestrophin family, which comprises four family members; each member forms a homotetrameric channel [9]. Interestingly, TMEM16A knockdown also inhibited the cGMP-dependent Ca2+-activated Cl− current, probably secondary to a suppression of bestrophin-3 [25]. The functional significance of this interaction is not clear. It has been suggested that bestrophin acts as a regulatory subunit of channels formed by TMEM16A to modify its biophysical properties [24].
Cl− also plays a role for sequestration of Ca2+ into the sarcoplasmic reticulum. Regulation of Ca2+ by the sarcoplasmic reticulum is essential for normal contractile function of muscle cells. In skeletal muscle, the sarcoplasmic reticulum maintains a large Ca2+ gradient, but there is no electrical potential between its lumen and the cytoplasm [79]. Its membrane is permeable to Cl−, and the entry of Cl− into sarcoplasmic reticulum along with Ca2+ is thought to prevent the development of an electrical potential opposing further Ca2+ accumulation. Currently, the molecular pathway for Cl− entry into the sarcoplasmic reticulum is not known, but bestrophin-1 has been suggested to form a Ca2+-dependent counter-anion channel to balance the transient membrane potential associated with Ca2+ release and store refill [8].
Cystic fibrosis transmembrane conductance regulator
The cystic fibrosis transmembrane conductance regulator (CFTR) is a cyclic AMP-regulated Cl− channel. Loss-of-function mutations in CFTR cause cystic fibrosis [91]. Human patients with cystic fibrosis have decreased blood pressures [60, 98], an observation also made in piglets and mice carrying the most common CFTR mutation, CFTR-F508del [42, 78]. CFTR protein has been detected by Western blotting and immunofluorescence in the mouse aorta and intrapulmonary arteries [85, 86]. Cultured aortic VSMCs from wild type but not CFTR-knockout mice responded with an increased iodide efflux to cAMP agonists as well as to the beta1/2 adrenoceptor agonist isoproterenol [85]. This effect was suppressed by the specific CFTR antagonist CFTRinh-172. Correspondingly, aortic rings from CFTR-knockout mice showed a much stronger, endothelium-independent vasoconstrictor response to elevated K+ than aortic rings from wild-type animals [85]. In addition, CFTR-knockout mice had a higher myogenic tone in pressurized isolated cerebral and mesenteric arteries [67]. Most of the inhibitory effects of CFTR activation on VSMC contraction were observed under depolarizing experimental conditions. Accordingly, activation of CFTR may predominantly operate at membrane potentials above the Cl− equilibrium potential to repolarize the VSMC explaining the surprisingly relaxing effects of CFTR activation (Fig. 1). Since CFTR currents have not been demonstrated yet in VSMCs, the hyperpolarizing effect may also be secondary to a regulatory effect of CFTR on other membrane channels.
Conclusion
The findings presented in this review strongly suggest that Cl− transporters and Cl− channels in VSMCs significantly contribute to the physiological regulation of vascular tone. The observation that mice with an inducible VSMC-specific knockout of TMEM16A are hypotensive without showing any measurable changes in renal function [45] further supports the concept that primary changes in vascular function can cause sustained changes in arterial blood pressure [68, 76]. However, one should bear in mind that renal hemodynamics affects water and electrolyte homeostasis. Long-term blood pressure is particularly sensitive to alterations in the renal medullary circulation, and it has been demonstrated that a selective increase in vascular resistance in this vascular bed is sufficient to cause a sustained increase of arterial blood pressure [23, 65]. Accordingly, a decreased vascular contractility caused by vascular disruption of TMEM16A may also operate through an altered regulation of the extracellular fluid balance to reduce blood pressure.
The role of Cl− for VSMC function is probably much more complex than the simple model that [Cl−] i above equilibrium just sets the driving force to boost contraction via Cl− channels. Cell-specific knockout mouse models will help to further advance the understanding of the role of Cl− for the regulation of arterial blood pressure. However, as VSMCs are highly heterogeneous and have markedly different functional properties in different organs [83], it is a difficult task if nearly impossible to predict the effects of knockout strategies on systemic blood pressure.
References
Adragna NC, White RE, Orlov SN, Lauf PK (2000) K-Cl cotransport in vascular smooth muscle and erythrocytes: possible implication in vasodilation. Am J Physiol Cell Physiol 278:C381–C390
Aickin CC, Vermue NA (1983) Microelectrode measurement of intracellular chloride activity in smooth muscle cells of guinea-pig ureter. Pflugers Arch 397:25–28
Aickin CC, Brading AF (1982) Measurement of intracellular chloride in guinea-pig vas deferens by ion analysis, 36chloride efflux and micro-electrodes. J Physiol 326:139–154
Alvarez BV, Gilmour GS, Mema SC, Martin BT, Shull GE, Casey JR, Sauve Y (2007) Blindness caused by deficiency in AE3 chloride/bicarbonate exchanger. PLoS One 2:e839
Anfinogenova YJ, Baskakov MB, Kovalev IV, Kilin AA, Dulin NO, Orlov SN (2004) Cell-volume-dependent vascular smooth muscle contraction: role of Na+, K+, 2Cl- cotransport, intracellular Cl- and L-type Ca2+ channels. Pflugers Arch 449:42–55
Bader CR, Bertrand D, Schwartz EA (1982) Voltage-activated and calcium-activated currents studied in solitary rod inner segments from the salamander retina. J Physiol 331:253–284
Barish ME (1983) A transient calcium-dependent chloride current in the immature Xenopus oocyte. J Physiol 342:309–325
Barro-Soria R, Aldehni F, Almaca J, Witzgall R, Schreiber R, Kunzelmann K (2010) ER-localized bestrophin 1 activates Ca2+-dependent ion channels TMEM16A and SK4 possibly by acting as a counterion channel. Pflugers Arch 459:485–497
Bharill S, Fu Z, Palty R, Isacoff EY (2014) Stoichiometry and specific assembly of best ion channels. Proc Natl Acad Sci U S A 111:6491–6496
Bianchi G, Ferrari P, Trizio D, Ferrandi M, Torielli L, Barber BR, Polli E (1985) Red blood cell abnormalities and spontaneous hypertension in the rat. A genetically determined link. Hypertension 7:319–325
Boettger T, Rust MB, Maier H, Seidenbecher T, Schweizer M, Keating DJ, Faulhaber J, Ehmke H, Pfeffer C, Scheel O, Lemcke B, Horst J, Leuwer R, Pape HC, Volkl H, Hübner CA, Jentsch TJ (2003) Loss of K-Cl co-transporter KCC3 causes deafness, neurodegeneration and reduced seizure threshold. EMBO J 22:5422–5434
Broegger T, Jacobsen JC, Secher Dam V, Boedtkjer DM, Kold-Petersen H, Pedersen FS, Aalkjaer C, Matchkov VV (2011) Bestrophin is important for the rhythmic but not the tonic contraction in rat mesenteric small arteries. Cardiovasc Res 91:685–693
Brosius FC 3rd, Pisoni RL, Cao X, Deshmukh G, Yannoukakos D, Stuart-Tilley AK, Haller C, Alper SL (1997) AE anion exchanger mRNA and protein expression in vascular smooth muscle cells, aorta, and renal microvessels. Am J Physiol 273:F1039–F1047
Bulley S, Neeb ZP, Burris SK, Bannister JP, Thomas-Gatewood CM, Jangsangthong W, Jaggar JH (2012) TMEM16A/ANO1 channels contribute to the myogenic response in cerebral arteries. Circ Res 111:1027–1036
Byrne NG, Large WA (1987) The action of noradrenaline on single smooth muscle cells freshly dispersed from the guinea-pig pulmonary artery. Br J Pharmacol 91:89–94
Byrne NG, Large WA (1988) Membrane ionic mechanisms activated by noradrenaline in cells isolated from the rabbit portal vein. J Physiol 404:557–573
Caputo A, Caci E, Ferrera L, Pedemonte N, Barsanti C, Sondo E, Pfeffer U, Ravazzolo R, Zegarra-Moran O, Galietta LJ (2008) TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science 322:590–594
Casteels R, Kitamura K, Kuriyama H, Suzuki H (1977) The membrane properties of the smooth muscle cells of the rabbit main pulmonary artery. J Physiol 271:41–61
Castrop H, Lorenz JN, Hansen PB, Friis U, Mizel D, Oppermann M, Jensen BL, Briggs J, Skott O, Schnermann J (2005) Contribution of the basolateral isoform of the Na-K-2Cl- cotransporter (NKCC1/BSC2) to renin secretion. Am J Physiol Renal Physiol 289:F1185–F1192
Chipperfield AR, Harper AA (2000) Chloride in smooth muscle. Prog Biophys Mol Biol 74:175–221
Cho HM, Lee HA, Kim HY, Han HS, Kim IK (2011) Expression of Na+-K+-2Cl- cotransporter 1 is epigenetically regulated during postnatal development of hypertension. Am J Hypertens 24:1286–1293
Coffman TM (2014) The inextricable role of the kidney in hypertension. J Clin Invest 124:2341–2347
Cowley AW, Roman RJ (1996) The role of the kidney in hypertension. JAMA 275:1581–1589
Dam VS, Boedtkjer DM, Aalkjaer C, Matchkov V (2014) The bestrophin- and TMEM16A-associated Ca2+- activated Cl- channels in vascular smooth muscles. Channels (Austin) 8:361–369
Dam VS, Boedtkjer DM, Nyvad J, Aalkjaer C, Matchkov V (2014) TMEM16A knockdown abrogates two different Ca2+-activated Cl- currents and contractility of smooth muscle in rat mesenteric small arteries. Pflugers Arch 466:1391–1409
Davis JP (1992) The effects of Na+-K+-Cl- co-transport and Cl–HCO3-exchange blockade on the membrane potential and intracellular chloride levels of rat arterial smooth muscle, in vitro. Exp Physiol 77:857–862
Davis AJ, Forrest AS, Jepps TA, Valencik ML, Wiwchar M, Singer CA, Sones WR, Greenwood IA, Leblanc N (2010) Expression profile and protein translation of TMEM16A in murine smooth muscle. Am J Physiol Cell Physiol 299:C948–C959
Davis AJ, Shi J, Pritchard HA, Chadha PS, Leblanc N, Vasilikostas G, Yao Z, Verkman AS, Albert AP, Greenwood IA (2013) Potent vasorelaxant activity of the TMEM16A inhibitor T16Ainh-A01. Br J Pharmacol 168:773–784
Davis JP, Chien PF, Chipperfield AR, Gordon A, Harper AA (2000) The three mechanisms of intracellular chloride accumulation in vascular smooth muscle of human umbilical and placental arteries. Pflugers Arch 441:150–154
Davis JP, Chipperfield AR, Harper AA (1993) Accumulation of intracellular chloride by (Na-K-Cl) co-transport in rat arterial smooth muscle is enhanced in deoxycorticosterone acetate (DOCA)/salt hypertension. J Mol Cell Cardiol 25:233–237
Doughty JM, Langton PD (2001) Measurement of chloride flux associated with the myogenic response in rat cerebral arteries. J Physiol 534:753–761
Duan DD (2011) The ClC-3 chloride channels in cardiovascular disease. Acta Pharmacol Sin 32:675–684
Duran C, Thompson CH, Xiao Q, Hartzell HC (2010) Chloride channels: often enigmatic, rarely predictable. Annu Rev Physiol 72:95–121
Forrest AS, Joyce TC, Huebner ML, Ayon RJ, Wiwchar M, Joyce J, Freitas N, Davis AJ, Ye L, Duan DD, Singer CA, Valencik ML, Greenwood IA, Leblanc N (2012) Increased TMEM16A-encoded calcium-activated chloride channel activity is associated with pulmonary hypertension. Am J Physiol Cell Physiol 303:C1229–C1243
Galmiche G, Pizard A, Gueret A, El Moghrabi S, Ouvrard-Pascaud A, Berger S, Challande P, Jaffe IZ, Labat C, Lacolley P, Jaisser F (2014) Smooth muscle cell mineralocorticoid receptors are mandatory for aldosterone-salt to induce vascular stiffness. Hypertension 63:520–526
Gamba G (2005) Molecular physiology and pathophysiology of electroneutral cation-chloride cotransporters. Physiol Rev 85:423–493
Garg P, Martin CF, Elms SC, Gordon FJ, Wall SM, Garland CJ, Sutliff RL, O’Neill WC (2007) Effect of the Na-K-2Cl cotransporter NKCC1 on systemic blood pressure and smooth muscle tone. Am J Physiol Heart Circ Physiol 292:H2100–H2105
Gawenis LR, Ledoussal C, Judd LM, Prasad V, Alper SL, Stuart-Tilley A, Woo AL, Grisham C, Sanford LP, Doetschman T, Miller ML, Shull GE (2004) Mice with a targeted disruption of the AE2 Cl-/HCO3 - exchanger are achlorhydric. J Biol Chem 279:30531–30539
Gerstheimer FP, Mühleisen M, Nehring D, Kreye VA (1987) A chloride-bicarbonate exchanging anion carrier in vascular smooth muscle of the rabbit. Pflugers Arch 409:60–66
Gomez-Pinilla PJ, Gibbons SJ, Bardsley MR, Lorincz A, Pozo MJ, Pasricha PJ, Van de Rijn M, West RB, Sarr MG, Kendrick ML, Cima RR, Dozois EJ, Larson DW, Ordog T, Farrugia G (2009) Ano1 is a selective marker of interstitial cells of Cajal in the human and mouse gastrointestinal tract. Am J Physiol Gastrointest Liver Physiol 296:G1370–G1381
Greenwood IA, Leblanc N (2007) Overlapping pharmacology of Ca2+-activated Cl- and K+ channels. Trends Pharmacol Sci 28:1–5
Guo JJ, Stoltz DA, Zhu V, Volk KA, Segar JL, McCray PB Jr, Roghair RD (2014) Genotype-specific alterations in vascular smooth muscle cell function in cystic fibrosis piglets. J Cyst Fibros 13:251–259
Guyton AC (1991) Blood pressure control—special role of the kidneys and body fluids. Science 252:1813–1816
Guyton AC, Coleman TG, Cowley AV Jr, Scheel KW, Manning RD Jr, Norman RA Jr (1972) Arterial pressure regulation. Overriding dominance of the kidneys in long-term regulation and in hypertension. Am J Med 52:584–594
Heinze C, Seniuk A, Sokolov MV, Huebner AK, Klementowicz AE, Szijarto IA, Schleifenbaum J, Vitzthum H, Gollasch M, Ehmke H, Schroeder BC, Hübner CA (2014) Disruption of vascular Ca2+-activated chloride currents lowers blood pressure. J Clin Invest 124:675–686
Hentschke M, Wiemann M, Hentschke S, Kurth I, Hermans-Borgmeyer I, Seidenbecher T, Jentsch TJ, Gal A, Hübner CA (2006) Mice with a targeted disruption of the Cl-/HCO3 - exchanger AE3 display a reduced seizure threshold. Mol Cell Biol 26:182–191
Hübner CA, Lorke DE, Hermans-Borgmeyer I (2001) Expression of the Na-K-2Cl-cotransporter NKCC1 during mouse development. Mech Dev 102:267–269
Hwang SJ, Blair PJ, Britton FC, O’Driscoll KE, Hennig G, Bayguinov YR, Rock JR, Harfe BD, Sanders KM, Ward SM (2009) Expression of anoctamin 1/TMEM16A by interstitial cells of Cajal is fundamental for slow wave activity in gastrointestinal muscles. J Physiol 587:4887–4904
Kaplan MR, Plotkin MD, Brown D, Hebert SC, Delpire E (1996) Expression of the mouse Na-K-2Cl cotransporter, mBSC2, in the terminal inner medullary collecting duct, the glomerular and extraglomerular mesangium, and the glomerular afferent arteriole. J Clin Invest 98:723–730
Kim SM, Eisner C, Faulhaber-Walter R, Mizel D, Wall SM, Briggs JP, Schnermann J (2008) Salt sensitivity of blood pressure in NKCC1-deficient mice. Am J Physiol Renal Physiol 295:F1230–F1238
Knot HJ, Nelson MT (1998) Regulation of arterial diameter and wall [Ca2+] in cerebral arteries of rat by membrane potential and intravascular pressure. J Physiol 508:199–209
Koltsova SV, Luneva OG, Lavoie JL, Tremblay J, Maksimov GV, Hamet P, Orlov SN (2009) HCO3-dependent impact of Na+, K+, 2Cl- cotransport in vascular smooth muscle excitation-contraction coupling. Cell Physiol Biochem 23:407–414
Koncz C, Daugirdas JT (1994) Use of MQAE for measurement of intracellular [Cl-] in cultured aortic smooth muscle cells. Am J Physiol 267:H2114–H2123
Korbmacher C, Helbig H, Stahl F, Wiederholt M (1988) Evidence for Na/H exchange and Cl/HCO3 exchange in A10 vascular smooth muscle cells. Pflugers Arch 412:29–36
Lamb FS, Clayton GH, Liu BX, Smith RL, Barna TJ, Schutte BC (1999) Expression of CLCN voltage-gated chloride channel genes in human blood vessels. J Mol Cell Cardiol 31:657–666
Lamb FS, Kooy NW, Lewis SJ (2000) Role of Cl- channels in alpha-adrenoceptor-mediated vasoconstriction in the anesthetized rat. Eur J Pharmacol 401:403–412
Large WA, Wang Q (1996) Characteristics and physiological role of the Ca2+-activated Cl- conductance in smooth muscle. Am J Physiol 271:C435–C454
Lawes CM, Vander Hoorn S, Rodgers A (2008) Global burden of blood-pressure-related disease, 2001. Lancet 371:1513–1518
Liang W, Ray JB, He JZ, Backx PH, Ward ME (2009) Regulation of proliferation and membrane potential by chloride currents in rat pulmonary artery smooth muscle cells. Hypertension 54:286–293
Lieberman J, Rodbard S (1975) Low blood pressure in young adults with cystic fibrosis: an effect of chronic salt loss in sweat? Ann Intern Med 82:806–808
Lifton RP, Gharavi AG, Geller DS (2001) Molecular mechanisms of human hypertension. Cell 104:545–556
Manoury B, Tamuleviciute A, Tammaro P (2010) TMEM16A/anoctamin 1 protein mediates calcium-activated chloride currents in pulmonary arterial smooth muscle cells. J Physiol 588:2305–2314
Matchkov VV, Aalkjaer C, Nilsson H (2005) Distribution of cGMP-dependent and cGMP-independent Ca2+-activated Cl- conductances in smooth muscle cells from different vascular beds and colon. Pflugers Arch 451:371–379
Matchkov VV, Larsen P, Bouzinova EV, Rojek A, Boedtkjer DM, Golubinskaya V, Pedersen FS, Aalkjaer C, Nilsson H (2008) Bestrophin-3 (vitelliform macular dystrophy 2-like 3 protein) is essential for the cGMP-dependent calcium-activated chloride conductance in vascular smooth muscle cells. Circ Res 103:864–872
Mattson DL, Lu S, Nakanishi K, Papanek PE, Cowley AW (1994) Effect of chronic renal medullary nitric oxide inhibition on blood pressure. Am J Physiol 266:H1918–H1926
McCurley A, Pires PW, Bender SB, Aronovitz M, Zhao MJ, Metzger D, Chambon P, Hill MA, Dorrance AM, Mendelsohn ME, Jaffe IZ (2012) Direct regulation of blood pressure by smooth muscle cell mineralocorticoid receptors. Nat Med 18:1429–1433
Meissner A, Yang J, Kroetsch JT, Sauve M, Dax H, Momen A, Noyan-Ashraf MH, Heximer S, Husain M, Lidington D, Bolz SS (2012) Tumor necrosis factor-alpha-mediated downregulation of the cystic fibrosis transmembrane conductance regulator drives pathological sphingosine-1-phosphate signaling in a mouse model of heart failure. Circulation 125:2739–2750
Mendelsohn ME (2005) In hypertension, the kidney is not always the heart of the matter. J Clin Invest 115:840–844
Meneton P, Jeunemaitre X, de Wardener HE, MacGregor GA (2005) Links between dietary salt intake, renal salt handling, blood pressure, and cardiovascular diseases. Physiol Rev 85:679–715
Meyer JW, Flagella M, Sutliff RL, Lorenz JN, Nieman ML, Weber CS, Paul RJ, Shull GE (2002) Decreased blood pressure and vascular smooth muscle tone in mice lacking basolateral Na+-K+-2Cl- cotransporter. Am J Physiol Heart Circ Physiol 283:H1846–H1855
Miledi R (1982) A calcium-dependent transient outward current in Xenopus laevis oocytes. Proc R Soc Lond B Biol Sci 215:491–497
Namkung W, Phuan PW, Verkman AS (2011) TMEM16A inhibitors reveal TMEM16A as a minor component of calcium-activated chloride channel conductance in airway and intestinal epithelial cells. J Biol Chem 286:2365–2374
Nelson MT, Conway MA, Knot HJ, Brayden JE (1997) Chloride channel blockers inhibit myogenic tone in rat cerebral arteries. J Physiol 502:259–264
Orlov SN, Koltsova SV, Tremblay J, Baskakov MB, Hamet P (2012) NKCC1 and hypertension: role in the regulation of vascular smooth muscle contractions and myogenic tone. Ann Med 44(Suppl 1):S111–S118
Owen NE (1984) Regulation of Na/K/Cl cotransport in vascular smooth muscle cells. Biochem Biophys Res Commun 125:500–508
Pao AC (2014) Update on the Guytonian view of hypertension. Curr Opin Nephrol Hypertens 23:391–398
Peng H, Matchkov V, Ivarsen A, Aalkjaer C, Nilsson H (2001) Hypothesis for the initiation of vasomotion. Circ Res 88:810–815
Peotta VA, Bhandary P, Ogu U, Volk KA, Roghair RD (2014) Reduced blood pressure of CFTR-F508del carriers correlates with diminished arterial reactivity rather than circulating blood volume in mice. PLoS One 9:e96756
Pollock NS, Kargacin ME, Kargacin GJ (1998) Chloride channel blockers inhibit Ca2+ uptake by the smooth muscle sarcoplasmic reticulum. Biophys J 75:1759–1766
Prasad V, Bodi I, Meyer JW, Wang Y, Ashraf M, Engle SJ, Doetschman T, Sisco K, Nieman ML, Miller ML, Lorenz JN, Shull GE (2008) Impaired cardiac contractility in mice lacking both the AE3 Cl-/HCO3 - exchanger and the NKCC1 Na+-K+-2Cl- cotransporter: effects on Ca2+ handling and protein phosphatases. J Biol Chem 283:31303–31314
Pritchard HA, Leblanc N, Albert AP, Greenwood IA (2014) Inhibitory role of phosphatidylinositol 4,5-bisphosphate on TMEM16A-encoded calcium-activated chloride channels in rat pulmonary artery. Br J Pharmacol 171:4311–4321
Qiu Z, Dubin AE, Mathur J, Tu B, Reddy K, Miraglia LJ, Reinhardt J, Orth AP, Patapoutian A (2014) SWELL1, a plasma membrane protein, is an essential component of volume-regulated anion channel. Cell 157:447–458
Reho JJ, Zheng X, Fisher SA (2014) Smooth muscle contractile diversity in the control of regional circulations. Am J Physiol Heart Circ Physiol 306:H163–H172
Howard HC, Mount DB, Rochefort D, Byun N, Dupré N, Lu J, Fan X, Song L, Rivière JB, Prévost C, Horst J, Simonati A, Lemcke B, Welch R, England R, Zhan FQ, Mercado A, Siesser WB, George AL Jr, McDonald MP, Bouchard JP, Mathieu J, Delpire E, Rouleau GA (2002) The K-Cl cotransporter KCC3 is mutant in a severe peripheral neuropathy associated with agenesis of the corpus callosum. Nat Genet 32(3):384–392
Robert R, Norez C, Becq F (2005) Disruption of CFTR chloride channel alters mechanical properties and cAMP-dependent Cl- transport of mouse aortic smooth muscle cells. J Physiol 568:483–495
Robert R, Savineau JP, Norez C, Becq F, Guibert C (2007) Expression and function of cystic fibrosis transmembrane conductance regulator in rat intrapulmonary arteries. Eur Respir J 30:857–864
Rust MB, Faulhaber J, Budack MK, Pfeffer C, Maritzen T, Didie M, Beck FX, Boettger T, Schubert R, Ehmke H, Jentsch TJ, Hübner CA (2006) Neurogenic mechanisms contribute to hypertension in mice with disruption of the K-Cl cotransporter KCC3. Circ Res 98:549–556
Saitta M, Cavalier S, Garay R, Cragoe E Jr, Hannaert P (1990) Evidence for a DIOA-sensitive [K+, Cl-]-cotransport system in cultured vascular smooth muscle cells. Am J Hypertens 3:939–942
Schleifenbaum J, Kassmann M, Szijarto IA, Hercule HC, Tano JY, Weinert S, Heidenreich M, Pathan AR, Anistan YM, Alenina N, Rusch NJ, Bader M, Jentsch TJ, Gollasch M (2014) Stretch-activation of angiotensin II type 1a receptors contributes to the myogenic response of mouse mesenteric and renal arteries. Circ Res 115:263–272
Schroeder BC, Cheng T, Jan YN, Jan LY (2008) Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell 134:1019–1029
Sheppard DN, Welsh MJ (1999) Structure and function of the CFTR chloride channel. Physiol Rev 79:S23–S45
Shi XL, Wang GL, Zhang Z, Liu YJ, Chen JH, Zhou JG, Qiu QY, Guan YY (2007) Alteration of volume-regulated chloride movement in rat cerebrovascular smooth muscle cells during hypertension. Hypertension 49:1371–1377
Simon DB, Karet FE, Hamdan JM, DiPietro A, Sanjad SA, Lifton RP (1996) Bartter’s syndrome, hypokalaemic alkalosis with hypercalciuria, is caused by mutations in the Na-K-2Cl cotransporter NKCC2. Nat Genet 13:183–188
Sober S, Org E, Kepp K, Juhanson P, Eyheramendy S, Gieger C, Lichtner P, Klopp N, Veldre G, Viigimaa M, Doring A, Kooperative Gesundheitsforschung in der Region, Augsburg S, Putku M, Kelgo P, Study HYE, Shaw-Hawkins S, Howard P, Onipinla A, Dobson RJ, Newhouse SJ, Brown M, Dominiczak A, Connell J, Samani N, Farrall M, Study MRCBGoH, Caulfield MJ, Munroe PB, Illig T, Wichmann HE, Meitinger T, Laan M (2009) Targeting 160 candidate genes for blood pressure regulation with a genome-wide genotyping array. PLoS One 4:e6034
Stauber T, Jentsch TJ (2013) Chloride in vesicular trafficking and function. Annu Rev Physiol 75:453–477
Stein V, Hermans-Borgmeyer I, Jentsch TJ, Hübner CA (2004) Expression of the KCl cotransporter KCC2 parallels neuronal maturation and the emergence of low intracellular chloride. J Comp Neurol 468:57–64
Sun H, Xia Y, Paudel O, Yang XR, Sham JS (2012) Chronic hypoxia-induced upregulation of Ca2+-activated Cl- channel in pulmonary arterial myocytes: a mechanism contributing to enhanced vasoreactivity. J Physiol 590:3507–3521
Super M, Irtiza-Ali A, Roberts SA, Schwarz M, Young M, Smith A, Roberts T, Hinks J, Heagerty A (2004) Blood pressure and the cystic fibrosis gene: evidence for lower pressure rises with age in female carriers. Hypertension 44:878–883
Terashima H, Picollo A, Accardi A (2013) Purified TMEM16A is sufficient to form Ca2+-activated Cl- channels. Proc Natl Acad Sci U S A 110:19354–19359
Thomas-Gatewood C, Neeb ZP, Bulley S, Adebiyi A, Bannister JP, Leo MD, Jaggar JH (2011) TMEM16A channels generate Ca2+-activated Cl- currents in cerebral artery smooth muscle cells. Am J Physiol Heart Circ Physiol 301:H1819–H1827
Vaughan-Jones RD (1979) Regulation of chloride in quiescent sheep-heart Purkinje fibres studied using intracellular chloride and pH-sensitive micro-electrodes. J Physiol 295:111–137
Verkman AS, Galietta LJ (2009) Chloride channels as drug targets. Nat Rev Drug Discov 8:153–171
Voss FK, Ullrich F, Munch J, Lazarow K, Lutter D, Mah N, Andrade-Navarro MA, von Kries JP, Stauber T, Jentsch TJ (2014) Identification of LRRC8 heteromers as an essential component of the volume-regulated anion channel VRAC. Science 344:634–638
Wahlstrom BA (1973) Ionic fluxes in the rat portal vein and the applicability of the Goldman equation in predicting the membrane potential from flux data. Acta Physiol Scand 89:436–448
Wahlstrom BA, Svennerholm B (1974) Potentiation and inhibition of noradrenaline induced contractions of the rat portal vein in anion substituted solutions. Acta Physiol Scand 92:404–411
Wall SM, Knepper MA, Hassell KA, Fischer MP, Shodeinde A, Shin W, Pham TD, Meyer JW, Lorenz JN, Beierwaltes WH, Dietz JR, Shull GE, Kim YH (2006) Hypotension in NKCC1 null mice: role of the kidneys. Am J Physiol Renal Physiol 290:F409–F416
Wang X, Breaks J, Loutzenhiser K, Loutzenhiser R (2007) Effects of inhibition of the Na+/K+/2Cl- cotransporter on myogenic and angiotensin II responses of the rat afferent arteriole. Am J Physiol Renal Physiol 292:F999–F1006
Wang M, Yang H, Zheng LY, Zhang Z, Tang YB, Wang GL, Du YH, Lv XF, Liu J, Zhou JG, Guan YY (2012) Downregulation of TMEM16A calcium-activated chloride channel contributes to cerebrovascular remodeling during hypertension by promoting basilar smooth muscle cell proliferation. Circulation 125:697–707
Welsh DG, Morielli AD, Nelson MT, Brayden JE (2002) Transient receptor potential channels regulate myogenic tone of resistance arteries. Circ Res 90:248–250
Yamazaki J, Duan D, Janiak R, Kuenzli K, Horowitz B, Hume JR (1998) Functional and molecular expression of volume-regulated chloride channels in canine vascular smooth muscle cells. J Physiol 507:729–736
Yang YD, Cho H, Koo JY, Tak MH, Cho Y, Shim WS, Park SP, Lee J, Lee B, Kim BM, Raouf R, Shin YK, Oh U (2008) TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature 455:1210–1215
Yu Y, Kuan AS, Chen TY (2014) Calcium-calmodulin does not alter the anion permeability of the mouse TMEM16A calcium-activated chloride channel. J Gen Physiol 144:115–124
Yu K, Zhu J, Qu Z, Cui YY, Hartzell HC (2014) Activation of the Ano1 (TMEM16A) chloride channel by calcium is not mediated by calmodulin. J Gen Physiol 143:253–267
Zhong XZ, Harhun MI, Olesen SP, Ohya S, Moffatt JD, Cole WC, Greenwood IA (2010) Participation of KCNQ (Kv7) potassium channels in myogenic control of cerebral arterial diameter. J Physiol 588:3277–3293
Lidington D, Schubert R, Bolz SS (2013) Capitalizing on diversity: an integrative approach towards the multiplicity of cellular mechanisms underlying myogenic responsiveness. Cardiovasc Res 97(3):404–412
Acknowledgments
This work was supported by grants of the DFG and the Else Kröner Fresenius-Stiftung to CAH, a grant of the Thyssen Stiftung to CAH and BCS, and a grant of the DZHK to HE.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Hübner, C.A., Schroeder, B.C. & Ehmke, H. Regulation of vascular tone and arterial blood pressure: role of chloride transport in vascular smooth muscle. Pflugers Arch - Eur J Physiol 467, 605–614 (2015). https://doi.org/10.1007/s00424-014-1684-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-014-1684-y