
Abstract
Syncope which occurs suddenly in the setting of recovery from exercise, known as post-exercise syncope, represents a failure of integrative physiology during recovery from exercise. We estimate that between 50 and 80 % of healthy individuals will develop pre-syncopal signs and symptoms if subjected to a 15-min head-up tilt following exercise. Post-exercise syncope is most often neurally mediated syncope during recovery from exercise, with a combination of factors associated with post-exercise hypotension and loss of the muscle pump contributing to the onset of the event. One can consider the initiating reduction in blood pressure as the tip of the proverbial iceberg. What is needed is a clear model of what lies under the surface; a model that puts the observational variations in context and provides a rational framework for developing strategic physical or pharmacological countermeasures to ultimately protect cerebral perfusion and avert loss of consciousness. This review summarizes the current mechanistic understanding of post-exercise syncope and attempts to categorize the variation of the physiological processes that arise in multiple exercise settings. Newer investigations into the basic integrative physiology of recovery from exercise provide insight into the mechanisms and potential interventions that could be developed as countermeasures against post-exercise syncope. While physical counter maneuvers designed to engage the muscle pump and augment venous return are often found to be beneficial in preventing a significant drop in blood pressure after exercise, countermeasures that target the respiratory pump and pharmacological countermeasures based on the involvement of histamine receptors show promise.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Post-exercise syncope can be defined as loss of consciousness or development of pre-syncopal signs and symptoms during recovery from a bout of physical activity or exercise. It is an alarming response to exercise which can occur in apparently healthy individuals, including athletes, or in individuals with autonomic disorders, and represents a failure of integrative physiology in the setting of recovery from exercise. It is associated with regulated reductions in blood pressure known as post-exercise hypotension, which can be large enough in magnitude to become symptomatic (Kenny and Seals 1993; Halliwill 2001; Halliwill et al. 2013). However, most reports of post-exercise syncope are likely to be incidents of neurally mediated syncope (formally called vasovagal syncope) (Freeman et al. 2011) that have occurred during recovery from exercise, with the underlying changes associated with post-exercise hypotension contributing to the onset of the event (Kosinski et al. 2000; Krediet et al. 2004; O’Connor et al. 2009).
Several case studies of individuals who have reportedly fainted following physical activity have been examined, yet the physiology remains poorly understood (see Table 1 below for an extensive listing). Important from these case studies is the recognition that cessation of exercise is a key step in the cascade leading to neurally mediated syncope in healthy individuals (Krediet et al. 2004), whereas hypotension during exercise is more likely to occur in individuals with autonomic disorders (Low et al. 2012). Characteristically, post-exercise syncopal events occur when an individual is standing motionless during the first 5–10 min after exercise (Krediet et al. 2004), a time when the muscle pump (with its implicit role of maintaining central venous pressure during exercise in the upright position) is no longer engaged.
Prior work on “the postural problem” had suggested a key role of postural muscles (activated tonically to offset gravity and rhythmically during postural sway) in aiding venous return to the heart during quiet standing, as highlighted by Amberson (1943). However, it was classic studies in the 1940s (Hellebrandt and Franseen 1943; Pollack and Wood 1949; Hellebrandt and Cary 1949; Barcroft and Dornhorst 1949) that coined the term “muscle pump” and demonstrated the profound power of skeletal muscle contractions to reduce both venous pressure and blood volume within the dependent exercising limbs, as well as the rapidity with which this beneficial effect of skeletal muscle contraction is lost when transitioning from exercise to quiet standing. Figure 1 illustrates some of these classic demonstrations. Indeed, this early work, as well as studies by Stegall (1966), show that rhythmic muscle contractions in the dependent limbs are capable of moving blood toward the heart against significant pressure gradients (i.e., >90 mmHg) so long as venous valves are competent.

Early observations of the muscle pump. a Changes in calf volume with muscle contraction as measured by plethysmography. E denotes the onset of exercise (plantar flexion at 1 Hz for 10 s); R denotes onset of rest; CP denotes inflation of a cuff above the knee to 90 mmHg for remainder of tracing. This early study shows the considerable volume of blood that can be mobilized from the calf in response to modest muscle contractions, as well as showing that the muscle pump is capable of moving blood against a considerable pressure gradient. (Reproduced from Barcroft and Dornhorst 1949). b Changes in saphenous vein pressure with walking on a treadmill at 1.7 mph. Step-by-step changes in minimum and maximum vein pressure at the level of the ankle during quiet standing (control), slow walking, and quiet standing after walking. This early study shows that dependent limb venous pressure can be markedly reduced within a few steps by engagement of the muscle pump, as well as the rate at which venous pressure will return that that predicted by gravitational forces when muscle pump activity ceases. (Reproduced from Pollack and Wood 1949)
Thus, with quiet standing after exercise, one can anticipate a significant rise in dependent limb venous pressure and blood volumes, and a consequent fall in venous return to the heart, when this second generator of forward blood flow is switched off. This shift in blood volume, along with sustained post-exercise vasodilation and other responses associated with post-exercise hypotension, set the stage for syncope in individuals who attempt to remain stationary and upright. Along these lines, we estimate that between 50 and 80 % of healthy individuals will develop pre-syncopal signs and symptoms if they are subjected to a head-up tilt for 15 min following a range of exercise activities (see Table 2 below). This is markedly higher than the incidence of ~8 % pre-syncope during similar head-up tilt in the general population (Kosinski and Grubb 1994; Natale et al. 1995).
This topic was last explored from a mechanistic or integrative physiology perspective a decade ago (Kosinski et al. 2000; Halliwill 2001; Krediet et al. 2004), but has been reviewed from a clinical assessment perspective in more recent years (Link and Estes 2007; O’Connor et al. 2009; Asplund et al. 2011; Hastings and Levine 2012). We believe that recent investigations into the basic integrative physiology of recovery from exercise provide new insight into the mechanisms (Halliwill et al. 2013) and potential interventions (Lacewell et al. 2013) that could be developed as countermeasures against post-exercise syncope.
One can consider the reduction in blood pressure associated with post-exercise hypotension and syncope as the tip of the proverbial iceberg. What we have endeavored to create is a clear model of what lies under the surface; a model that puts the observational variations in context and provides a rational framework for developing strategic physical or pharmacological countermeasures. Part one of this review summarizes the current understanding of the mechanisms that contribute to post-exercise hypotension and syncope in humans. Part two of this review explores and attempts to categorize the variations on these physiological processes that arise in the settings of (1) moderate-intensity exercise, (2) high-intensity exercise, (3) exercise in the heat, (4) exercise at altitude, and (5) resistance exercise. Part three of this review explores and attempts to categorize targets of intervention including physical and pharmacological countermeasures against post-exercise syncope.
Part 1: review of the mechanisms underlying post-exercise hypotension and syncope
The observation that “arterial pressure becomes depressed below normal resting pressure after severe muscular work” (Hill 1898) is nearly as old as the sphygmomanometer itself. There are two distinct phenomena to consider in this context. Post-exercise hypotension represents a well-regulated reduction in arterial pressure lasting several hours following a single bout of exercise. It is well regulated in that the hemodynamic state is stable, the reduction in pressure is similar in magnitude whether the individual is studied upright or supine, and it is driven by modest quantitative changes in autonomic control of arterial pressure. In contrast, post-exercise syncope represents a failure of the normal regulation of pressure and or cerebral perfusion such that symptoms develop which are syncopal in nature. It is an unstable hemodynamic state in which there is either a failure to compensate for symptomatic hypotension or in which a qualitative change in autonomic control of arterial pressure results in paradoxical vasodilatory and bradycardic responses. With a basic definition of these two phenomena, let us explore the known mechanisms, which link them to recovery from exercise.
As recently reviewed (Halliwill et al. 2013), the hemodynamic patterns most readily observed following exercise (e.g., reduced blood pressure, increased heart rate) are part of a larger pattern of hemodynamic responses and underlying mechanisms. Work over the last few years has generated a number of important mechanistic insights into post-exercise hypotension and identified the related phenomenon of sustained post-exercise vasodilation and its primary cause. During the exercise recovery period, the combination of centrally mediated decreases in sympathetic nerve activity, in addition to a reduced signal transduction from sympathetic nerve activation into vasoconstriction, and local vasodilator mechanisms contribute to the fall in arterial blood pressure seen after exercise, which in some cases may contribute to the development of pre-syncopal signs or symptoms.
Post-exercise baroreflex resetting and blunted transduction
After exercise, the arterial baroreflex is reset to defend lower blood pressures in humans (Halliwill et al. 1996) and in rats (Kajekar et al. 2002; Miki et al. 2003), resulting in reduced sympathetic outflow (Floras et al. 1989; Halliwill et al. 1996; Kulics et al. 1999; Kajekar et al. 2002; Miki et al. 2003). In this discussion, baroreflex resetting is used to indicate a shift in the operating point of the baroreceptor reflex in absence of any change in gain or sensitivity of the control of sympathetic outflow. In humans, the sensitivity of baroreflex control of sympathetic outflow is unaltered during post-exercise resetting (Halliwill et al. 1996) but it appears blunted in rats (Kajekar et al. 2002; Miki et al. 2003). Baroreflex control of heart rate has proven to be more variable in its response to exercise, perhaps due to differences in methodology, idiosyncrasies resulting from the dual autonomic control of the heart, or thermal influences on heart rate. The current model for the mechanism of post-exercise baroreflex resetting is based on studies conducted by Chen and colleagues (reviewed in Chen and Bonham 2010), in which muscle afferents to the nucleus tractus solitarii (NTS) provide the key modulation of sympathetic output from the cardiovascular control centers in the medulla. In brief, muscle afferents activated in response to muscle contraction release substance-P at neurokinin-1 receptors on GABAergic interneurons in the caudal (NTS). These GABAergic interneurons inhibit second-order barosensitive neurons within the NTS which convey information from baroreceptor afferents to the caudal ventrolateral medulla. GABA reduces their excitability, resulting in less inhibition of sympathetic neurons in the rostral medulla, greater firing of sympathetic vasoconstrictor neurons during exercise, and resetting of the baroreflex to higher pressures during exercise. As exercise continues, neurokinin-1 receptors internalize on the GABA interneuron so that after exercise, neurokinin-1 receptors are less available for binding (Chen et al. 2009). As a result, the GABAergic interneurons exert less inhibitory influence on the second-order barosensitive neurons, leading to an overall decrease in sympathetic outflow from the rostral ventrolateral medulla after exercise. When neurokinin-1 receptors are blocked prior to exercise, post-exercise hypotension is attenuated (Chen et al. 2002). Likewise, GABAa receptor antagonism in the rostral ventrolateral medulla prevents post-exercise hypotension (Kajekar et al. 2002).
In young healthy humans exercised in thermoneutral conditions, neither the splanchnic nor renal vasculatures demonstrate changes in vascular conductance from resting pre-exercise levels to post-exercise recovery (although both vasoconstrict during exercise). This is an interesting contrast to the well-known vasoconstriction that occurs in these vascular beds in response to hypotension in general, and has been interpreted to be another representation of the central resetting of the arterial baroreflex (Pricher et al. 2004). Hence, these vascular beds do not directly contribute to post-exercise hypotension, yet they do not attenuate it. Likewise, cutaneous vascular conductance quickly returns to pre-exercise levels despite continued elevations in body core temperature (Wilkins et al. 2004). This appears to be an expression of a shift in threshold of the thermoreflexes, which allow core temperature to remain higher during recovery from exercise.
In addition to the sympathoinhibition that results from baroreflex resetting, post-exercise hypotension is associated with a blunted transduction of sympathetic outflow into vasoconstriction. For comparable levels of muscle sympathetic nerve activity, there is a reduced vascular resistance after exercise in the previously active skeletal muscles (Halliwill et al. 1996). It is unlikely that a post-junctional mechanism is responsible for this blunted sensitivity to sympathetic outflow, as both α1- and α2-adrenergic responsiveness remain intact following exercise (Halliwill et al. 2003). Pre-synaptic inhibition of norepinephrine release or increased reuptake of norepinephrine are possible explanations for the blunted transduction.
Post-exercise vasodilation
There are two recognized vasodilatory phenomena during recovery from exercise: (1) immediate post-exercise hyperemia, and (2) sustained post-exercise vasodilation of the previously active skeletal muscle vascular beds (Laughlin et al. 2012). The immediate post-exercise hyperemia can last from several seconds up to 20 min. The magnitude and duration of the hyperemia is dependent on the time, type, and intensity of exercise, but is not fully explained by mechanisms related to oxygen consumption within the previously active skeletal muscle (Morganroth et al. 1975; Bangsbo and Hellsten 1998). It has been suggested that the same vasodilator signals that drive exercise hyperemia contribute to immediate post-exercise hyperemia (Bangsbo and Hellsten 1998; Halliwill et al. 2013), but there is no consensus on its cause which is likely to be multi-factorial.
In contrast to the short-lived immediate post-exercise hyperemia, sustained post-exercise vasodilation typically lasts upwards of 2 h following moderate-intensity aerobic exercise. It is dependent on the activation of histamine H1- and H2-receptors, as combined H1- and H2-receptor antagonism reduces post-exercise vasodilation by ~80 % following 60 min of moderate-intensity cycle ergometry in both sedentary and endurance-trained athletes (Lockwood et al. 2005; McCord and Halliwill 2006; McCord et al. 2006). Further, sustained post-exercise vasodilation following 60 min of moderate-intensity unilateral dynamic knee-extension exercise is abolished by H1- and H2-receptor antagonism (Barrett-O’Keefe et al. 2013). Several possible mechanisms may increase intramuscular histamine during recovery from exercise. Mast cells located within the connective tissue layer surrounding skeletal muscle fascicles or near blood vessels may degranulate, releasing histamine locally (Metcalfe et al. 1997). Exercise-related factors that have been associated with mast cell degranulation in other contexts include reactive oxygen species (Son et al. 2006), a variety of cytokines (and perhaps myokines), increases in temperature, and vibration (Grabbe 2001). Alternatively, histamine can be formed de novo without storage in mast cells through histidine decarboxylase (Watanabe and Ohtsu 2002). Along these lines, histidine decarboxylase mRNA expression (Endo et al. 1998) and enzyme activity increase with prolonged exercise in mice (but not rats) (Graham et al. 1964; Ayada et al. 2000). There appear to be links between oxidative stress (Höcker et al. 1998), hypoxia inducible factor-1α (Jeong et al. 2009), and histidine decarboxylase transcription. There is also older evidence that increased shear stress promotes histamine formation in large vessels such as the aorta (DeForrest and Hollis 1978). Future studies are needed to define the cascade of events that result in post-exercise histamine-receptor activation. In particular, studies must identify the exercise-related signal (e.g., oxidative stress, increased muscle temperature) that activates the histaminergic pathway, determine the source of histamine released by this signal (i.e., mast cell degranulation and/or de novo formation), and the timing or dose–response of this system in relation to exercise.
Obligatory versus situational influences
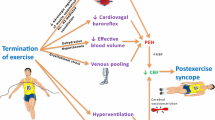
As a way to comprehend varied outcomes during recovery from exercise, we identified mechanisms that appear to be obligatory versus situational influences that vary from case to case. Along these lines, prolonged muscle afferent activation and consequent resetting of the baroreflex, resetting of thermoreflexes (Journeay et al. 2006), pre-synaptic inhibition of sympathetic vasoconstrictor nerves, and activation of H1- and H2-receptors in the previously active muscle all appear to be obligatory components of post-exercise hypotension. Of these factors, histaminergic vasodilation may be the most important contributor to the fall in pressure in the normotensive and recreationally active population, as blockade of histamine receptors (Lockwood et al. 2005; McCord and Halliwill 2006; McCord et al. 2006; Barrett-O’Keefe et al. 2013), but not removal of the sympathetic component (Halliwill et al. 2000), reduces post-exercise hypotension. Situational influences include the fluid status of the individual, heat balance with the environment, and the presence or absence of gravitational stress. We have explored how combinations of obligatory mechanisms and situational influences produce the integrated responses observed, as illustrated conceptually in Fig. 2 (reproduced from Halliwill et al. 2013).

Integrated hemodynamic responses following exercise. Regulated reductions in blood pressure known as post-exercise hypotension can be large enough in magnitude to induce pre-syncopal symptoms and lead to post-exercise syncope. The expression of post-exercise hypotension is the integration of a variety of obligatory and situational components. Obligatory components (indicated in pale blue) include (1) a sustained histaminergic vasodilation of the previously exercise skeletal muscle vascular beds, (2) resetting of the baroreflex (which generally results in sympathoinhibition of sympathetic nerves to muscle vascular beds), (3) pre-synaptic inhibition of norepinephrine release from sympathetic nerves to the exercised muscle, and (4) resetting of thermoreflexes. These changes manifest as a rise in vascular conductance of the previously exercised muscle, inhibition of sympathetic vasoconstrictor nerve activity to previously active muscle vascular beds, and little or no change in cutaneous vascular conductance despite higher core temperatures. Situational components (indicated in light yellow) include the impact of whether or not significant fluid loss, gravitational pooling of blood, and hyperthermia are present. These components can greatly impact on the extent to which cardiac output is elevated. (Reproduced from Halliwill et al. 2013)
The majority of studies report an elevated cardiac output concurrent with post-exercise hypotension, at least when subjects are studied in the supine position (Halliwill 2001); however, this is not always the case. Reductions in cardiac output are commonly reported following exercise in endurance-trained men even in the supine position (but not in endurance-trained women, for unknown reasons) (Senitko et al. 2002). Heart rate is consistently elevated following exercise, and this is likely the product of higher temperatures affecting the SA node pacemaker. This may confound many of the studies exploring baroreflex control of heart rate following exercise.
Stroke volume is generally well maintained, despite reductions in central venous pressure (Halliwill et al. 2000). Stroke volume appears highly sensitive to both core temperature and fluid status but seems to be supported by enhanced contractility of unknown origin. Again, endurance-trained men stand alone as a group that exhibits a reduced stroke volume following exercise (Senitko et al. 2002; Lynn et al. 2009). Ironically, it may be that the greater heat storage seen in less fit men (a response which allows for heat dissipation via physical mechanisms when physiological mechanisms are limited), leads to an enhanced cardiac contractility which is absent in highly fit men, and the absence of this effect leads to the marked fall in stroke volume in endurance-trained men after exercise (Lynn et al. 2009).
It is worth noting that the majority of studies reporting cardiac output and stroke volume have been performed with subjects in a supine position, and one would predict that upright recovery, via the increase in gravitational pooling of blood, would shift the system toward lower stroke volumes and cardiac outputs. While this is often observed (McCord et al. 2008), there are also examples in which cardiac output remained surprisingly high during upright tilt after exercise (Lacewell et al. 2013). These differences are likely related to the different exercise stimuli in these studies which may result in differing thermal, autonomic, and hormonal influences on the heart. Further, the impact of changes in plasma volume, which decreases ~10 % in the upright position, may be markedly impacted by exercise or thermal stress, and may change in response to the vascular responses in liver, skin, and muscle vascular beds.
Despite all these potential variations in the underlying physiology, in most cases where measurements are made upright following moderate-intensity exercise, post-exercise blood pressure remains lower than pre-exercise, yet is still well regulated in the upright position (Senitko et al. 2002).
Post-exercise syncope
Post-exercise syncope has an unknown rate of occurrence. There is likely an overlap between severe post-exercise hypotension, the development of post-exercise syncope, and the phenomenon of exercise-induced collapse, particularly when collapse occurs immediately after exercise and/or completion of athletic performance (e.g., crossing the finish line of a marathon) (Noakes 1988; Holtzhausen et al. 1994; Holtzhausen and Noakes 1995, 1997; Kenefick and Sawka 2007). While it is likely that the regulated reductions in blood pressure known as post-exercise hypotension can be large enough in magnitude as to become symptomatic under some circumstances, most reports of post-exercise syncope are likely incidences of neurally mediated syncope (formerly called vasovagal syncope) during recovery from exercise, with the underlying changes associated with post-exercise hypotension contributing to the onset of the event (Krediet et al. 2004). Generally speaking, when standing upright following exercise, elevations in skeletal muscle blood flow combined with an inactive muscle pump can lead to marked reductions in venous return to the heart, a response which can be exacerbated in the heat (Krediet et al. 2004; McCord et al. 2008; Kenney et al. 2013). By mechanisms which remain surprisingly murky (Noakes 2003; Hainsworth 2003; Joyner 2009), the situation of high cardiac contractility and compromised venous return may trigger both paradoxical reductions in sympathetic vasoconstrictor nerve activity and increases in parasympathetic cardiac nerve activity. The mechanism or trigger for such neurally mediated syncope is often attributed to ventricular mechanoreceptors firing in response to a misinterpreted tissue distortion and producing a response that is akin to the Bezold-Jarish reflex. Others have argued that the trigger is more likely to be generated within the central nervous system rather than within the ventricles (Hainsworth 2003), and may involve serotonin receptors in the rostral ventrolateral medulla (Dean and Bago 2002). This is a neglected topic of investigation. Regardless of where the origins of the response resides, the shift toward greater vasodilation (due to sympathetic withdrawal) and a relative or frank bradycardia leads to a rapid collapse of arterial pressure and cerebral perfusion.
It is possible that some cases of post-exercise syncope are related to alterations in cerebral blood flow independent of vasovagal reactions or frank hypotension. For example, hypocapnia induced by hyperventilation during recovery from exercise could produce a cerebral vasoconstriction and diminish cerebral oxygen delivery in the absence of any change in perfusion pressure. Along these lines, Rasmussen et al. (2006) showed that cerebral CO2 vascular reactivity following strenuous exercise is increased such that arterial CO2 becomes the primary factor influencing changes in cerebral blood flow during and after prolonged exercise.
The potential for changes in cerebral autoregulation following exercise has also been explored. While cerebral autoregulation is well maintained following moderate-intensity exercise (Ogoh et al. 2007; Murrell et al. 2007; Willie et al. 2013), two studies suggest that more intense exercise may produce a lasting cerebral autoregulatory deficit. Specifically, Ogoh et al. (2005) found reduced dynamic cerebral autoregulation during heavy cycle exercise and Bailey et al. (2011) found reduced dynamic cerebral autoregulation during recovery from a recumbent cycle peak test. Thus, it seems likely that cerebral perfusion is less protected from sudden onset hypotension following more intense exercise.
Table 1 summarizes case reports and case series of post-exercise syncope in which individual patient information on age and sex were provided. It is worth noting that many cases include asystole, evidence of the vagal component of neurally mediated syncope. However, depending on the timing of events and the progression toward symptoms, bradycardia is not always evident.
In reviewing the case reports and case series, some interesting patterns are evident regarding age and sex of individual patients, as indicated in Fig. 3. First, in contrast to conventional wisdom, the majority of cases of post-exercise syncope occur in men. In general, women have decreased orthostatic tolerance compared to men, and this holds true in response to heat stress (Meendering et al. 2005), but not recovery from exercise (Lacewell et al. 2013). It is reasonable to suggest that the higher number of cases in men is a spurious result, perhaps due to greater historical participation in physical activities, but the pattern holds true in several prospective studies (Fleg and Lakatta 1986; Lacewell et al. 2013). Second, the cases represent a broad range of ages, with nearly every decade of life represented with a higher proportion of cases below the age of 50.

Sex and age of patients evaluated with post-exercise syncope. a The percentage of male and female patients in case reports. b The distribution of case reports on post-exercise syncope by age. Both figures are based on data from references listed in Table 1 (n = 27)
There are several other notable case series related to post-exercise syncope. First, as part of the Baltimore Longitudinal Study on Aging, Fleg and Lakatta (1986) analyzed post-exercise blood pressure responses in a large cohort of healthy volunteers. During seated recovery from a graded maximal treadmill test, 3.1 % of subjects younger than 55 years exhibited a marked hypotension that was associated with pre-syncopal symptoms. The incidence was lower in women than men, and was only 0.3 % for subjects older than 55 years. Long-term investigation of subjects who became symptomatically hypotensive following exercise did not indicate any increased health risks, supporting the notion that the prognosis related to post-exercise syncope is generally benign. Second, Sakaguchi et al. (1995) reported on a group of 12 patients who were referred for exercise-related syncope. They noted a general susceptibility to neurally mediated syncope in these individuals, fostering the concept that most cases of syncope following exercise are incidences of neurally mediated syncope. Third, Holtzhausen et al. (1994) reported on 46 male athletes who collapsed during or after a 56-km ultramarathon, finding that 85 % of these cases occurred after the finish line (i.e., post-exercise), and that hydration status (a surrogate for circulating blood volume) and core temperature in those who collapsed post-race are no different than control athletes who do not collapse. In other words, traditional contributing factors such as dehydration, hypovolemia, and hyperthermia are not necessary for post-exercise syncope following longer duration athletic events. Finally, an interesting report by Eckart et al. (2010) suggests that the use of ergogenic supplements (e.g., stimulants such as ephedra and ephedra-substitutes) can increase the risk of post-exercise syncope threefold.
While the long-term prognosis related to post-exercise syncope is generally benign, it is important that other more serious causes be ruled out by thorough clinical evaluation, as syncope after exercise can be a sign of a significant pathophysiology (Kramer et al. 1988; Smith et al. 1993; Krediet et al. 2004; O’Connor et al. 2009; Hastings and Levine 2012).
Part 2: exploration of different exercise models that generate post-exercise syncope
Much of our understanding of the mechanisms of post-exercise hypotension come from a single model of exercise, namely, moderate-intensity exercise of medium-duration (e.g., up to 60 min) performed in a thermoneutral environment. However, the cases of post-exercise syncope do not fall neatly into this model, but come from a range of exercise activities which may differentially affect arterial pressure regulation or control of cerebral oxygen delivery. Let us explore five archetypal exercise models, with the understanding that when individuals exercise, it may be on a spectrum that combines elements of these five models. Table 2 summarizes prospective studies on post-exercise syncope, as they relate to these models, in which pre-syncopal rates during head-up tilt (or tolerance to lower body negative pressure) were assessed during recovery from exercise.
Moderate-intensity exercise
In young healthy normotensive individuals, moderate-intensity exercise for 30–60 min produces modest post-exercise hypotension via the mechanisms described above, but is generally insufficient to elicit post-exercise syncope during seated recovery or brief (5 min) head-up tilt when performed in a thermoneutral environment (Raine et al. 2001; Senitko et al. 2002). However, the incidence of orthostatic intolerance can be surprisingly high in response to even mild exercise when orthostatic stress is more prolonged (15 min) (Mayerson 1944). Further, when moderate-intensity exercise is of extended duration, or under moderately warm conditions, the pre-syncopal incidence can reach as high as 54–81 % (Eichna et al. 1947a; Murrell et al. 2009, 2011), as shown in Table 2.
This model of post-exercise syncope is ostensibly explained by the effects of the sustained post-exercise vasodilation on reducing total peripheral resistance and reducing venous return to the heart. Noakes and colleagues (Noakes 1988, 2007; Holtzhausen and Noakes 1997) have argued that hyperthermia and dehydration play little or no role in the collapse of athletes at the end of marathons, whereas sustained peripheral vasodilation and loss of the muscle pump appear sufficient to explain many cases of post-exercise syncope following moderate-intensity exercise of extended duration. It is worth noting that the early work by Eichna et al. (1947a) found the increased incidence of pre-syncope during head-up tilt persists for 1–2 h following exercise. This is a timeline that is similar to the duration of the sustained post-exercise vasodilation that is mediated by histamine-receptor activation (Emhoff et al. 2011).
High-intensity exercise
In contrast to moderate-intensity exercise, there are fewer studies investigating post-exercise hypotension and related phenomenon following short-duration high-intensity exercise. However, there is anecdotal evidence that the Wingate test of anaerobic power, a staple of undergraduate exercise physiology labs, results in pre-syncopal symptoms in many individuals. Likewise, the classic study by Eichna et al. (1947a) found that more than half of healthy individuals will become pre-syncopal during a 5-min head-up tilt after completion of maximal exercise. More recently, Lacewell et al. (2013) developed a modified Wingate test which produces post-exercise syncope in many individuals. Such maximal or supra-maximal efforts when followed by head-up tilt or active standing elicit pre-syncopal responses in 58–100 % of individuals, as shown in Table 2.
What is interesting in the study by Lacewell et al. (2013) is that arterial pressure was often well maintained at or above pre-exercise levels until a sudden shift to hypotension and development of pre-syncopal symptoms. In other words, arterial pressure did not demonstrate post-exercise hypotension prior to post-exercise syncope, rather pressure was well maintained (until it was not).
One of the characteristics of recovery from high-intensity exercise is marked hyperventilation which can reduce arterial CO2 to values that would likely induce prominent cerebral vasoconstriction. For example, after a modified Wingate test, end-tidal CO2 decreased 14 mmHg to levels that would be associated with a ~40 % reduction in cerebral perfusion (Lacewell et al. 2013). There are cases of athletes developing syncope as a result of brief vigorous hyperventilation (Buja et al. 1989).
The potential role of autoregulation in this model also merits discussion. While cerebral autoregulation is well maintained following moderate-intensity exercise (Ogoh et al. 2007; Murrell et al. 2007; Willie et al. 2013), two studies suggest that more intense exercise may produce a lasting cerebral autoregulatory deficit. Specifically, Ogoh et al. (2005) found reduced dynamic cerebral autoregulation (as assessed by transfer function) during heavy cycle exercise and Bailey et al. (2011) found reduced dynamic cerebral autoregulation (as assessed by thigh-cuff release) during recovery from a recumbent cycle peak test. Thus, it seems likely that cerebral perfusion is less protected from sudden onset hypotension following high-intensity exercise.
Exercise in the heat
Exercise in the heat adds another level of complexity to these integrated responses. This model is exemplified by moderate-intensity exercise of moderate to long duration that is performed in a hot environment, which could be either arid or humid. Thus, the situational component that adds complexity to this model is the greater heat load, secondary to the reduced ability to dissipate metabolic heat to the environment. When individuals are exercising hard enough or long enough to generate significant increases in core temperature, then the physiological accommodations to heat stress such as cutaneous vasodilation and sweating can lead to reductions in central venous pressure, dehydration, and hypovolemia.
As recently reviewed by Kenney et al. (2013), there is a commensal use of cardiac output to serve both the metabolic demands of the exercising muscle and thermoregulatory need for heat dissipation (Rowell 1977). While skin blood flow may increase to more than 7 L/min (more than half of available cardiac output) during passive heat stress (Rowell 1986; Crandall et al. 2010), in normal exercising individuals the blood flow needed to maintain heat dissipation and muscular activity is not a great enough demand to limit either of these vasculatures or to compromise blood pressure regulation (Kenney et al. 2013). That said, the non-competing division of cardiac output may deteriorate into competition for limited cardiac output at the end of exercise (when the muscle pump is lost), but some surprising observations have been made.
While the greater fluid loss associated with exercise in the heat can be predicted to decrease cardiac preload, Lynn et al. (2009) showed that greater elevations in heart rate following exercise in the heat are associated with an augmentation of cardiac output that is as great as what can be produced by fluid replacement, at least when subjects are supine. However, it is clear that the skin, when vasodilated in response to passive heat stress, is insufficiently vasoconstricted with orthostatic stress to support cardiac preload and, therefore, predisposes individuals to the development of syncope in the heat (Crandall 2008; Crandall et al. 2010; Pearson et al. 2013).
Following exercise, other vascular beds likely vasoconstrict to maintain pressure in response to orthostatic challenge. While the arterial baroreflex is reset to maintain arterial pressure at lower levels following exercise, sensitivity of baroreflex control of sympathetic outflow is augmented after exercise in the heat, possibly due to greater reductions in central venous pressure that result in decreased cardiopulmonary baroreceptor activation (Charkoudian et al. 2003). However, it is likely that the vasoconstrictor reserve is reached for important vasculatures such as the splanchnic region during this combination of stressors (Crandall 2008; Crandall et al. 2010; Pearson et al. 2013), leaving little if any margin for maintaining cerebral perfusion in the upright position. Further, there is some evidence that exercise in the heat may produce a lasting deficit in the cerebral autoregulation which is observable after the return to resting body temperatures (Carter et al. 2006). Thus, it is not surprising that classic studies documented a decreased orthostatic tolerance following exercise in the heat (Greenleaf et al. 1974; Shvartz et al. 1977), and brief high-intensity (Greenleaf et al. 1974), prolonged low-intensity (Shvartz et al. 1977), and medium-duration moderate-intensity (McCord et al. 2008) exercise in the heat have all been used as prospective models for studying post-exercise syncope.
Exercise at altitude
Anecdotes of altitude-related syncope both during and after exercise abound, but few (if any) have been documented in the literature (Hultgren 1997). That said, it is well established that neurally mediated syncope can be produced in most individuals by having them breathe low O2 levels (<8 %), and that some individuals will become syncopal while breathing moderately hypoxic O2 mixtures (13–14 % O2) similar to the O2 levels at altitudes of 3,000–4,000 m. At these altitudes, orthostatic tolerance is reduced (Halliwill 2003).
Early hemodynamic studies highlighted a potential role of exaggerated circulating epinephrine levels in precipitating hypoxic syncope (Halliwill 2003; Halliwill and Minson 2005). Dinenno et al. (2003) were able to obtain an arterial blood sample from a hypoxic individual at the onset of hypotension and bradycardia and found fourfold higher epinephrine than other subjects exposed to the same degree of hypoxia. This observation is consistent with the notion of exaggerated circulating epinephrine in hypoxic syncope. In this individual, high epinephrine was associated with progressive skeletal muscle vasodilation, but part of the hypotension may have been linked to hypoxic vasodilation of the splanchnic circulation (Rowell and Blackmon 1989; Westendorp et al. 1997). One case report suggests that β-adrenergic blockade may prevent hypoxic syncope (Freitas et al. 1996).
With this background on hypoxic syncope, one can surmise that post-exercise syncope at altitude is multi-factorial, combining the elements of the other exercise models with epinephrine-induced vasodilation and a possible splanchnic steal of blood flow. These factors, which acting alone can compromise cerebral perfusion, would act in combination with reduced arterial oxygen content to diminish cerebral oxygenation.
Resistance exercise
While most of the focus of this review is on the response to dynamic whole-body exercise, it is worth mentioning that resistance exercise is also associated with post-exercise syncopal episodes. Often referred to as “weightlifter’s blackout,” post-resistance exercise syncope occurs during exercise or within seconds of completion (Compton et al. 1973).
Unlike the moderate blood pressure increase that occurs during aerobic exercise, arterial blood pressure is profoundly elevated during resistance exercise. Arterial blood pressure has been reported to reach 320/250 mmHg during maximal double-leg press (Macdougall et al. 1985), although the rise in pressure during submaximal resistance exercise does not approach these levels (Edwards et al. 2002; Romero and Cooke 2007). In the face of increased perfusion pressure, cerebral perfusion (measured via transcranial Doppler) is unchanged (Edwards et al. 2002) or slightly elevated (Romero and Cooke 2007) during moderate-intensity resistance exercise, but is reduced during high-intensity resistance exercise (Dickerman et al. 2000; Perry et al. 2013) perhaps as protection against hypertension (Tzeng and Ainslie 2013).
Subsequent to the marked elevation in blood pressure during resistance exercise, pressure falls rapidly to below pre-exercise levels upon the completion of the lift (Macdougall et al. 1985; Edwards et al. 2002; Pott et al. 2003; Romero and Cooke 2007). As a result, cerebral perfusion falls below pre-exercise levels immediately following exercise (Edwards et al. 2002; Romero and Cooke 2007). The precipitous fall in arterial blood pressure immediately following resistance exercise may be a sufficient challenge to the cerebrovascular regulatory mechanisms to produce syncope (i.e., the large and rapid “swing” in pressure may be beyond the upper and lower limits of cerebral autoregulation) (Macdougall et al. 1985; Dickerman et al. 2000).
Several additional mechanisms may exacerbate this state and contribute to post-resistance exercise syncope. First, voluntary or involuntary hyperventilation before or during resistance exercise may induce cerebral vasoconstriction (Compton et al. 1973; Romero and Cooke 2007), similar to what we have discussed above for high-intensity exercise. Second, Valsalva straining during lifting may produce sudden blood pressure transitions both during and immediately after straining (Compton et al. 1973). Third, dehydration is a common practice among competitive weightlifters and other weight-class athletes (e.g., wrestling and boxing), and the reduced circulating blood volume associated with hypohydration can further reduce tolerance to post-exercise orthostatic challenges (Hand 1997; Moralez et al. 2012).
In summary, one can consider these five archetypal exercise models as situational variations on the underlying physiology that may increase or decrease the predisposition toward post-exercise syncopal events. Some of these models are well developed, but some remain poorly investigated.
Part 3: classifications of potential countermeasures
The majority of evidence indicates that post-exercise syncope, in the absence of other medical conditions, is a nuisance. One to three percent of emergency department visits in the United States, with an estimated cost of $5,400 per patient and totaling $2.5 billion a year, are due to syncope (Sun et al. 2005). It is important to recognize that post-exercise syncope can be situationally life-threatening (e.g., cycling, climbing, firefighting, and military operations), and several case reports on post-exercise syncope involve firefighters (Osswald et al. 1994; van den Berg et al. 1995; Krediet et al. 2006). Thus, the development of countermeasures which can protect against post-exercise syncope should focus on ways to protect the individual from loss of consciousness during recovery from exercise. The current arsenal of countermeasures falls into three basic categories: behavioral, pharmacological, and pacemakers. The evidence for these existing countermeasures is almost exclusively anecdotal. Table 3 summarizes case reports of post-exercise syncope which included an intervention or treatment and follow-up.
To the best of our knowledge, there is one randomized controlled trial for post-collapse treatment (but not prevention) (Anley et al. 2011), one randomized controlled trial for prevention in athletes with post-exercise syncope (Privett et al. 2010b), and two randomized controlled trials for prevention in models of post-exercise syncope performed in healthy subjects (McCord et al. 2008; Lacewell et al. 2013), as summarized in Table 4; Fig. 4.
Behavioral
It is disappointing that one of the most common medical recommendations to patients with post-exercise syncope is to simply avoid vigorous exercise. Considering the numerous health benefits of routine vigorous exercise, this is not an acceptable remedy. Rather, avoidance of exercise should be seen as a last resort when all other countermeasures have failed to protect the individual.
In contrast to preaching avoidance, a number of easily learned physical countermeasures designed to engage the muscle pump and augment venous return are often found to be beneficial in preventing a significant drop in blood pressure after exercise. A classic study by Eichna et al. (1947a) showed that alternate movement of the legs during orthostatic hypotension following exhaustive physical work in young, healthy soldiers raised blood pressure markedly. Likewise, Krediet et al. (2005) demonstrated that a simple squatting maneuver could ameliorate pre-syncopal symptoms and normalize blood pressure in a patient in whom neurally mediated syncope could be consistently triggered by moderate cycle exercise. One can consider such countermeasures, along with the advice to “keep moving” after exercise, as a simple and effective means to avoid many cases of post-exercise syncope, as active recovery has been reported to be beneficial in some cases (Tamura et al. 1990; Krediet et al. 2006). However, there are likely limitations inherent in asking an exhausted athlete to rely on their fatigued muscles to protect them from post-race collapse, and it is likely that the finish-line chute standstill has contributed to some cases of syncope (Holtzhausen and Noakes 1995; Anon 2004). If active recovery is not practical, then use of the Trendelenberg position (or simply lifting the legs of the supine individual) may be an effective way to rectify venous return to the heart (Anley et al. 2011).
Lacewell et al. (2013) recently demonstrated the benefits of using an inspiratory resistance device to alleviate post-exercise syncope following high-intensity exercise, as shown in Fig. 4b. Originally developed for the treatment of severe hemorrhage and shock, an impedance threshold device generates resistance during inspiration which may augment the function of the respiratory pump in mobilizing venous blood toward the heart. However, it is possible that the main benefit of the device following high-intensity exercise is the attenuation of hypocapnic cerebral vasoconstriction or enhancement of a cerebral siphon effect.

Survival time during head-up tilt following exercise—two models and two countermeasures. The proportion of subjects remaining in the tilted position as shown by survival function curve. a Effect of H1-receptor blockade on head-up tilt after 45 min running in the heat. (Modified from McCord et al. 2008). b Effect of inspiratory resistance on head-up tile after 1 min high-intensity exercise. (Modified from Lacewell et al. 2013). Solid line represents the control day; dotted line countermeasure day
Another behavioral approach with promise is the use of water ingestion (Thijs et al. 2003), not in the prevention of dehydration, but owing to the pressor response elicited by ingestion of modest to large volumes of hypotonic fluid. In other studies, fluid ingestion to replace that lost during exercise has been shown to reduce cardiovascular strain during lower body negative pressure (Davis and Fortney 1997). However, when treating post-competition collapse following endurance events Anley et al. (2011) found that use of the Trendelenberg position was as effective as intravenous fluids.
Lastly, compression garments can be of benefit in reducing the likelihood of post-exercise syncope (Privett et al. 2010b). Use of compression garments of various types are seeing more widespread use by the athletic community, so routine use of such garments in an individual with recurring symptoms during recovery from exercise may be an appealing alternative to some of the more invasive countermeasures described below.
Pharmacological
It is clear that many of the clinical approaches that are employed to treat patients with post-exercise syncope have been adapted from those tools that have also seen routine use in treating neurally mediated syncope. Thus, the use of β-adrenergic blocking drugs is the most common pharmacological countermeasure (see Table 3), followed by the anti-arrhythmic agent disopyramide (which is used in part because of its vagolytic effects). These standard pharmacological approaches remain controversial in treatment of neurally mediated syncope, where anecdotal evidence and case reports are promising but evidence from controlled trials is often inconclusive (Kaufmann and Freeman 2004).
It is important to recognize that none of the common pharmacological approaches, such as β-adrenergic blocking drugs, act directly on the underlying physiology related to post-exercise hypotension or sustained post-exercise vasodilation (except perhaps in the setting of altitude, where circulating epinephrine is an issue). However, McCord et al. (2008), tested the effectiveness of H1-receptor antagonism in preventing post-exercise syncope induced by head-up tilt following exercise in the heat. They found the magnitude of the arterial pressure drop during head-up tilt was blunted by administration of an H1-receptor antagonist, and subjects were able to stand longer before the development of pre-syncopal signs and symptoms, as shown in Fig. 4a. In essence, this is the first pharmacological countermeasure which directly targets the underlying physiology of recovery from exercise.
Pacemakers
In a number of cases, pacemakers have been implanted in patients with post-exercise syncope. Like the use of β-adrenergic blocking drugs, this is a countermeasure adapted from the clinical toolbox routinely used to deal with neurally mediated syncope. While a number of case reports (see Table 3) suggest that pacemakers are effective, such anecdotes are far from definitive and the use of pacemakers for the treatment of neurally mediated syncope remains controversial (Petersen and Sutton 1997; Romme et al. 2011). As reviewed here, knowledge of the physiology of post-exercise hypotension, sustained post-exercise vasodilation, and post-exercise syncope do not provide a rationale for the use of implanted pacemakers, when other less-invasive countermeasures are likely to be beneficial.
Other directions
We believe there are other potential directions for interventions, and that a systematic view of the underlying physiology of post-exercise hypotension and syncope can be used as a roadmap. Specifically, each of the five archetypal exercise models may respond to targeted interventions with greater efficacy than a “one size fits all” approach, but some interventions may prove universally beneficial, but perhaps to lesser effect. The ideal solution, if it exists, would be a non-invasive countermeasure that does not require the individual to medicate daily, and that can be deployed “as needed” without the need to carry equipment or wear special clothing.
One approach would be countermeasures that increase total peripheral resistance. In hot conditions, rapid skin cooling may prove an effective means of reducing cutaneous vascular conductance and improving hemodynamic stability. Along these lines, Wilson et al. (2002) demonstrated that, in a model of passive heat stress, rapid skin cooling protected arterial pressure, cerebral perfusion, and decreased pre-syncopal symptoms during head-up tilt. In other situations, sympathoexcitatory maneuvers such as bolus fluid ingestion of water or hand immersion in ice water to elicit a cold pressor response might be sufficient to increase total peripheral resistance and provide benefit. Pharmacological interventions that target specific vascular beds such as the highly compliant splanchnic circulation may prove effective in enhancing total peripheral resistance (Jarvis et al. 2012), and could be very advantageous for exercise at altitude.
Another approach would be countermeasures designed to augment cardiac preload, beyond administering fluids and engaging the muscle pump (Tamura et al. 1990; Krediet et al. 2006). Augmentation of the respiratory pump via inspiratory resistance shows some promise in this area (Lacewell et al. 2013), and it is possible that some of the sympathoexcitatory maneuvers or rapid skin cooling also function to augment cardiac preload. External compression by medical compression garments or other means may be viable, but perhaps less than ideal.
The least explored approach would be countermeasures designed to protect cerebral blood flow directly, by either causing cerebral vasodilation or boosting cerebral perfusion pressure gradients. Along these lines, when hypocapnia is a likely contributing factor (high-intensity exercise or resistance exercise), it is possible that something as simple as rebreathing from a paper bag might help protect cerebral blood flow by maintaining higher arterial CO2 levels. To the extent that the cerebral siphon effect is present in upright humans (which remains highly debated), breathing maneuvers which engage the respiratory pump may prove effective in augmenting the cerebral perfusion pressure gradient (as discussed in Lacewell et al. 2013). Lastly, supplemental oxygen could also be beneficial, but it seems to be seldom deployed during recovery from exercise aside from NFL games played at modest altitudes (~1,610 m).
One interesting potential countermeasure that may need to be re-explored is the role of acclimatization to heat. Early work by Greenleaf et al. (1974) found that orthostatic tolerance following exercise in the heat was, surprisingly, made worse by prior heat acclimatization. It is unclear how acclimatization, which greatly improves the ability to exercise in the heat and is associated with plasma volume expansion, would prove to be deleterious in the setting of post-exercise syncope.
Summary and conclusions
Blood pressure regulation after exercise is about much more than a simple loss of the muscle pump. Newer investigations on the recovery from exercise provide insight into the mechanisms of baroreflex resetting, the sustained histamine-mediated vasodilation, and changes in cerebral autoregulation. Thus, obligatory alterations in the control of sympathetic vascular tone and skeletal muscle blood flow interact with situational effects like cutaneous vasodilation in the heat, dehydration, hypoxic vasodilation, and hypocapnic cerebral vasoconstriction to produce the post-exercise hypotension, and in some cases post-exercise syncope. Mining of case and prospective studies hints at additional influences regarding who is likely to suffer from pre-syncopal symptoms after exercise and under what circumstances. Together, this information creates a framework for challenging some of the traditional treatment strategies and for developing novel countermeasures against post-exercise syncope.
References
Amberson WR (1943) Physiological adjustments to the standing position. Univ Md Sch Med Bull 27:127–145
Anley C, Noakes TD, Collins M, Schwellnus MP (2011) A comparison of two treatment protocols in the management of exercise-associated postural hypotension: a randomised clinical trial. Br J Sports Med 45:1113–1118
Anon (2004). A knockout finish. Runner’s World (March)
Arad M, Solomon A, Roth A, Atsmon J, Rabinowitz B (1993) Postexercise syncope: evidence for increased activity of the sympathetic nervous system. Cardiology 83:121–123
Asplund CA, O’Connor FG, Noakes TD (2011) Exercise-associated collapse: an evidence-based review and primer for clinicians. Br J Sports Med 45:1157–1162
Ayada K, Watanabe M, Endo Y (2000) Elevation of histidine decarboxylase activity in skeletal muscles and stomach in mice by stress and exercise. Am J Physiol Regul Integr Comp Physiol 279:R2042–R2047
Bailey DM, Evans KA, McEneny J, Young IS, Hullin DA, James PE, Ogoh S, Ainslie PN, Lucchesi C, Rockenbauer A, Culcasi M, Pietri S (2011) Exercise-induced oxidative-nitrosative stress is associated with impaired dynamic cerebral autoregulation and blood-brain barrier leakage. Exp Physiol 96:1196–1207
Bangsbo J, Hellsten Y (1998) Muscle blood flow and oxygen uptake in recovery from exercise. Acta Physiol Scand 162:305–312
Barcroft H, Dornhorst AC (1949) Demonstration of the muscle pump in the human leg. J Physiol 108:39P
Barrett-O’Keefe Z, Kaplon RE, Halliwill JR (2013) Sustained postexercise vasodilatation and histamine receptor activation following small muscle-mass exercise in humans. Exp Physiol 98:268–277
Bjurstedt H, Rosenhamer G, Balldin U, Katkov V (1983) Orthostatic reactions during recovery from exhaustive exercise of short duration. Acta Physiol Scand 119:25–31
Brogdon E, Hellebrandt FA (1940) Post-exercise orthostatic collapse. Am J Physiol 129:P318
Buja G, Folino AF, Bittante M, Canciani B, Martini B, Miorelli M, Tognin D, Corrado D, Nava A (1989) Asystole with syncope secondary to hyperventilation in three young athletes. PACE 12:406–412
Carter RI, Cheuvront SN, Vernieuw CR, Sawka MN (2006) Hypohydration and prior heat stress exacerbates decreases in cerebral blood flow velocity during standing. J Appl Physiol 101:1744–1750
Charkoudian N, Halliwill JR, Morgan BJ, Eisenach JH, Joyner MJ (2003) Influences of hydration on post-exercise cardiovascular control in humans. J Physiol 552:635–644
Chen CY, Bonham AC (2010) Postexercise hypotension: central mechanisms. Exerc Sport Sci Rev 38:122–127
Chen CY, Munch PA, Quail AW, Bonham AC (2002) Postexercise hypotension in conscious SHR is attenuated by blockade of substance P receptors in NTS. Am J Physiol Heart Circ Physiol 283:H1856–H1862
Chen CY, Bechtold AG, Tabor J, Bonham AC (2009) Exercise reduces GABA synaptic input onto nucleus tractus solitarii baroreceptor second-order neurons via NK1 receptor internalization in spontaneously hypertensive rats. J Neurosci 29:2754–2761
Compton D, Hill PM, Sinclair JD (1973) Weight-lifters’ blackout. Lancet 2:1234–1237
Crandall CG (2008) Heat stress and baroreflex regulation of blood pressure. Med Sci Sports Exerc 40:2063–2070
Crandall CG, Shibasaki M, Wilson TE (2010) Insufficient cutaneous vasoconstriction leading up to and during syncopal symptoms in the heat stressed human. Am J Physiol Heart Circ Physiol 299:H1168–H1173
Crisafulli A, Melis F, Orrù V, Lener R, Lai C, Concu A (2000) Hemodynamic during a postexertional asystolia in a healthy athlete: a case study. Med Sci Sports Exerc 32:4–9
Davis JE, Fortney SM (1997) Effect of fluid ingestion on orthostatic responses following acute exercise. Int J Sports Med 18:174–178
Dean C, Bago M (2002) Renal sympathoinhibition mediated by 5-HT1Areceptors in the RVLM during severe hemorrhage in rats. Am J Physiol Regul Integr Comp Physiol 282:R122–R130
DeForrest JM, Hollis TM (1978) Shear stress and aortic histamine synthesis. Am J Physiol Heart Circ Physiol 3:H701–H705
Dickerman RD, McConathy WJ, Smith GH, East JW, Rudder L (2000) Middle cerebral artery blood flow velocity in elite power athletes during maximal weight-lifting. Neurol Res 22:337–340
Dinenno FA, Joyner MJ, Halliwill JR (2003) Failure of systemic hypoxia to blunt alpha-adrenergic vasoconstriction in the human forearm. J Physiol 549:985–994
Dockery BK, Newman KP (2007) Exercise-induced asystole with syncope in a healthy young man. Am J Med Sci 334:145–148
Eckart RE, Gentlesk PJ, Shry EA (2010) Differential manifestation of cardiovascular complaints as a function of utilization of ergogenic supplements. PACE 33:286–289
Edwards MR, Martin DH, Hughson RL (2002) Cerebral hemodynamics and resistance exercise. Med Sci Sports Exerc 34:1207–1211
Eichna LW, Horvath SM, Bean WB (1947a) Post-exertional orthostatic hypotension. Am J Med Sci 213:641–654
Eichna LW, Horvath SM, Bean WB (1947b) Cardiac asystole in a normal young man following physical effort. Am Heart J 33:254–262
Emhoff CA, Barrett-O’Keefe Z, Padgett RC, Hawn JA, Halliwill JR (2011) Histamine-receptor blockade reduces blood flow but not muscle glucose uptake during postexercise recovery in humans. Exp Physiol 96:664–673
Endo Y, Tabata T, Kuroda H, Tadano T, Matsushima K, Watanabe M (1998) Induction of histidine decarboxylase in skeletal muscle in mice by electrical stimulation, prolonged walking and interleukin-1. J Physiol 509:587–598
Fleg JL, Asante AV (1983) Asystole following treadmill exercise in a man without organic heart disease. Arch Int Med 143:1821–1822
Fleg JL, Lakatta EG (1986) Prevalence and significance of postexercise hypotension in apparently healthy subjects. Am J Cardiol 57:1380–1384
Floras JS, Sinkey CA, Aylward PE, Seals DR, Thoren PN, Mark AL (1989) Postexercise hypotension and sympathoinhibition in borderline hypertensive men. Hypertension 14:28–35
Freeman R et al (2011) Consensus statement on the definition of orthostatic hypotension, neurally mediated syncope and the postural tachycardia syndrome. Clin Auto Res 21:69–72
Freitas J, Costa O, Carvalho MJ, de Freitas AF (1996) High altitude-related neurocardiogenic syncope. Am J Cardiol 77:1021
Grabbe J (2001) Pathomechanisms in physical urticaria. J Invest Dermatol Symp Proc 6:135–136
Graham P, Kahlson G, Rosengren E (1964) Histamine formation in physical exercise, anoxia and under the influence of adrenaline and related substances. J Physiol 172:174–188
Gratze G, Mayer H, Skrabal F (2008) Sympathetic reserve, serum potassium, and orthostatic intolerance after endurance exercise and implications for neurocardiogenic syncope. Eur Heart J 29:1531–1541
Greenleaf JR, Bosco JS, Matter M (1974) Orthostatic tolerance in dehydrated, heat-acclimated men following exercise in the heat. Aerospace Med 45:491–497
Hainsworth R (2003) Syncope: what is the trigger? Heart 89:123–124
Halliwill JR (2001) Mechanisms and clinical implications of post-exercise hypotension in humans. Exerc Sport Sci Rev 29:65–70
Halliwill JR (2003) Hypoxic regulation of blood flow in humans. Skeletal muscle circulation and the role of epinephrine. Adv Exper Med Biol 543:223–236
Halliwill JR, Minson CT (2005) Cardiovagal regulation during combined hypoxic and orthostatic stress: fainters vs. nonfainters. J Appl Physiol 98:1050–1056
Halliwill JR, Taylor JA, Eckberg DL (1996) Impaired sympathetic vascular regulation in humans after acute dynamic exercise. J Physiol 495:279–288
Halliwill JR, Minson CT, Joyner MJ (2000) Effect of systemic nitric oxide synthase inhibition on postexercise hypotension in humans. J Appl Physiol 89:1830–1836
Halliwill JR, Dinenno FA, Dietz NM (2003) Alpha-adrenergic vascular responsiveness during postexercise hypotension in humans. J Physiol 550:279–286
Halliwill JR, Buck TM, Lacewell AN, Romero SA (2013) Postexercise hypotension and sustained postexercise vasodilation: what happens after we exercise? Exp Physiol 98:7–18
Hand J (1997) Exercise-induced vasodepressor syncope in a collegiate wrestler: a case study. J Ath Train 32:359–362
Hastings JL, Levine BD (2012) Syncope in the Athletic Patient. Prog Cardiovasc Dis 54:438–444
Hellebrandt FA, Cary MK (1949) Relative importance of the muscle pump in the prevention of gravity shock. Phys Ther Rev 29:12–22
Hellebrandt FA, Franseen EB (1943) Physiological study of the vertical stance of man. Physiol Rev 23:220–255
Hill L (1898) Arterial pressure in man while sleeping, resting, working and bathing. J Physiol 22:xxvi-xxx
Hirata T, Yano K, Okui T, Mitsuoka T, Hashiba K (1987) Asystole with syncope following strenuous exercise in a man without organic heart disease. J Electrocardiol 20:280–283
Höcker M, Rosenberg I, Xavier R, Henihan RJ, Wiedenmann B, Rosewicz S, Podolsky DK, Wang TC (1998) Oxidative stress activates the human histidine decarboxylase promoter in AGS gastric cancer cells. J Biol Chem 273:23046–23054
Holtzhausen LM, Noakes TD (1995) The prevalence and significance of post-exercise (postural) hypotension in ultramarathon runners. Med Sci Sports Exerc 27:1595–1601
Holtzhausen LM, Noakes TD (1997) Collapsed ultraendurance athlete: proposed mechanisms and an approach to management. Clin J Sport Med 7:292–301
Holtzhausen LM, Noakes TD, Kroning B, de Klerk M, Roberts M, Emsley R (1994) Clinical and biochemical characteristics of collapsed ultra-marathon runners. Med Sci Sports Exerc 26:1095–1101
Hultgren H (1997) Syncope. In: High altitude medicine. Hultgren Publications, Stanford, pp 419–423
Huycke EC, Card HG, Sobol SM, Nguyen NX, Sung RJ (1987) Postexertional cardiac asystole in a young man without organic heart disease. Ann Int Med 106:844–845
Jarvis SS, Florian JP, Curren MJ, Pawelczyk JA (2012) A somatostatin analog improves tilt table tolerance by decreasing splanchnic vascular conductance. J Appl Physiol 112:1504–1511
Jeong HJ, Moon PD, Kim SJ, Seo JU, Kang TH, Kim JJ, Kang IC, Um JY, Kim HM, Hong SH (2009) Activation of hypoxia-inducible factor-1 regulates human histidine decarboxylase expression. Cell Mol Life Sci 66:1309–1319
Journeay WS, Carter R, Kenny GP (2006) Thermoregulatory control following dynamic exercise. Aviat Space Environ Med 77:1174–1182
Joyner MJ (2009) Orthostatic stress, haemorrhage and a bankrupt cardiovascular system. J Physiol 587:5015–5016
Kajekar R, Chen CY, Mutoh T, Bonham AC (2002) GABA(A) receptor activation at medullary sympathetic neurons contributes to postexercise hypotension. Am J Physiol Heart Circ Physiol 282:H1615–H1624
Kapoor WN (1989) Syncope with abrupt termination of exercise. Am J Med 87:597–599
Karadag B, Aliyev F, Duman D, Ongen Z, Vural VA (2007) Post-exercise cardiac asystole in persons without underlying heart disease: management and follow-up of three cases. J Cardiol Med 8:855–858
Kaufmann H, Freeman R (2004) Pharmacological treatment of reflex syncope. Clin Auto Res 14(Suppl 1):71–75
Kenefick RW, Sawka MN (2007) Heat exhaustion and dehydration as causes of marathon collapse. Sports Med 37:378–381
Kenney WL, Stanhewicz AE, Bruning RS, Alexander LM (2013) Blood pressure regulation III: what happens when one system must serve two masters: temperature and pressure regulation? Eur J Appl Physiol. doi:10.1007/s00421-013-2652-5
Kenny MJ, Seals DR (1993) Postexercise hypotension: key features, mechanisms, and clinical significance. Hypertension 22:653–664
Kosinski DJ, Grubb BP (1994) Neurally mediated syncope with an update on indications and usefulness of head-upright tilt table testing and pharmacologic therapy. Curr Opin Cardiol 9:53–64
Kosinski D, Grubb BP, Karas BJ, Frederick S (2000) Exercise-induced neurocardiogenic syncope: clinical data, pathophysiological aspects, and potential role of tilt table testing. Europace 2:77–82
Kramer R, Drori Y, Lev B (1988) Sudden death in young soldiers. High incidence of syncope prior to death. Chest 93:345–347
Krediet CTP, Wilde AAM, Wieling W, Halliwill JR (2004) Exercise related syncope, when it’s not the heart. Clin Auto Res 14(Suppl):25–36
Krediet CTP, Wilde AAM, Halliwill JR, Wieling W (2005) Syncope during exercise, documented with continuous blood pressure monitoring during ergometer testing. Clin Auto Res 15:59–62
Krediet CTP, Wilde AAM, Halliwill JR, Wieling W (2006) Post-exercise vasovagal syncope. In: Civera R, Esquivias G, Bland J, Brignole M, Mitjans A, Granell R, Wieling W (eds) Syncope Cases. Blackwell, Malden, pp 46–48
Kulics JM, Collins HL, DiCarlo SE (1999) Postexercise hypotension is mediated by reductions in sympathetic nerve activity. Am J Physiol Heart Circ Physiol 45:H27–H32
Lacewell AN, Buck TM, Romero SA, Halliwill JR (2013) Post-exercise syncope: wingate syncope test and effective countermeasure. Exp Physiol. doi:10.1113/expphysiol.2013.075333
Laughlin MH, Davis MJ, Secher NH, van Lieshout JJ, Arce-esquivel AA, Simmons GH, Bender SB, Padilla J, Bache RJ, Merkus D, Duncker DJ (2012) Peripheral circulation. Compr Physiol 2:321–447
Link MS, Estes NAM (2007) How to manage athletes with syncope. Cardiol Clin 25:457–466
Lockwood JM, Wilkins BW, Halliwill JR (2005) H1 receptor-mediated vasodilatation contributes to postexercise hypotension. J Physiol 563:633–642
Low DA, da Nóbrega AC, Mathias CJ (2012) Exercise-induced hypotension in autonomic disorders. Auton Neurosci 171:66–78
Lowe MD, Petch MC, Everard P (2000) Syncope after effort. Postgrad Med J 76:164–165
Luft UC, Myhre LG, Loeppky JA, Venters MD (1976) A study of factors affecting tolerance of gravitational stress simulated by lower body negative pressure. Lovelace Foundation. NASA Contract NAS 9-14472. Albuquerque
Lynn BM, Minson CT, Halliwill JR (2009) Fluid replacement and heat stress during exercise alter post-exercise cardiac haemodynamics in endurance exercise-trained men. J Physiol 587:3605–3617
Macdougall JD, Tuxen D, Sale DG, Moroz JR, Sutton JR (1985) Arterial blood pressure response to heavy resistance exercise. J Appl Physiol 58:785–790
Mayerson HS (1944) A cardiovascular “blackout” test. J Aviation Med 15:304–315
McCord JL, Halliwill JR (2006) H1 and H2 receptors mediate postexercise hyperemia in sedentary and endurance exercise-trained men and women. J Appl Physiol 101:1693–1701
McCord JL, Beasley JM, Halliwill JR (2006) H2-receptor-mediated vasodilation contributes to postexercise hypotension. J Appl Physiol 100:67–75
McCord JL, Pellinger TK, Lynn BM, Halliwill JR (2008) Potential benefit from an H1-receptor antagonist on postexercise syncope in the heat. Med Sci Sports Exerc 40:1953–1961
Meendering JR, Torgrimson BN, Houghton BL, Halliwill JR, Minson CT (2005) Menstrual cycle and sex affect hemodynamic responses to combined orthostatic and heat stress. Am J Physiol Heart Circ Physiol 289:H631–H642
Metcalfe D, Baram D, Mekori Y (1997) Mast cells. Physiol Rev 77:1033–1079
Miki K, Yoshimoto M, Tanimizu M (2003) Acute shifts of baroreflex control of renal sympathetic nerve activity induced by treadmill exercise in rats. J Physiol 548:313–322
Moralez G, Romero SA, Rickards CA, Ryan KL, Convertino VA, Cooke WH (2012) Effects of dehydration on cerebrovascular control during standing after heavy resistance exercise. J Appl Physiol 112:1875–1883
Morganroth M, Mohrman D, Sparks H (1975) Prolonged vasodilation following fatiguing exercise of dog skeletal muscle. Am J Physiol 229:38–43
Murrell C, Wilson L, Cotter JD, Lucas S, Ogoh S, George K, Ainslie PN (2007) Alterations in autonomic function and cerebral hemodynamics to orthostatic challenge following a mountain marathon. J Appl Physiol 103:88–96
Murrell C, Cotter JD, George K, Shave R, Wilson L, Thomas K, Williams MJA, Lowe T, Ainslie PN (2009) Influence of age on syncope following prolonged exercise: differential responses but similar orthostatic intolerance. J Physiol 587:5959–5969
Murrell CJ, Cotter JD, George K, Shave R, Wilson L, Thomas K, Williams MJA, Ainslie PN (2011) Syncope is unrelated to supine and postural hypotension following prolonged exercise. Eur J Appl Physiol 111:469–476
Natale A, Akhtar M, Jazayeri M, Dhala A, Blanck Z, Deshpande S, Krebs A, Sra JS (1995) Provocation of hypotension during head-up tilt testing in subjects with no history of syncope or presyncope. Circulation 92:54–58
Noakes TD (1988) Why marathon runners collapse. S Afr Med J 73:569–571
Noakes TD (2003) The forgotten Barcroft/Edholm reflex: potential role in exercise associated collapse. Br J Sports Med 37:277–282
Noakes TD (2007) Reduced peripheral resistance and other factors in marathon collapse. Sports Med 37:382–385
O’Connor FG, Levine BD, Childress MA, Asplundh CA, Oriscello RG (2009) Practical management: a systematic approach to the evaluation of exercise-related syncope in athletes. Clin J Sport Med 19:429–434
Ogoh S, Dalsgaard MK, Yoshiga CC, Dawson EA, Keller DM, Raven PB, Secher NH (2005) Dynamic cerebral autoregulation during exhaustive exercise in humans. Am J Physiol Heart Circ Physiol 288:H1461–H1467
Ogoh S, Fisher JP, Purkayastha S, Dawson EA, Fadel PJ, White MJ, Zhang R, Secher NH, Raven PB (2007) Regulation of middle cerebral artery blood velocity during recovery from dynamic exercise in humans. J Appl Physiol 102:713–721
Osswald S, Brooks R, O’Nunain SS, Curwin JH, Roelke M, Radvany P, Ruskin JN, McGovern BA (1994) Asystole after exercise in healthy persons. Ann Int Med 120:1008–1011
Pearson J, Lucas RAI, Crandall CG (2013) Elevated local skin temperature impairs cutaneous vasoconstrictor responses to a simulated haemorrhagic challenge while heat stressed. Exp Physiol 98:444–450
Pedersen WR, Janosik DL, Goldenberg IF, Stevens LL, Redd RM (1989) Post-exercise asystolic arrest in a young man without organic heart disease: utility of head-up tilt testing in guiding therapy. Am Heart J 118:410–413
Perry BG, Schlader ZJ, Barnes MJ, Cochrane DJ, Lucas SJE, Mündel T (2013) Hemodynamic response to upright resistance exercise: effect of load and repetition. Med Sci Sports Exerc. doi:10.1249/MSS.0b013e3182a7980f
Petersen ME, Sutton R (1997) Cardiac pacing for vasovagal syncope: a reasonable therapeutic option? PACE 20:824–826
Pollack AA, Wood EH (1949) Venous pressure in the saphenous vein at the ankle in man during exercise and changes in posture. J Appl Physiol 1:649–662
Pott F, Van Lieshout JJ, Ide K, Madsen P, Secher NH (2003) Middle cerebral artery blood velocity during intense static exercise is dominated by a Valsalva maneuver. J Appl Physiol 94:1335–1344
Pricher MP, Holowatz LA, Williams JT, Lockwood JM, Halliwill JR (2004) Regional hemodynamics during postexercise hypotension. I. Splanchnic and renal circulations. J Appl Physiol 97:2065–2070
Privett SE, George KP, Middleton N, Whyte GP, Cable NT (2010a) The effect of prolonged endurance exercise upon blood pressure regulation during a postexercise orthostatic challenge. Br J Sports Med 44:720–724
Privett SE, George KP, Whyte GP, Cable NT (2010b) The effectiveness of compression garments and lower limb exercise on post-exercise blood pressure regulation in orthostatically intolerant athletes. Clin J Sport Med 20:362–367
Raine NM, Cable NT, George KP, Campbell IG (2001) The influence of recovery posture on post-exercise hypotension in normotensive men. Med Sci Sports Exerc 33:404–412
Rasmussen P, Stie H, Nielsen B, Nybo L (2006) Enhanced cerebral CO2 reactivity during strenuous exercise in man. Eur J Appl Physiol 96:299–304
Romero SA, Cooke WH (2007) Hyperventilation before resistance exercise: cerebral hemodynamics and orthostasis. Med Sci Sports Exerc 39:1302–1307
Romme JJ, Reitsma JB, Black CN, Colman N, Scholten RJ, Wieling W, Van Dijk N (2011) Drugs and pacemakers for vasovagal, carotid sinus and situational syncope. Cochrane Database Syst Rev 5(10):CD004194
Rowell LB (1977) Competition between skin and muscle for blood flow during exercise. In: Nadel ER (ed) Problems with temperature regulation during exercise. Academic Press, New York, pp 49–77
Rowell LB (1986) Human circulation: Regulation during physical stress. Oxford University Press, New York
Rowell LB, Blackmon JR (1989) Hypotension induced by central hypovolaemia and hypoxaemia. Clin Physiol 9:269–277
Sakaguchi S, Shultz JJ, Remole SC, Adler SW, Lurie KG, Benditt DG (1995) Syncope associated with exercise, a manifestation of neurally mediated syncope. Am J Cardiol 75:476–481
Schlesinger Z (1973) Letter: life-threatening “vagal reaction” to physical fitness test. JAMA 226:1119
Senitko AN, Charkoudian N, Halliwill JR (2002) Influence of endurance exercise training status and gender on postexercise hypotension. J Appl Physiol 92:2368–2374
Shvartz E, Meroz A, Magazanik A, Shoenfeld Y, Shapiro Y (1977) Exercise and heat orthostatism and the effect of heat acclimation and physical fitness. Aviat Space Environ Med 48:836–842
Smith GD, Bannister R, Mathias CJ (1993) Post-exertion dizziness as the sole presenting symptom of autonomic failure. Br Heart J 69:359–361
Son A, Nakamura H, Kondo N, Matsuo Y, Liu W, Oka S, Ishii Y, Yodoi J (2006) Redox regulation of mast cell histamine release in thioredoxin-1 (TRX) transgenic mice. Cell Res 16:230–239
Stegall HF (1966) Muscle pumping in the dependent leg. Circ Res 19:180–190
Sun BC, Emond JA, Camargo CA (2005) Direct medical costs of syncope-related hospitalizations in the United States. Am J Cardiol 95:668–671
Tamura Y, Onodera O, Kodera K, Igarashi Y, Miida T, Aizawa Y, Izumi T, Shibata A, Takano S (1990) Atrial standstill after treadmill exercise test and unique response to isoproterenol infusion in recurrent postexercise syncope. Am J Cardiol 65:533–535
Thijs RD, Reijntjes RHAM, van Dijk JG (2003) Water drinking as a potential treatment for idiopathic exercise-related syncope: a case report. Clin Auto Res 13:103–105
Tse HF, Lau CP (1995) Exercise-associated cardiac asystole in persons without structural heart disease. Chest 107:572–576
Tsutsumi E, Hara H, RP B (1979) Syncope after running. Br Med J 280:1480
Tzeng Y-C, Ainslie PN (2013). Blood pressure regulation IX: cerebral autoregulation under blood pressure challenges. Eur J Appl Physiol
Ueyama T, Hano T, Kasamatsu K, Mohara O, Shima H, Ueshima K, Fukuda K, Ueno Y, Nishio I (1994) A case of orthostatic and postexertional hypotension with ischemic electrocardiographic changes. Jap Heart J 35:375–382
Van den Berg MP, Crijns HJ, Bouwmeester TR, Smit AJ, Lie KI (1995) Cardiac asystole post-exercise: a report of two cases. Int J Cardiol 51:296–300
Watanabe T, Ohtsu H (2002) l-histidine decarboxylase as a probe in studies on histamine. Chem Rec 2:369–376
Westendorp RG, Blauw GJ, Frölich M, Simons R (1997) Hypoxic syncope. Aviat Space Environ Med 68:410–414
Wilkins BW, Minson CT, Halliwill JR (2004) Regional hemodynamics during postexercise hypotension. II. Cutaneous circulation. J Appl Physiol 97:2071–2076
Willie CK, Ainslie PN, Taylor CE, Eves ND, Tzeng Y-C (2013) Maintained cerebrovascular function during post-exercise hypotension. Eur J Appl Physiol 113:1597–1604
Wilson TE, Cui J, Zhang R, Witkowski S, Crandall CG (2002) Skin cooling maintains cerebral blood flow velocity and orthostatic tolerance during tilting in heated humans. J Appl Physiol 93:85–91
Ziegelstein RC (2004) Near-syncope after exercise. JAMA 292:1221–1226
Acknowledgments
This research was supported in part by grants from the American Heart Association, Grant-in-Aid 11GRNT5490000, and National Institutes of Health Grant HL115027.
Author contributions
All authors contributed directly to the writing of this review. Each drafted a substantial section, assisted in revising the entire review, and approved the final version.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Nigel A. S. Taylor.
Rights and permissions
About this article
Cite this article
Halliwill, J.R., Sieck, D.C., Romero, S.A. et al. Blood pressure regulation X: what happens when the muscle pump is lost? Post-exercise hypotension and syncope. Eur J Appl Physiol 114, 561–578 (2014). https://doi.org/10.1007/s00421-013-2761-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00421-013-2761-1