
Abstract
Reactive oxygen species (ROS), including superoxide (·O2−), hydrogen peroxide (H2O2), and hydroxyl anion (OH-), and reactive nitrogen species, such as nitric oxide (NO) and peroxynitrite (ONOO−), are biologically important O2 derivatives that are increasingly recognized to be important in vascular biology through their oxidation/reduction (redox) potential. All vascular cell types (endothelial cells, vascular smooth muscle cells, and adventitial fibroblasts) produce ROS, primarily via cell membrane-associated NAD(P)H oxidase. Reactive oxygen species regulate vascular function by modulating cell growth, apoptosis/anoikis, migration, inflammation, secretion, and extracellular matrix protein production. An imbalance in redox state where pro-oxidants overwhelm anti-oxidant capacity results in oxidative stress. Oxidative stress and associated oxidative damage are mediators of vascular injury and inflammation in many cardiovascular diseases, including hypertension, hyperlipidemia, and diabetes. Increased generation of ROS has been demonstrated in experimental and human hypertension. Anti-oxidants and agents that interrupt NAD(P)H oxidase-driven ·O2− production regress vascular remodeling, improve endothelial function, reduce inflammation, and decrease blood pressure in hypertensive models. This experimental evidence has evoked considerable interest because of the possibilities that therapies targeted against reactive oxygen intermediates, by decreasing generation of ROS and/or by increasing availability of antioxidants, may be useful in minimizing vascular injury and hypertensive end organ damage. The present chapter focuses on the importance of ROS in vascular biology and discusses the role of oxidative stress in vascular damage in hypertension.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Reactive oxygen species (ROS) are ubiquitous reactive derivatives of O2 metabolism found in the environment and in all biological systems. Reactive oxygen species from mitochondria and other cellular sources were traditionally considered as injurious cellular by-products with the potential to damage lipids, proteins and DNA (Freeman and Crapo 1982). However, there is now convincing evidence that ROS are not only toxic consequences of cellular metabolism but also essential participants in cell signaling and regulation (Griendling et al. 2000a; Sauer et al. 2001; Reth 2002; Chiarugi and Cirri 2003). Reactive oxygen species are implicated in many intracellular signaling pathways leading to changes in gene transcription and protein synthesis and consequently in cell function.
Within the cardiovascular system, ROS play an essential physiological role in maintaining cardiac and vascular integrity and a pathophysiological role in cardiovascular dysfunction associated with conditions such as hypertension, diabetes, atherosclerosis, ischemia–reperfusion injury, ischemic heart disease, and congestive cardiac failure (Landmesser and Harrison 2001; Zalba et al. 2001a). Among the major ROS important in these processes are superoxide anion (·O2−), hydrogen peroxide (H2O2), hydroxyl radical (·OH), and the reactive nitrogen species, nitric oxide (NO) and peroxynitrite (ONOO−). Under physiological conditions, ROS are produced in a controlled manner at low concentrations and function as signaling molecules regulating vascular smooth muscle cell (VSMC) contraction–relaxation and VSMC growth (Rao and Berk 1992; Cosentino et al. 1994; Zafari et al. 1998; Touyz and Schiffrin 1999). Under pathological conditions increased ROS production leads to endothelial dysfunction, increased contractility, VSMC growth and apoptosis, monocyte migration, lipid peroxidation, inflammation, and increased deposition of extracellular matrix proteins, major processes contributing to vascular damage in cardiovascular disease (Rao and Berk 1992; Harrison 1997).
In experimental and clinical hypertension, ROS generation is increased (Kerr et al. 1999; Romero and Reckelhoff 1999; Schnackenberg et al. 1999; Chen et al. 2001). Treatment with antioxidants improves vascular function and structure, prevents target-organ damage, and reduces blood pressure in animal models (Romero and Reckelhoff 1999; Schnackenberg et al. 1999; Chen et al. 2001; Hoagland et al. 2003) and in human hypertension (Sharma et al. 1996; Duffy et al. 1999; Fotheby et al. 2000; Boshtam et al. 2002; Mullan et al. 2002). Mouse models deficient in ROS-forming enzymes have lower blood pressure compared with wild-type counterparts and angiotensin II (Ang II) infusion in these mice fails to induce hypertension (Bendall et al. 2002; Li and Shah 2003). Furthermore, in cultured VSMCs and isolated arteries from hypertensive rats and humans, ROS production is augmented and antioxidant capacity is reduced (Schnackenberg et al. 1999; Chen et al. 2001; Touyz and Schiffrin 2001). Accordingly, evidence at both experimental and clinical levels supports a pathophysiological role for ROS and oxidative stress in the development and progression of hypertension and its associated end-organ damage.
In the present review, we focus on the role of ROS in vascular biology and implications of oxidative stress in hypertensive vascular damage. Although the cardiac, renal, endocrine, and central nervous systems are also major targets for oxidative damage by ROS, these systems will not be discussed here and the reader is referred to excellent recent reviews on these systems (Wilcox 2002; Zanzinger 2002; Cantor et al. 2003). In this chapter we discuss mechanisms whereby ROS are formed in vascular cells, especially relating to non-phagocytic NAD(P)H oxidase, how ROS influence vascular function, and what the implications of oxidative stress are in vascular injury in hypertension.
Reactive oxygen species and oxidative stress in the vasculature
Reactive oxygen species are formed as intermediates in reduction–oxidation (redox) processes, leading from oxygen to water (Fridovich 1997). The univalent reduction of oxygen, in the presence of a free electron (e), yields ·O2−, H2O2, and ·OH. Superoxide has an unpaired electron, which imparts high reactivity and renders it unstable and short-lived. Superoxide is water soluble and acts either as an oxidizing agent, where it is reduced to H2O2, or as a reducing agent, where it donates its extra electron to form ONOO− with NO (Darley-Usmar et al. 1995; Fridovich 1997). In physiological conditions in aqueous solutions at a neutral pH, the favored reaction of ·O2− is the dismutation reaction yielding H2O2. However, when produced in excess, a significant amount of ·O2− reacts with NO to produce ONOO− (Darley-Usmar et al. 1995). Superoxide is membrane-impermeable, but can cross cell membranes via anion channels (Schafer and Beuttner 2001; Han et al. 2003).
Hydrogen peroxide is produced mainly from dismutation of ·O2−. This reaction can be spontaneous or it can be catalyzed by superoxide dismutase (SOD), of which there are three isoforms, CuZnSOD, MnSOD, and extracellular SOD (EC-SOD) (Fridovich 1997). The SOD-catalyzed dismutation is favored when the concentration of ·O2− is low and when the concentration of SOD is high, which occurs under physiological conditions. Unlike ·O2−,H2O2 is not a free radical and is a much more stable molecule. Hydrogen peroxide is lipid soluble, crosses cell membranes, and has a longer half-life than ·O2−. In biological systems, it is scavenged by catalase and by glutathione peroxidase (Schafer and Beuttner 2001). Hydrogen peroxide can also be reduced to generate the highly reactive ·OH (Haber-Weiss or Fenton reaction) in the presence of metal-containing molecules such as Fe2+ (Fridovich 1997). Hydroxyl radical is extremely reactive and unlike ·O2− and H2O2, which travel some distance from their site of generation, ·OH induces local damage where it is formed.
In the vasculature, ·O2−, H2O2, NO, OONO−,and ·OH are all produced to varying degrees (Fig. 1). These pro-oxidants are tightly regulated by anti-oxidants such as SOD, catalase, thioredoxin, glutathione, anti-oxidant vitamins, and other small molecules (Stralin et al. 1995; Halliwell 1999; Channon and Guzik 2002; Yamawaki et al. 2003). Under normal conditions, the rate of ROS production is balanced by the rate of elimination. However, a mismatch between ROS formation and the ability to defend against them by antioxidants results in increased bioavailability of ROS leading to a state of oxidative stress (Griendling et al. 2000a; Landmesser and Harrison 2001; Zalba et al. 2001a). The pathogenic outcome of oxidative stress is oxidative damage (Griendling et al. 2000a; Schafer and Buettner 2001; Zalba et al. 2001a), a major cause of vascular injury in hypertension.

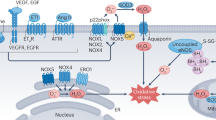
Generation of superoxide (·O2−) and H2O2 from O2 in vascular cells. Many enzyme systems, including NAD(P)H oxidase, xanthine oxidase, and uncoupled nitric oxide synthase (NOS) among others, have the potential to generate reactive oxygen species (ROS). Superoxide acts either as an oxidizing agent, where it is reduced to H2O2 by superoxide dismutase (SOD), or as a reducing agent, where it donates its extra electron (e−) to form ONOO− with NO. Hydrogen peroxide is scavenged by catalase, glutathione and thioredoxin systems, and can also be reduced to generate ·OH in the presence of Fe2+
Production of ROS in vessels
Non-phagocytic, vascular NAD(P)H oxidase(s)
Vascular ROS are produced in endothelial, adventitial, and VSMCs (Stralin et al. 1995; Rajagopalan et al. 1996a; Halliwell 1999; Channon and Guzik 2002; Sorescu et al. 2002; Yamawaki et al. 2003) and derived predominantly from NAD(P)H oxidase, which is a multi-subunit enzyme (Jones et al. 1996; Azumimi et al. 1999; Griendling et al. 2000b; Lassegue and Clempus 2003) that catalyzes the production of ·O2− by the one electron reduction of oxygen using NAD(P)H as the electron donor: 2O2 + NAD(P)H → 2O2− + NAD(P)H + H+. The prototypical and best characterized NAD(P)H oxidase is that found in phagocytes (neutrophilic and eosinophilic granulocytes, monocytes, and macrophages; Leusen et al. 1996; Babior et al. 2002; Vignais 2002). Phagocytic NAD(P)H oxidase comprises at least five components: (phox for PHagocyte OXidase), p47phox, p67phox, p40phox, p22phox, and gp91phox (Babior et al. 2002; Vignais 2002). Additional components include the small G proteins Rac2 (Rac1 in some cells) and Rap1A. In unstimulated cells, p40phox, p47phox, and p67phox exist in the cytosol, whereas p22phox and gp91phox are located in the membranes, where they occur as a heterodimeric flavoprotein, cytochrome b558. Upon cell stimulation, p47phox becomes phosphorylated, the cytosolic subunits form a complex, which then migrates to the membrane where it associates with cytochrome b558 to assemble the active oxidase, which now transfers electrons from the substrate to O2 leading to ·O2− generation (Leusen et al. 1996; Babior et al. 2002). Whereas phosphorylation of p47phox and p67phox is critically involved in the activation of NAD(P)H oxidase (Touyz et al. 2003a), phosphorylated p40phox is not essential for activation and has recently been reported to be a negative regulator of the oxidase (Lopes et al. 2004).
It is now clearly established that NAD(P)H oxidase is also functionally important in non-phagocytic cells. In fact NAD(P)H oxidase is the primary source of ·O2− in the vasculature (Berry et al. 2000; Channon and Guzik 2002; Touyz et al. 2002a; Lassegue and Clempus 2003) and is functionally active in all layers of the vessel wall: in the endothelium (Muzaffar et al. 2003), the media (Berry et al. 2000; Touyz et al. 2002a), the adventitia (Rey and Pagano 2002), and in cultured VSMCs, fibroblasts, and endothelial cells (Griendling et al. 1994; Seshiah et al. 2002; Touyz et al. 2002a). Unlike phagocytic NAD(P)H oxidase, which is activated only upon stimulation and which generates ·O2− in a burst-like manner extracellularly (De Leo et al. 1996; Babior et al. 2002), vascular oxidases are constitutively active, preassembled, and produce ·O2− intracellularly in a slow and sustained fashion and act as intracellular signaling molecules (Li and Shah 2002; Lassegue and Clempus 2003; Table 1).
All of the phagocytic NAD(P)H oxidase subunits are expressed, to varying degrees, in vascular cells. In endothelial and adventitial cells p47phox, p67phox, p22phox, and gp91phox are present (Rey and Pagano 2002; Lassegue and Clempus 2003; Touyz et al. 2003a). The situation is more complex in VSMCs, where the major subunits are not always detected. Only p47phox and p22phox seem to be consistently expressed (Lassegue and Clempus 2003). In rat aortic VSMCs, p22phox and p47phox, but not gp91phox, are present, whereas in human resistance arteries, all of the major subunits, including gp91phox, are expressed (Azumimi et al. 1999; Touyz et al. 2002a; Yamawaki et al. 2003). Recent studies demonstrated that the newly discovered gp91phox (Nox2) homologs, Nox1, Nox4, and Nox5 (Nox for NAD(P)H Oxidase) are found in the vasculature (Suh et al. 1999; Cheng et al. 2001; Ago et al. 2004; Hilenski et al. 2004). Nox1 mRNA is expressed in rat aortic VSMCs and may be a substitute for gp91phox in these cells (Suh et al. 1999; Griendling et al. 2000b; Touyz et al. 2002a). Although initial studies suggested that Nox1 is a subunit-independent low capacity ·O2−-generating enzyme involved in the regulation of mitogenesis (Banfi et al. 2003), recent data indicate that Nox1 requires p47phox and p67phox and that it is regulated by NoxO1 (Nox organizer 1) and NoxA1 (Nox activator 1) (Banfi et al. 2003). The exact role of NoxO1 and NoxA1 in vascular cells is currently unknown. Nox1 may be important in pathological processes as it is significantly upregulated in vascular injury (Lassegue and Clempus 2003). Nox4 appears to be abundantly expressed in all vascular cell types (Wingler et al. 2001; Yamawaki et al. 2003) and may play an important role in constitutive production of ·O2− in non-proliferating cells (Lassegue et al. 2001; Wingler et al. 2001). Ago et al. (2004) recently reported that Nox4 is the major catalytic component of endothelial NAD(P)H oxidase. A unique p67phox homolog has also been identified, but it is not yet known whether this isoform is present in vascular cells (Gauss et al. 2002). The functional significance of NAD(P)H oxidase subunit homologs in the vasculature is presently unclear and awaits further clarification.
Vascular NAD(P)H oxidase is regulated by many humoral factors, including cytokines, growth factors, and vasoactive agents (Lassegue and Clempus 2003; Fig. 1, 2). Physical factors, such as stretch, pulsatile strain, and shear stress also stimulate NAD(P)H oxidase activation (Grote et al. 2003; Lassegue and Clempus 2003). Of particular importance, with respect to hypertension, is Ang II, which stimulates activation of NAD(P)H oxidase, increases expression of NAD(P)H oxidase subunits, and induces ROS production in cultured VSMCs, endothelial cells, adventitial fibroblasts (Lassegue and Clempus 2003), and in intact arteries (Griendling et al. 1994; Seshiah et al. 2002; Touyz et al. 2002a; Yamawaki et al. 2003). Oxidase activation occurs acutely by stimulation of intracellular signaling molecules (Griendling et al. 2000b; Lassegue and Clempus 2003) and chronically by upregulation of NAD(P)H oxidase subunits (Touyz et al. 2002a; Lassegue and Clempus 2003). These effects are mediated via AT1 receptors (Privratsky et al. 2003). Interestingly ROS regulate AT1 receptor gene expression, which in turn modulates ROS formation (Nickenig et al. 2000).

Upstream regulators of NAD(P)H oxidase in vascular cells. Vasoactive agents, such as angiotensin II, which signal through G protein-coupled receptors (GPCR), growth factors and cytokines, which signal through receptor tyrosine kinases and physical factors, such as stretch and pressure, stimulate NAD(P)H oxidase through multiple signaling cascades. AA Arachidonic acid, PLA2 phospholipase A2, PLD phospholipase D, PKC protein kinase C
Mechanisms linking Ang II/AT1 to NAD(P)H oxidase and upstream signaling molecules regulating the oxidase in vascular cells have not been fully elucidated, but PLD, PLA, PKC, c-Src, PI3 K, RhoA, and Rac have been demonstrated to be implicated in AT1 signaling to NAD(P)H oxidase (Seshiah et al. 2002; Touyz et al. 2003a; Fig. 2). Platelet-derived growth factor (PDGF), transforming growth factor-β (TGF-β), tumor necrosis factor (TNF)-α and thrombin also activate NAD(P)H oxidase in VSMCs (Marumo et al. 1997; De Keulenaer et al. 1998; Gorlach et al. 2001; Brandes et al. 2002). Endothelin-1 increases NAD(P)H oxidase activity in human endothelial cells (Duerrschmidt et al. 2000) and, in intact vessels, this effect is mediated via ETA receptors (Li et al. 2003a). Activators of peroxisome proliferator-activated receptors (PPARs), statins, and antihypertensive drugs, such as β-blockers, Ca2+ channel blockers, ACE inhibitors, and AT1 receptor blockers, downregulate expression of oxidase subunits and decrease NAD(P)H oxidase activity (Dandona et al. 2000; Dhalla et al. 2000; Mantle et al. 2000; Ohtahara et al. 2001; Taddei et al. 2001; Diep et al. 2002; Wassman et al. 2002). These actions may have therapeutic potential in cardiovascular disease.
Other enzymatic sources of ROS in the vasculature
Nitric oxide synthase (NOS), the enzyme primarily responsible for NO production, can also generate ·O2− in conditions of substrate (arginine) or cofactor (tetrahydrobiopterin; BH4) deficiency (Milstien and Katusic 1999; Cosentino et al. 2001). These findings have led to the concept of “NOS uncoupling”, where the activity of the enzyme for NO production is decreased in association with an increase in NOS-dependent ·O2− formation. eNOS uncoupling has been demonstrated in atherosclerosis (Vasquez-Vivar et al. 2002), diabetes (Bagi and Koller 2003), hyperhomocysteinemia (Virdis et al. 2003), and hypertension (Landmesser et al. 2003; Podjarny et al. 2003). Landmesser et al. (2003) reported that, in hypertension, increased NAD(P)H oxidase-derived ·O2− leads to augmented ROS bioavailability, which causes oxidation of BH4 and consequent uncoupling of eNOS, further contributing to ROS production. Gene transfer of GTP cyclohydrolase (GTPCH) I, the enzyme responsible for regenerating BH4, restored arterial GTPCH I activity and BH4 levels, reduced ROS, and improved endothelium-dependent relaxation and NO release in DOCA-salt hypertensive rats, in which endothelial dysfunction results from NAD(P)H-dependent oxidant excess (Zheng et al. 2003). The potential role of uncoupling of NOS as a source of ROS in hypertension is also supported in human studies where increased endothelial ·O2− production in vessels from diabetic and hypertensive patients is inhibited by sepiapterin, a precursor of BH4 (Guzik et al. 2002; Higashi et al. 2002a). The relative importance of NOS- versus NAD(P)H oxidase-mediated ·O2− generation in hypertension probably relates, in part, to the magnitude of endothelial dysfunction, since most conditions in which ·O2− is derived from NOS are associated with marked endothelial dysfunction (Channon and Guzik 2002.
Other enzymatic sources capable of generating ROS in the vasculature are xanthine oxidase, cytochrome P450, mitochondrial respiratory chain enzymes, and phagocyte-derived myeloperoxidase (Taniyama and Griendling 2003; Fig. 1). However, the contribution of these enzymes to vascular generation of ROS is relatively minor compared with NAD(P)H oxidase.
Signaling pathways and molecular targets of ROS
Although ROS have been shown to be involved in many signal transduction pathways (Fig. 3), the exact molecular targets have not yet been clearly defined. Addition of exogenous ROS activates mitogen-activated protein (MAP) kinases, including ERK1/2, p38MAP kinase, JNK, and ERK5, important in cell growth, inflammation, apoptosis, and cell differentiation, respectively (Torres 2003). In cultured VSMCs, generation of endogenous ROS by Ang II influences activation of p38MAP kinase, JNK, and ERK5, but not of ERK1/2 (Ushio-Fukai et al. 1998; Viedt et al. 2000; Touyz et al. 2003b, 2004). However, serotonin-mediated ERK1/2 activation in smooth muscle cells is redox-sensitive, but in fibroblasts it is not (Lee et al. 1999), suggesting that redox-regulation of MAP kinases may be ligand- and cell-specific. Although MAP kinases are redox-sensitive, they are probably not direct substrates of ·O2− and H2O2. Upstream modulators such as MEKs, tyrosine kinases, and phosphatases are likely direct targets (Lee and Esselman 2002).

Redox-dependent signaling pathways in vascular cells. Intracellular ROS influence the activity of protein tyrosine phosphatases (PTP) by modifying cysteine residues. Oxidation of the cysteine residue to sulfenic acid by H2O2 renders PTPs inactive, whereas reduction renders PTPs active. Activated PTP decreases activity of protein tyrosine kinases (PTK) and mitogen-activated kinases (MAPK), whereas inactivated PTP have opposite actions. ROS also influence gene and protein expression by activating transcription factors, such as NFκB and AP-1. ROS stimulate ion channels, such as plasma membrane Ca2+ and K+ channels, leading to changes in cation concentration and matrix metalloproteinases (MMPs), which influence extracellular matrix proteins (ECM) degradation. Activation of these redox-sensitive pathways results in many cellular responses, which, if uncontrolled, could contribute to altered vascular tone, increased vascular smooth muscle cell (VSMC) growth, inflammation, and increased deposition of extracellular matrix protein, leading to vascular remodeling in hypertension. ↓ Decreased effect, ↑ increased effect
Receptor and non-receptor tyrosine kinases are also influenced by ROS (Yang et al. 2000). Exogenous H2O2 induces tyrosine phosphorylation and activation of PDGFR and EGFR, probably due to ROS-mediated inhibition of dephosphorylation of PDGFR and EGFR by inactivation of membrane-associated protein tyrosine phosphatases (Yang et al. 2000; Droge 2001). Oxygen intermediates, which are produced in response to tyrosine kinase receptor activation, are also involved in transactivation of PDGFR and EGFR by Ang II. Under pathological conditions associated with oxidative stress, such as hypertension, ROS may directly activate cell surface receptors, thereby amplifying the process of ·O2− generation. Non-receptor tyrosine kinases such as Src, JAK2, Pyk2, and Akt, all of which have been implicated in cardiovascular remodeling and vascular damage, are also regulated by ROS (Griendling et al. 2000a; Touyz and Schiffrin 2000; Droge 2001; Touyz et al. 2002b).
The best-established direct targets of ROS signaling are protein tyrosine phosphatases (Lee et al. 1998; Meng et al. 2002) and transcription factors (Haddad 2002; Turpaev 2002). All tyrosine phosphatases have a conserved 230-amino acid domain that contains a reactive and redox-regulated cysteine, which catalyses the hydrolysis of protein phosphotyrosine residues by the formation of a cysteinyl-phosphate intermediate (Brigelius-Flohe et al. 2004). Oxidation of this cysteine residue to sulfenic acid by H2O2 renders the tyrosine phosphatase inactive (Brigelius-Flohe et al. 2004). Thus ROS inhibit activity of tyrosine phosphatases, resulting in increased tyrosine phosphorylation, which influences oxidative stress-induced activation of receptor protein tyrosine kinases, such as the EGFR, IGF-1R, and PDGFR (Kamata et al. 2000), and non-receptor tyrosine kinases, such as Src, FAK, PI3 K, and JAK2.
Transcription factors, including nuclear factor κB (NFκB), activator protein 1 (AP-1), c-Myb, Sp-1, p53, early growth response-1 (egr-1), and hypoxia-inducible factor (HIF-1) are directly activated by ROS (Haddad 2002; Turpaev 2002). NFκB and AP-1 induce expression of pro-inflammatory genes, including monocyte chemotactic protein-1 (MCP-1), adhesion molecules, and interleukins (Brigelius-Flohe et al. 2004), that play a role in vascular inflammation associated with hypertension and atherosclerosis. Most redox-sensitive transcription factors possess conserved cysteines, which are susceptible to oxidative modification (Haddad 2002). It has been suggested that the reactive cysteines constitute redox-sulfhydryl switches that directly regulate gene expression (Kamata et al. 2000). Another mode of redox regulation of transcription factor activity is by the redox sensitivity of protein degradation. Increased activation of vascular NFκB and AP-1 and associated inflammatory and mitogenic responses have been demonstrated in hypertensive rats (Taddei et al. 2001). These actions have been attributed, in part, to oxidative excess.
In addition to influencing cellular processes associated with growth and inflammation, ROS modulate intracellular free Ca2+ concentration ([Ca2+]i), a major determinant of vascular contraction/dilation. Superoxide and H2O2 increase [Ca2+]i in VSMCs and endothelial cells (Lounsbury et al. 2000). These effects have been attributed to redox-dependent inositol trisphosphate-induced Ca2+ mobilization, increased Ca2+ influx, and decreased Ca2+-ATPase activation (Lounsbury et al. 2000; Ermak and Davies 2001). Plasma membrane K+ channels in VSMCs controlling hyperpolarization-elicited relaxation are opened by mechanisms associated with thiol oxidation by ROS (Touyz and Schiffrin 2000; Droge 2001; Ermak and Davies 2001; Touyz et al. 2002b). These redox-regulated Ca2+ processes may be more important in stress responses than in receptor-mediated signaling by growth factors or cytokines and may play a role in altered vascular contractility in hypertension. Contractile responses to H2O2 are exaggerated in arteries from spontaneously hypertensive rats (SHR) compared with normotensive counterparts (Gao and Lee 2001), suggesting that in addition to impaired endothelium-dependent vasodilation (due to increased quenching of NO by ·O2−), redox-sensitive Ca2+ changes could contribute to altered vascular contractility in hypertension.
Vascular effects of ROS
Vascular growth and inflammation
Reactive oxygen species stimulate growth factor-like cellular responses, such as intracellular alkalinization, MAP kinase phosphorylation, tyrosine kinase activation, DNA synthesis, and increased expression of proto-oncogenes (Rao and Berk 1992; Droge 2001). During vascular damage in hypertension when oxidative stress is increased, redox-sensitive growth processes may lead to accelerated proliferation and hypertrophy, further contributing to vascular injury and remodeling (Rao and Berk 1992; Griendling et al. 2000a; Touyz 2003a; Fig. 4). In addition to growth-promoting actions, ROS induce apoptosis and differentiation under certain circumstances. This differential response appears to relate to the specific species generated, the concentration of ROS, and the cellular localization of ROS (Deshpande et al. 2002; Li et al. 2003b). At high concentrations (>100 μmol/l) H2O2 and peroxynitrite are pro-apoptotic and induce anoikis (cell detachment and shedding), whereas at lower concentrations they stimulate growth and differentiation (Deshpande et al. 2002; Li et al. 1999, 2003b).

Vascular effects of reactive oxygen species (ROS). Increased bioavailability of ROS influences cellular processes leading to vascular smooth muscle cell (VSMC) growth, inflammation, migration, and extracellular matrix (ECM) protein deposition as well as endothelial damage. MMP Matrix metalloproteinases
Reactive oxygen species also modulate vascular structure in hypertension by increasing deposition of extracellular matrix proteins, such as collagen and fibronectin. Superoxide anion and H2O2 influence activity of vascular MMP2 and MMP9, which promote degradation of basement membrane and elastin, respectively (Rajagopalan et al. 1996b). Redox-sensitive inflammatory processes, including expression of proinflammatory molecules, such as MCP-1, osteopontin, and interleukin-6, expression of adhesion molecules, including vascular cell adhesion molecule-1 (VCAM-1) and intracellular adhesion molecule-1 (ICAM-1), lipid peroxidation, and cell migration, further contribute to vascular remodeling in hypertension (Muller et al. 2000; Luft 2001; Suematsu et al. 2002).
Oxygen radicals induce endothelial permeability with extravasation of plasma proteins and other macromolecules, and recruitment of inflammatory proteins and cells, which also impair endothelial function and aggravate vascular damage (Alexander 1995; Kristal et al. 1998). Peripheral polymorphonuclear leukocytes, which generate ·O2−, participate in oxidative stress and inflammation in patients with hypertension (Alexander 1995; Rajagopalan et al. 1996b; Kristal et al. 1998). The co-existence of an inflammatory reaction with oxidative stress induces endothelial dysfunction. Many of the redox-sensitive vascular changes that occur in hypertension also exist in atherosclerotic vessels.
Vascular contraction/dilation
Impaired endothelium-mediated vasodilation in hypertension and hypercholesterolemia has been linked to decreased NO bioavailability. This may be secondary to decreased synthesis of NO and/or to increased degradation of NO because of its interaction with ·O2− to form ONOO− (Tschudi et al. 1996; List et al. 1997; Somers and Harrison 1999). Peroxynitrite is a weak vasodilator compared with NO and has pro-inflammatory properties (Szabo 2003). In experimental models of hypertension, hypercholesterolemia, and diabetes and in hypertensive patients, endothelial function is improved by anti-oxidant vitamins, probucol, SOD, or sepiapterin (a stable precursor of BH4) (Virdis et al. 2003; Mitchell et al. 2004).
Vasomotor tone may also be modulated through direct ROS effects. Reactive oxygen species appear to elicit both contraction and dilation, depending on the vascular bed and type of species generated. Hydrogen peroxide causes vasodilation of pulmonary, coronary, and mesenteric arteries and has been considered to be an endothelium-derived relaxing factor (Somers and Harrison 1999; Yada et al. 2003). In rat aorta, Ang II stimulates vasoconstriction via H2O2-dependent mechanisms (Torrecillas et al. 2001), whereas in human and porcine vessels, acute vasoconstriction by Ang II is not mediated via ROS (Schuijt et al. 2003; Touyz 2003b). In aortic and mesenteric arteries from SHR, redox-mediated contractile effects are enhanced (Gao and Lee 2001). Major factors underlying these differential vascular responses to activated oxygen metabolites could relate to the blood vessel studied, the presence or absence of the endothelium, the concentration and species of free radical studied, and the compartment in which ·O2− or H2O2 predominate (Touyz 2003b). At present it is still unclear exactly what the functions of ·O2− and H2O2 are with respect to vascular contraction/dilation in physiological and pathophysiological conditions.
Reactive oxygen species in hypertension
Oxidative stress in genetic models of hypertension
Reactive oxygen species play an important pathophysiological role in hypertension. This is evidenced by findings that oxidative stress is increased in hypertension and that treatment with antioxidants or agents that inhibit NAD(P)H oxidase-driven generation of ROS reduces, and may even prevent, blood pressure elevation in hypertensive animals. Genetic models of hypertension, such as SHR (Zalba et al. 2000) and stroke-prone SHR (SHRSP) (Chen et al. 2001) exhibit enhanced NAD(P)H oxidase-mediated ·O2− generation in resistance arteries (mesenteric), conduit vessels (aorta), and kidneys (Zalba et al. 2000; Chen et al. 2001; Fig. 5). These processes are associated with increased expression of NAD(P)H oxidase subunits, particularly p22phox and p47phox, and increased activity of the enzyme (Lassegue and Clempus 2003). 8-Hydroxy-2′-deoxyguanosine, a marker for oxidative stress-induced DNA damage, and protein carbonylation, a marker for oxidation status of proteins that are enhanced in aorta, heart, and kidney, are markedly suppressed in SHR and SHRSP compared with normotensive Wistar Kyoto rats, as is the expression of the redox regulating protein thioredoxin (Tanito et al. 2004). Male SHR have a higher vascular ·O2− concentration than female counterparts, a phenomenon that has been linked to upregulation of AT1 receptors in male SHR arteries (Dantas et al. 2004). Several polymorphisms in the promoter region of the p22phox gene have been identified in SHR, which could contribute to enhanced NAD(P)H oxidase activity (Zalba et al. 2001b). These findings may have clinical relevance since an association between a p22phox gene polymorphism and NAD(P)H oxidase-mediated ·O2− production in the vascular wall of patients with atherosclerosis and hypertension has been described (Diez et al. 2003; Moreno et al. 2003). Increased expression of p47phox has been demonstrated in the renal vasculature, macula densa, and distal nephron from young SHR, suggesting that upregulation of renal NAD(P)H precedes development of hypertension (Chabrashvili et al. 2002). The importance of p47phox was demonstrated in p47phox−/− mice, which failed to develop hypertension in response to Ang II infusion (Landmesser et al. 2002). Diminished NO bioavailability as a consequence of enhanced vascular ·O2− generation may also contribute to oxidative stress in SHR and SHRSP. Treatment with antioxidant vitamins, NAD(P)H oxidase inhibitors, SOD mimetics, BH4, and AT1 receptor blockers decrease vascular ·O2− production and attenuate, to varying degrees, the development of hypertension in these genetic models of hypertension (Sharma et al. 1996; Schnackenberg et al. 1999; Chen et al. 2001; Hong et al. 2001). Lifelong treatment with antioxidants can even prevent development of hypertension in SHR (Zhan et al. 2004).

Detection of vascular superoxide by dihydroethidine fluorescence in hypertensive rats. Shown are baseline superoxide levels in aorta from normotensive control Wistar Kyoto rats (WKY) and stroke-prone spontaneously hypertensive rats (SHR-SP) treated or not with the NAD(P)H oxidase inhibitor, apocynin. The increase in superoxide involves all layers within the vessel wall. E Endothelium, M media, A adventitia
Oxidative stress and experimentally induced hypertension
Oxidative excess has been demonstrated in various models of experimental hypertension, including Ang II-induced hypertension (Laursen et al. 1997; Virdis et al. 2004), Dahl-salt-sensitive hypertension (Tojo et al. 2002), lead-induced hypertension (Ding et al. 2001), obesity-associated hypertension (Dobrian et al. 2001), mineralocorticoid hypertension (Wu et al. 2001; Virdis et al. 2002), 2-kidney, 1-clip hypertension (Welch et al. 2003), and postmenopausal hypertension (Fortepiani et al. 2003; Table 2). Increased activation of vascular NAD(P)H oxidase (Lassegue and Clempus 2003) and xanthine oxidase (Fortepiani et al. 2003) and uncoupling of eNOS (Milstien and Katusik 1999; Cosentino et al. 2001; Vasquez-Vivar et al. 2002) have been implicated in enhanced ·O2− generation in experimental hypertension. Inhibition of ROS generation with apocynin or allopurinol and scavenging of free radicals with antioxidants or SOD mimetics decreases blood pressure and prevents development of hypertension in most models of experimental hypertension (Sharma et al. 1996; Chen et al. 2001; Frenoux et al. 2002; Park et al. 2002; Tanito et al. 2004). These beneficial effects have been attributed to improved endothelial function, vascular regression, and reduced vascular inflammation (Touyz 2000; Wilcox 2002). Interestingly, norepinephrine-induced hypertension is not associated with enhanced vascular oxidative stress and SOD does not decrease blood pressure in this model (Laursen et al. 1997). These findings suggest that blood pressure itself may not be the primary cause of oxidative excess in hypertension.
Oxidative stress in human hypertension
Clinical studies have demonstrated that essential hypertensive patients produce excessive amounts of ROS (Prabha et al. 1990; Sagar et al. 1992; Lacy et al. 2000; Minuz et al. 2002; Stojiljkovic et al. 2002) and have decreased antioxidant capacity (Russo et al. 1998). In most of these studies, hypertensive patients are salt-sensitive and exhibit some degree of renal dysfunction (Manning et al. 2003). Oxidative stress has also been demonstrated in patients with renovascular hypertension (Higashi et al. 2002b), malignant hypertension (Lip et al. 2002), and in pre-eclampsia (Lee et al. 2003). Most of these findings are based on increased levels of plasma and urine TBARS and 8-epi-isoprostanes, systemic markers of lipid peroxidation and oxidative stress (Sagar et al. 1992; Minuz et al. 2002; Stojiljkovic et al. 2002). In never-treated mild-to-moderate hypertension lipid peroxidation is not increased (Cracowski et al. 2003), suggesting that oxidative stress may not be important in mild hypertension.
Decreased antioxidant activity and reduced levels of ROS scavengers such as vitamin E, glutathione, and SOD (Sagar et al. 1992) and increased activation of vascular NAD(P)H oxidase may contribute to oxidative excess in hypertensive patients (Berry et al. 2000; Bengtsson et al. 2003). Activation of the renin–angiotensin system has been proposed as a major mediator of NAD(P)H oxidase activation and ROS production in human hypertension (Touyz 2003a). In fact some of the therapeutic blood pressure-lowering effects of AT1 receptor blockers and ACE inhibitors have been attributed to inhibition of NAD(P)H oxidase activity and decreased ROS production (Ghiadoni et al. 2003). It has also been suggested that p22phox polymorphisms may play a role in altered NAD(P)H oxidase-generated ·O2− production in human cardiovascular disease (Schachinger et al. 2001; Zalba et al. 2001b; Moreno et al. 2003; Dantas et al. 2004). In particular the -930(A/G) polymorphism in the p22(phox) promoter may be a novel genetic marker associated with hypertension (Zalba et al. 2001b). However, to confirm that these polymorphisms are indeed markers for hypertension, studies in large populations are necessary.
Based on experimental evidence and clinical studies that oxidative stress plays a key role in vascular damage, there has been great interest in developing strategies that target ROS in the treatment of hypertension and other cardiovascular diseases. Therapeutic approaches that have been considered include mechanisms to increase antioxidant bioavailability through diet or supplementation and/or to reduce generation of ROS by decreasing activity of ·O2−-generating enzymes and by increasing levels of BH4 (Brown and Hu 2001; Cai et al. 2003). Findings from clinical trials have been conflicting. Until definitive data become available, antioxidants should not be recommended in the prevention and management of hypertension (Galley et al. 1997; Taddei et al. 1998; Chappell et al. 1999; HOPE Investigators 2000; Brown and Hu 2001; Digiesi et al. 2001; Khaw et al. 2001; Kim et al. 2002; Wu et al. 2002; Vivekananthan et al. 2003).
Conclusions
Reactive oxygen species are produced in the vessel wall in a controlled and tightly regulated manner. Superoxide and H2O2 have important signaling properties, mainly through oxidative modification of proteins and activation of transcription factors that maintain vascular function and structure. In hypertension, dysregulation of enzymes such as NAD(P)H oxidase, NOS, xanthine oxidase, mitochondrial enzymes, or SOD that generate ·O2−, H2O2, and ·OH, altered thioredoxin and glutathione systems, or reduced scavenging by anti-oxidants, results in increased formation of ROS, which has damaging actions on the vasculature. Reactive oxygen species in hypertension contribute to vascular injury by promoting VSMC growth, extracellular matrix protein deposition, activation of matrix metalloproteinases, inflammation, endothelial dysfunction, and increased vascular tone. In experimental hypertension oxidative stress is increased. Clinical data suggest that hypertensive patients, especially those with severe hypertension, salt-sensitive hypertension, and renovascular hypertension, exhibit oxidative excess. Although inconclusive at present, treatment strategies to alter ROS bioavailability by decreasing production and/or by increasing radical scavenging, may regress vascular remodeling, prevent further vascular injury, and reduce blood pressure and associated target organ damage in hypertensive patients. With greater insights and understanding of processes regulating vascular ROS metabolism and identification of molecular pathways that tip the equilibrium to states of oxidative stress which cause vascular damage, it may be possible to target therapies more effectively so that detrimental actions of vascular oxygen free radicals can be reduced and beneficial effects of NO· can be enhanced. Such therapies may be useful in the treatment of hypertension and in the prevention of target-organ damage.
References
Ago T, Kitazono T, Ooboshi H, Iyama T, Han YH, Takada J, Wakisaka M, Ibayashi S, Utsumi H, Iida M (2004) Nox4 as the major catalytic component of an endothelial NAD(P)H oxidase. Circulation 109:227–233
Alexander RW (1995) Hypertension and the pathogenesis of atherosclerosis. Oxidative stress and the mediation of arterial inflammatory response: a new perspective. Hypertension 25:155–161
Azumimi H, Inoue N, Takeshita S (1999) Expression of NADH/NADPH oxidase p22phox in human coronary arteries. Circulation 100:1494–1498
Babior BM, Lambeth JD, Nauseef W (2002) The neutrophil NADPH oxidase. Arch Biochem Biophys 397:342–344
Bagi Z, Koller A (2003) Lack of nitric oxide mediation of flow-dependent arteriolar dilation in type I diabetes is restored by sepiapterin. J Vasc Res 40:47–57
Banfi B, Clark RA, Steger K, Krause K-H (2003) Two novel proteins activate superoxide generation by the NADPH oxidase Nox1. J Biol Chem 278:3510–3513
Bendall JK, Cave AC, Heymes C, Gall N, Shah AM (2002) Pivotal role of a gp91(phox)-containing NADPH oxidase in angiotensin II-induced cardiac hypertrophy in mice. Circulation 105:293–296
Bengtsson SH, Gulluyan LM, Dusting GJ, Drummond GR (2003) Novel isoforms of NADPH oxidase in vascular physiology and pathophysiology. Clin Exp Pharmacol Physiol 30:849–854
Berry C, Hamilton CA, Brosnan MJ, Magill FG, Berg GA, McMurray JJ, Dominiczak AF (2000) Investigation into the sources of superoxide in human blood vessels: angiotensin II increases superoxide production in human internal mammary arteries. Circulation 101:2206–2212
Boshtam M, Rafiei M, Sadeghi K, Sarraf-Zadegan N (2002) Vitamin E can reduce blood pressure in mild hypertensives. Int J Vitam Nutr Res 72:309–314
Brandes RP, Miller FJ, Beer S, Haendeler J, Hoffmann J, Ha T, Holland SM, Gorlach A, Busse R (2002) The vascular NADPH oxidase subunit p47phox is involved in redox-mediated gene expression. Free Radic Biol Med 32:1116–1122
Brigelius-Flohe R, Banning A, Kny M, Bol GF (2004) Redox events in interleukin-1 signaling. Arch Biochem Biophys 423:66–73
Brown AA, Hu FB (2001) Dietary modulation of endothelial function: implications for cardiovascular disease. Am J Clin Nutr. 73:673–686
Cai H, Griendling KK, Harrison DG (2003) The vascular NAD(P)H oxidases as therapeutic targets in cardiovascular diseases. Trends Pharmacol Sci 24:471–478
Cantor EJ, Mancini EV, Seth R, Yao XH, Netticadan T (2003) Oxidative stress and heart disease: cardiac dysfunction, nutrition, and gene therapy. Curr Hypertens Rep 5:215–220
Chabrashvili T, Tojo A, Onozato ML, Kitiyakara C, Quinn MT, Fujita T, Welch WJ, Wilcox CS (2002) Expression and cellular localization of classic NADPH oxidase subunits in the spontaneously hypertensive rat kidney. Hypertension 39:269–274
Channon KM, Guzik TJ (2002) Mechanisms of superoxide production in human blood vessels: relationship to endothelial dysfunction, clinical and genetic risk factors. J Physiol Pharmacol 53:515–524
Chappell LC, Seed PT, Briley AL, Kelly FJ, Lee R, et al (1999) Effect of antioxidants on the occurrence of pre-eclampsia in women at increased risk: a randomized trial. Lancet 354:810–815
Chen X, Touyz RM, Park JB, Schiffrin EL (2001) Antioxidant effects of vitamins C and E are associated with altered activation of vascular NAD(P)H oxidase and superoxide dismutase in stroke-prone SHR. Hypertension 38:606–611
Cheng G, Cao Z, Xu X, van Meir EG, Lambeth JD (2001) Homologs of gp91phox: cloning and tissue expression of Nox3, Nox4, and Nox5. Gene 269:131–140
Chiarugi P, Cirri P (2003) Redox regulation of protein tyrosine phosphatases during receptor tyrosine kinase signal transduction. Trends Biochem Sci 28:509–514
Cosentino F, Sill JC, Katusic ZS (1994) Role of superoxide anions in the mediation of endothelium-dependent contractions. Hypertension 23:229–235
Cosentino F, Barker JE, Brand MP, Heales SJ, Werner ER, Tippins JR, West N, Channon KM, Volpe M, Luscher TF (2001) Reactive oxygen species mediate endothelium-dependent relaxations in tetrahydrobiopterin-deficient mice. Arterioscler Thromb Vasc Biol 21:496–502
Cracowski JL, Baguet JP, Ormezzano O, Bessard J, Stanke-Labesque F, Bessard G, Mallion JM (2003) Lipid peroxidation is not increased in patients with untreated mild-to-moderate hypertension. Hypertension 41:286–288
Dandona P, Karne R, Ghanim H, Hamouda W, Aljada A, Magsino CH (2000) Carvedilol inhibits reactive oxygen species generation by leukocytes and oxidative damage to amino acids. Circulation 101:122–124
Dantas AP, Franco Mdo C, Silva-Antonialli MM, Tostes RC, Fortes ZB, Nigro D, Carvalho MH (2004) Gender differences in superoxide generation in microvessels of hypertensive rats: role of NAD(P)H-oxidase. Cardiovasc Res 61:22–29
Darley-Usmar V, Wiseman H, Halliwell B (1995) Nitric oxide and oxygen radicals, a question of balance. FEBS Lett 369:13–15
De Keulenaer GW, Alexander RW, Ushio-Fukai M, Ishizaka N, Griendling KK (1998) Tumour necrosis factor alpha activates a p22phox-based NADH oxidase in vascular smooth muscle. Biochem J 329:653–657
De Leo FR, Ulman KV, Davis AR,Jutila KL, Quinn MT (1996) Assembly of the human neutrophil NADPH oxidase involves binding of p67phox and flavocytochrome b to a common functional domain in p47phox. J Biol Chem 271:17013–17020
Deshpande NN, Sorescu D, Seshiah P, Ushio-Fukai M, Akers M, Yin Q, Griendling KK (2002) Mechanism of hydrogen peroxide-induced cell cycle arrest in vascular smooth muscle. Antioxid Redox Signal 4:845–854
Dhalla SN, Temsah RM, Netticadan T (2000) Role of oxidative stress in cardiovascular diseases. J Hypertens 18:655–673
Diep QN, Amiri F, Touyz RM, Cohn JS, Endemann D, Neves MF, Schiffrin EL (2002) PPARalpha activator effects on Ang II-induced vascular oxidative stress and inflammation. Hypertension 40:866–871
Diez J, Laviades C, Orbe J, Zalba G, Lopez B, Gonzalez A, Mayor G, Paramo JA, Beloqui O (2003) The A1166C polymorphism of the AT1 receptor gene is associated with collagen type I synthesis and myocardial stiffness in hypertensives. J Hypertens 21:2085–2092
Digiesi D, Lenuzza M, Digiese G (2001) Prospects for the use of antioxidant therapy in hypertension. Ann Ital Med Int 16:93–100
Ding Y, Gonick HC, Vaziri ND, Liang K, Wei L (2001) Lead-induced hypertension. III. Increased hydroxyl radical production. Am J Hypertens 14:169–173
Dobrian AD, Davies MJ, Schriver SD, Lauterio TJ, Prewitt RL (2001) Oxidative stress in a rat model of obesity-induced hypertension. Hypertension 37:554–560
Droge W (2001) Free radicals in the physiological control of cell function. Physiol Rev 82:47–95
Duerrschmidt N, Wippich N, Goettsch W, Broemme HJ, Morawietz H (2000) Endothelin-1 induces NAD(P)H oxidase in human endothelial cells. Biochem Biophys Res Commun 269:713–717
Duffy SJ, Gokce N, Holbrook M, Huang A, Frei B, Keaney JF, Vita JA (1999) Treatment of hypertension with ascorbic acid. Lancet 354:2048–2049
Ermak G, Davies KJA (2001) Calcium and oxidative stress: from cell signaling to cell death. Mol Immunol 38:713–721
Fortepiani LA, Zhang H, Racusen L, Roberts LJ II, Reckelhoff JF (2003) Characterization of an animal model of postmenopausal hypertension in spontaneously hypertensive rats. Hypertension 41:640–645
Fotheby MD, Williams JC, Forster LA, Craner P, Ferns GA (2000) Effect of vitamin C on ambulatory blood pressure and plasma lipids in older patients. J Hypertens 18:411–415
Freeman BA, Crapo JD (1982) Biology of disease: free radicals and tissue injury. Lab Invest 47:412–426
Frenoux JM, Noirot B, Prost ED, Madani S, Blond JP, Belleville JL, Prost JL (2002) Very high alpha-tocopherol diet diminishes oxidative stress and hypercoagulation in hypertensive rats but not in normotensive rats. Med Sci Monit 8:BR401–BR407
Fridovich I (1997) Superoxide anion radical, superoxide dismutases, and related matters. J Biol Chem 272:18515–18517
Galley HF, Thornton J, Howdle PD, Walker BE, Webster NR (1997) Combination oral antioxidant supplementation reduces blood pressure. Clin Sci (Lond) 92:361–365
Gao YJ, Lee RM (2001) Hydrogen peroxide induces a greater contraction in mesenteric arteries of spontaneously hypertensive rats through thromboxane A(2) production. Br J Pharmacol 134:1639–1646
Gauss KA, Mascolo PL, Siemsen DW, Nelson LK, Bunger PL, Pagano PJ, Quinn MT (2002) Cloning and sequencing of rabbit leukocyte NADPH oxidase genes reveals a unique p67(phox) homolog. J Leukoc Biol 71:319–328
Ghiadoni L, Magagna A, Versari D, Kardasz I, Huang Y, Taddei S, Salvetti A (2003) Different effect of antihypertensive drugs on conduit artery endothelial function. Hypertension 41:1281–1286
Gorlach A, Diebold I, Schini-Kerth VB, Berchner-Pfannschmidt U, Roth U, Brandes RP, Kietzmann T, Busse R (2001) Thrombin activates the hypoxia-inducible factor-1 signaling pathway in vascular smooth muscle cells: role of the p22(phox)-containing NADPH oxidase. Circ Res 89:47–54
Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW (1994) Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res 74:1141–1148
Griendling KK, Sorescu D, Lassegue B, Ushio-Fukai M (2000a) Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathophysiology. Arterioscler Thromb Vasc Biol 20:2175–2183
Griendling KK, Sorescu D, Ushio-Fukai M (2000b) NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res 86:494–501
Grote K, Flach I, Luchtefeld M, Akin E, Holland SM, Drexler H, Schieffer B (2003) Mechanical stretch enhances mRNA expression and proenzyme release of matrix metalloproteinase-2 (MMP-2) via NAD(P)H oxidase-derived reactive oxygen species. Circ Res 92:e80–e86
Guzik TJ, Mussa S, Gastaldi D, Sadowski J, Ratnatunga C, Pillai R, Channon KM (2002) Mechanisms of increased vascular superoxide production in human diabetes mellitus: role of NAD(P)H oxidase and endothelial nitric oxide synthase. Circulation 105:1656–1562
Haddad JJ (2002) Antioxidant and prooxidant mechanisms in the regulation of redox(y)-sensitive transcription factors. Cell Signal 14:879–897
Halliwell B (1999) Antioxidant defence mechanisms: from the beginning to the end (of the beginning). Free Radic Res 31:261–272
Han D, Antunes F, Canali R, Rettori D, Cadenas E (2003) Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J Biol Chem 278:5557–5563
Harrison DG (1997) Cellular and molecular mechanisms of endothelial cell dysfunction. J Clin Invest 1997:2153–2157
Higashi Y, Sasaki S, Nakagawa K, Fukuda Y, Matsuura H, Oshima T, Chayama K (2002a) Tetrahydrobiopterin enhances forearm vascular response to acetylcholine in both normotensive and hypertensive individuals. Am J Hypertens 15:326–332
Higashi Y, Sasaki S, Nakagawa K, et al (2002b) Endothelial function and oxidative stress in renovascular hypertension. N Engl J Med 346:1954–1962
Hilenski LL, Clempus RE, Quinn MT, Lambeth JD, Griendling KK (2004) Distinct subcellular localizations of Nox1 and Nox4 in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 24:1–8
Hoagland KM, Maier KG, Roman RJ (2003) Contributions of 20-HETE to the antihypertensive effects of Tempol in Dahl salt-sensitive rats. Hypertension 41:697–702
Hong HJ, Hsiao G, Cheng TH, Yen MH (2001) Supplementation with tetrahydrobiopterin suppresses the development of hypertension in spontaneously hypertensive rats. Hypertension 38:1044–1048
HOPE Investigators (2000) Vitamin E supplementation and cardiovascular events in high risk patients. N Engl J Med 342:154–160
Jones A, O’Donnell VB, Wood JD (1996) Expression of phagocyte NADPH oxidase components in human endothelial cells. Am J Physiol H1626–H1634
Kamata H, Shibukawa Y, Oka S-I, Hirata H (2000) Epidermal growth factor receptor is modulated by redox through multiple mechanisms. Effects of reductants and H2O2. Eur J Biochem 267:1933–1944
Kerr S, Brosnan J, McIntyre M, Reid JL, Dominiczak AF, Hamilton CA (1999) Superoxide anion production is increased in a model of genetic hypertension. Role of endothelium. Hypertension 33:1353–1358
Khaw K-T, Bingham S, Welch A, Luben R, Wareham N, Oakes S, et al (2001) Relation between plasma ascorbic acid and mortality in men and women in EPIC-Norfolk prospective study: a prospective population study. Lancet 357:657–663
Kim MY, Sasaki S, Sasazuki S, Okubo S, Hayashi M, Tsugane S (2002) Lack of long-term effect of vitamin C supplementation on blood pressure. Hypertension 40:797–803
Kristal B, Shurta-Swirrski R, Chezar J (1998) Participation of peripheral polymorphonuclear leukocytes in the oxidative stress and inflammation in patients with essential hypertension. Am J Hypertens 11:921–928
Lacy F, Kailasam MT, O’Connor DT, Schmid-Schonbein GW, Parmer RJ (2000) Plasma hydrogen peroxide production in human essential hypertension: role of heredity, gender, and ethnicity. Hypertension 36:878–884
Landmesser U, Harrison DG (2001) Oxidative stress and vascular damage in hypertension. Coron Artery Dis 12:455–461
Landmesser U, Cai H, Dikalov S, McCann L, Hwang J, Jo H, Holland SM, Harrison DG (2002) Role of p47(phox) in vascular oxidative stress and hypertension caused by angiotensin II. Hypertension 40:511–515
Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, Mitch WE, Harrison DG (2003) Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest 111:1201–1209
Lassegue B, Clempus RE (2003) Vascular NAD(P)H oxidases: specific features, expression, and regulation. Am J Physiol Regul Integr Comp Physiol 285:R277–R297
Lassegue B, Sorescu D, Szocs K, Yin Q, Akers M, Zhang Y, Grant SL, Lambeth JD, Griendling KK (2001) Novel gp91(phox) homologues in vascular smooth muscle cells: Nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ Res 88:888–894
Laursen JB, Rajagopalan S, Galis Z, Tarpey M, Freeman BA, Harrison DG (1997) Role of superoxide in angiotensin II-induced but not catecholamine-induced hypertension. Circulation 95:588–593
Lee K, Esselman WJ (2002) Inhibition of PTPS by H2O2 regulates the activation of distinct MAPK pathways. Free Radic Biol Med 33:1121–1132
Lee SR, Kwon KS, Kim SR, Rhee SG (1998) Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J Biol Chem 273:15366–15372
Lee SL, Wang WW, Finlay GA, Fanburg BL (1999) Serotonin stimulates MAP kinase activity through the formation of superoxide anion. Am J Physiol Lung Cell Mol Physiol 277:L282–L291
Lee VM, Quinn PA, Jennings SC, Ng LL (2003) Neutrophil activation and production of reactive oxygen species in pre-eclampsia. J Hypertens 21:395–402
Leusen JHW, Verhoeven AJ, Roos D (1996) Interactions between the components of the human NADPH oxidase: a review about the intrigues in the phox family. Front Biosci 1:72–90
Li JM, Shah AM (2002) Intracellular localization and preassembly of the NADPH oxidase complex in cultured endothelial cells. J Biol Chem 277:19952–19960
Li JM, Shah AM (2003) Mechanism of endothelial cell NADPH oxidase activation by angiotensin II. Role of the p47phox subunit. J Biol Chem 278:12094–12100
Li AE, Ito H, Rovira II, Kim KS, Takeda K, Yu ZY, Ferrans VJ, Finkel T (1999) A role for reactive oxygen species in endothelial cell anoikis. Circ Res 85:304–310
Li L, Fink GD, Watts SW, Northcott CA, Galligan JJ, Pagano PJ, Chen AF (2003a) Endothelin-1 increases vascular superoxide via endothelin(A)-NADPH oxidase pathway in low-renin hypertension. Circulation 107:1053–1058
Li J, Li W, Su J, Liu W, Altura BT, Altura BM (2003b) Hydrogen peroxide induces apoptosis in cerebral vascular smooth muscle cells: possible relation to neurodegenerative diseases and strokes. Brain Res Bull 62:101–106
Lip GY, Edmunds E, Nuttall SL, Landray MJ, Blann AD, Beevers DG (2002) Oxidative stress in malignant and non-malignant phase hypertension. J Hum Hypertens 16:333–336
List BM, Klosch B, Volker C, Gorren AC, Sessa WC, Werner ER, Kukovetz WR, Schmidt K, Mayer B (1997) Characterization of bovine endothelial nitric oxide synthase as a homodimer with down-regulated uncoupled NADPH oxidase activity: tetrahydrobiopterin binding kinetics and role of haem in dimerization. Biochem J 323:159–165
Lopes LR, Dagher MC, Gutierrez A, Young B, Bouin A-P, Fuchs A, Babior BM (2004) Phosphorylated p40phox as a negative regulator of NADPH oxidase. Biochemistry 43:3723–3730
Lounsbury KM, Hu Q, Ziegelstein RC (2000) Calcium signaling and oxidant stress in the vasculature. Free Radic Biol Med 28:1362–1369
Luft FC (2001) Mechanisms and cardiovascular damage in hypertension. Hypertension 37:594–598
Manning RD Jr, Meng S, Tian N (2003) Renal and vascular oxidative stress and salt-sensitivity of arterial pressure. Acta Physiol Scand 179:243–250
Mantle D, Patel VB, Why HJ, Ahmed S, Rahman I, MacNee W, Wassif WS, Richardson PJ, Preedy VR (2000) Effects of lisinopril and amlodipine on antioxidant status in experimental hypertension. Clin Chim Acta 299:1–10
Marumo T, Schini-Kerth VB, Fisslthaler B, Busse R (1997) Platelet-derived growth factor-stimulated superoxide anion production modulates activation of transcription factor NF-kappaB and expression of monocyte chemoattractant protein 1 in human aortic smooth muscle cells. Circulation 96:2361–2367
Meng TC, Fukada T, Tonks NK (2002) Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol Cell 9:387–399
Milstien S, Katusic Z (1999) Oxidation of tetrahydrobiopterin by peroxynitrite: implications for vascular endothelial function. Biochem Biophys Res Commun 263:681–684
Minuz P, Patrignani P, Gaino S, Degan M, Menapace L, Tommasoli R (2002) Increased oxidative stress and platelet activation in patients with hypertension and renovascular disease. Circulation 106:2800–2805
Mitchell BM, Dorrance AM, Ergul A, Webb RC (2004) Sepiapterin decreases vasorelaxation in nitric oxide synthase inhibition-induced hypertension. J Cardiovasc Pharmacol 43:93–98
Moreno MU, San Jose G, Orbe J, Paramo JA, Beloqui O, Diez J, Zalba G (2003) Preliminary characterisation of the promoter of the human p22(phox) gene: identification of a new polymorphism associated with hypertension. FEBS Lett 542:27–31
Mullan B, Young IS, Fee H, McCance DR (2002) Ascorbic acid reduces blood pressure and arterial stiffness in type 2 diabetes. Hypertension 40:804–809
Muller DN, Dechend R, Mervaala EMA, Park JK, Schmidt F, Fiebeler, et al (2000) NFκB inhibition ameliorates angiotensin II-induced inflammatory damage in rats. Hypertension 35:193–201
Muzaffar S, Jeremy JY, Angelini GD, Stuart-Smith K, Shukla N (2003) Role of the endothelium and nitric oxide synthases in modulating superoxide formation induced by endotoxin and cytokines in porcine pulmonary arteries. Thorax 58:598–604
Nickenig G, Strehlow K, Baumer AT, Baudler S, Wassmann S, Sauer H, Bohm M (2000) Negative feedback regulation of reactive oxygen species on AT1 receptor gene expression. Br J Pharmacol 131:795–803
Ohtahara A, Hisatome I, Yamamoto Y, Furuse M, Sonoyama K, Furuse Y (2001) The release of the substrate for xanthine oxidase in hypertensive patients was suppressed by angiotensin converting enzyme inhibitors and α1-blockers. J Hypertens 19:575–582
Park JB, Touyz RM, Chen X, Schiffrin EL (2002) Chronic treatment with a superoxide dismutase mimetic prevents vascular remodeling and progression of hypertension in salt-loaded stroke-prone spontaneously hypertensive rats. Am J Hypertens 15:78–84
Podjarny E, Benchetrit S, Rathaus M, Pomeranz A, Rashid G, Shapira J, Bernheim J (2003) Effect of tetrahydrobiopterin on blood pressure in rats after subtotal nephrectomy. Nephron Physiol 94:6–9
Prabha PS, Das UN, Koratkar R, Sagar PS, Ramesh G (1990) Free radical generation, lipid peroxidation and essential fatty acids in uncontrolled essential hypertension. Prostaglandins Leukot Essent Fatty Acids 41:27–33
Privratsky JR, Wold LE, Sowers JR, Quinn MT, Ren J (2003) AT1 blockade prevents glucose-induced cardiac dysfunction in ventricular myocytes: role of the AT1 receptor and NADPH oxidase. Hypertension 42:206–212
Rajagopalan S, Kurz S, Munzel T (1996a) Angiotensin II mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation: contribution to alterations of vasomotor tone. J Clin Invest 97:1916–1923
Rajagopalan S, Meng XP, Ramasamy S, Harrison DG, Galis ZS (1996b) Reactive oxygen species produced by macrophage-derived foam cells regulate the activity of vascular matrix metalloproteinases in vitro. J Clin Invest 98:2572–2579
Rao GN, Berk BC (1992) Active oxygen species stimulate vascular smooth muscle cell growth and proto-oncogene expression. Circ Res 70:593–599
Reth M (2002) Hydrogen peroxide as second messenger in lymphocyte activation. Nat Immunol 3:1129–1134
Rey FE, Pagano PJ (2002) The reactive adventitia: fibroblast oxidase in vascular function. Arterioscler Thromb Vasc Biol 22:1962–1971
Romero JC, Reckelhoff JF (1999) Role of angiotensin and oxidative stress in essential hypertension. Hypertension 34:943–949
Russo C, Olivieri O, Girelli D, Faccini G, Zenari ML, Lombardi S, Corrocher R (1998) Anti-oxidant status and lipid peroxidation in patients with essential hypertension. J Hypertens 16:1267–1271
Sagar S, Kallo IJ, Kaul N, Ganguly NK, Sharma BK (1992) Oxygen free radicals in essential hypertension. Mol Cell Biochem 111:103–108
Sauer H, Wartenberg M, Hescheler J (2001) Reactive oxygen species as intracellular messengers during cell growth and differentiation. Cell Physiol Biochem 11:173–186
Schachinger V, Britten MB, Dimmeler S, Zeiher AM (2001) NADH/NADPH oxidase p22 phox gene polymorphism is associated with improved coronary endothelial vasodilator function. Eur Heart J 22:96–101
Schafer FQ, Buettner GR (2001) Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med 30:1191–1212
Schnackenberg CG, Welch W, Wilcox CS (1999) Normalization of blood pressure and renal vascular resistance in SHR with a membrane-permeable superoxide dismutase mimetic. Role of nitric oxide. Hypertension 32:59–64
Schuijt MP, Tom B, De Vries R, Saxena PR, Sluiter W, Van Kats JP, Danser AH (2003) Superoxide does not mediate the acute vasoconstrictor effects of angiotensin II: a study in human and porcine arteries. J Hypertens 21:2335–2344
Seshiah PN, Weber DS, Rocic P, Valppu L, Taniyama Y, Griendling KK (2002) Angiotensin II stimulation of NAD(P)H oxidase activity. Upstream mediators. Circ Res 91:406–413
Sharma RC, Hodis HN, Mack WJ (1996) Probucol suppresses oxidant stress in hypertensive arteries. Immunohistochemical evidence. Am J Hypertens 9:577–590
Somers MJ, Harrison DG (1999) Reactive oxygen species and the control of vasomotor tone. Curr Hypertens Rep 1:102–108
Sorescu D, Weiss D, Lassegue B, Clempus RE, Szocs K, Sorescu GP, Valppu L, Quinn MT, Lambeth JD, Vega JD, Taylor WR, Griendling KK (2002) Superoxide production and expression of Nox family proteins in human atherosclerosis. Circulation 105:1429–1435
Stojiljkovic MP, Lopes HF, Zhang D, Morrow JD, Goodfriend TL, Egan BM (2002) Increasing plasma fatty acids elevates F2-isoprostanes in humans: implications for the cardiovascular risk factor cluster. J Hypertens 20:1215–1221
Stralin P, Karlsson K, Johannson BO, Marklund SL (1995) The interstitium of the human arterial wall contains very large amounts of extracellular superoxide dismutase. Arterioscler Thromb Vasc Biol 15:2032–2036
Suematsu M, Suzuki H, Delano FA, Schmid-Schonbein GW (2002) The inflammatory aspect of the microcirculation in hypertension: oxidative stress, leukocytes/endothelial interaction, apoptosis. Microcirculation 9:259–276
Suh YA, Arnold RS, Lassegue B (1999) Cell transformation by the superoxide-generating Mox-1. Nature 410:79–82
Szabo C (2003) Multiple pathways of peroxynitrite cytotoxicity. Toxicol Lett 140:105–112
Taddei S, Virdis A, Ghiadoni L, Magagna A, Salvetti A (1998) Vitamin C improves endothelium-dependent vasodilation by restoring nitric oxide activity in essential hypertension. Circulation 97:2222–2229
Taddei S, Virdis A, Ghiadoni L, Magagna A, Pasini AF, Garbin U, et al (2001) Effect of calcium antagonist or beta blockade treatment on nitric oxide-dependent vasodilation and oxidative stress in essential hypertensive patients. J Hypertens 19:1379–1386
Tanito M, Nakamura H, Kwon YW, Teratani A, Masutani H, Shioji K, Kishimoto C, Ohira A, Horie R, Yodoi J (2004) Enhanced oxidative stress and impaired thioredoxin expression in spontaneously hypertensive rats. Antioxid Redox Signal 6:89–97
Taniyama Y, Griendling KK (2003) Reactive oxygen species in the vasculature: molecular and cellular mechanisms. Hypertension 42:1075–1081
Tojo A, Onozato ML, Kobayashi N, Goto A, Matsuoka H, Fujita T (2002) Angiotensin II and oxidative stress in Dahl salt-sensitive rat with heart failure. Hypertension 40:834–839
Torrecillas G, Boyano-Adanez MC, Medina J, Parra T, Griera M, Lopez-Ongil S, Arilla E, Rodriguez-Puyol M, Rodriguez-Puyol D (2001) The role of hydrogen peroxide in the contractile response to angiotensin II. Mol Pharmacol 9:104–112
Torres M (2003) Mitogen-activated protein kinase pathway in redox signaling. Front Biosci 8:369–391
Touyz RM (2000) Oxidative stress and vascular damage in hypertension. Curr Hypertens Rep 2:98–105
Touyz RM (2003a) Recent advances in intracellular signalling in hypertension. Curr Opin Nephrol Hypertens 12:165–174
Touyz RM (2003b) Activated oxygen metabolites: do they really play a role in angiotensin II-regulated vascular tone? J Hypertens 21:2235–2238
Touyz RM, Schiffrin EL (1999) Ang II-stimulated superoxide production is mediated via phospholipase D in human vascular smooth muscle cells. Hypertension 34:976–982
Touyz RM, Schiffrin EL (2000) Signal transduction mechanisms mediating the physiological and pathophysiological actions of angiotensin II in vascular smooth muscle cells. Pharmacol Rev 52:639–672
Touyz RM, Schiffrin EL (2001) Increased generation of superoxide by angiotensin II in smooth muscle cells from resistance arteries of hypertensive patients: role of phospholipase D-dependent NAD(P)H oxidase-sensitive pathways. J Hypertens 19:1245–1254
Touyz RM, Chen X, He G, Quinn MT, Schiffrin EL (2002a) Expression of a gp91phox-containing leukocyte-type NADPH oxidase in human vascular smooth muscle cells: modulation by Ang II. Circ Res 90:1205–1213
Touyz RM, Wu XH, He G, Salomon S, Schiffrin EL (2002b) Increased angiotensin II-mediated Src signaling via epidermal growth factor receptor transactivation is associated with decreased C-terminal Src kinase activity in vascular smooth muscle cells from spontaneously hypertensive rats. Hypertension 39:479–485
Touyz RM, Yao G, Schiffrin EL (2003a) c-Src induces phosphorylation and translocation of p47phox: role in superoxide generation by angiotensin II in human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 23:981–987
Touyz RM, Cruzado M, Tabet F, Yao G, Salomon S, Schiffrin EL (2003b) Redox-dependent MAP kinase signaling by Ang II in vascular smooth muscle cells: role of receptor tyrosine kinase transactivation. Can J Physiol Pharmacol 81:159–167
Touyz RM, Yao G, Viel E, Amiri F, Schiffrin EL (2004) Angiotensin II and endothelin-1 regulate MAP kinases through different redox-dependent mechanisms in human vascular smooth muscle cells. J Hypertens (in press)
Tschudi M, Mesaros S, Luscher TF, Malinski T (1996) Direct in situ measurement of nitric oxide in mesenteric resistance arteries: increased decomposition by superoxide in hypertension. Hypertension 27:32–35
Turpaev KT (2002) Reactive oxygen species and regulation of gene expression. Biochemistry 67:281–292
Ushio-Fukai M, Alexander RW, Akers M, Griendling KK (1998) p38 mitogen-activated protein kinase is a critical component of the redox-sensitive signaling pathways activated by angiotensin II. Role in vascular smooth muscle cell hypertrophy. J Biol Chem 273:15022–15029
Vasquez-Vivar J, Duquaine D, Whitsett J, Kalyanaraman B, Rajagopalan S (2002) Altered tetrahydrobiopterin metabolism in atherosclerosis: implications for use of oxidized tetrahydrobiopterin analogues and thiol antioxidants. Arterioscler Thromb Vasc Biol 22:1655–1661
Viedt C, Soto U, Krieger-Brauer HI, Fei J, Elsing C, Kubler W, Kreuzer J (2000) Differential activation of mitogen-activated protein kinases in smooth muscle cells by angiotensin II: involvement of p22phox and reactive oxygen species. Arterioscler Thromb Vasc Biol 20:940–948
Vignais PV (2002) The superoxide-generating NADPH oxidase: structural aspects and activation mechanism. Cell Mol Life Sci 59:1428–1459
Virdis A, Fritsch Neves M, Amiri F, Viel E, Touyz RM, Schiffrin EL (2002) Spironolactone improves angiotensin-induced vascular changes and oxidative stress. Hypertension 40:504–510
Virdis A, Iglarz M, Neves MF, Amiri F, Touyz RM, Rozen R, Schiffrin EL (2003) Effect of hyperhomocystinemia and hypertension on endothelial function in methylenetetrahydrofolate reductase-deficient mice. Arterioscler Thromb Vasc Biol 23:1352–1357
Virdis A, Neves MF, Amiri F, Touyz RM, Schiffrin EL (2004) Role of NAD(P)H oxidase on vascular alterations in angiotensin II-infused mice. J Hypertens 22:535–542
Vivekananthan DP, Penn MS, Sapp SK, Hsu A, Topol EJ (2003) Use of antioxidant vitamins for the prevention of cardiovascular disease: meta-analysis of randomised trials. Lancet 361:2017–2023
Wassmann S, Laufs U, Muller K, Konkol C, Ahlbory K, Baumer AT, Linz W, Bohm M, Nickenig G (2002) Cellular antioxidant effects of atorvastatin in vitro and in vivo. Arterioscler Thromb Vasc Biol 22:300–305
Welch WJ, Mendonca M, Aslam S, Wilcox CS (2003) Roles of oxidative stress and AT1 receptors in renal hemodynamics and oxygenation in the postclipped 2 K,1C kidney. Hypertension 41:692–696
Wilcox CS (2002) Reactive oxygen species: roles in blood pressure and kidney function. Curr Hypert Rep 4:160–166
Wingler K, Wunsch S, Kreutz R, Rothermund L, Paul M, Schmidt HH (2001) Upregulation of the vascular NAD(P)H-oxidase isoforms Nox1 and Nox4 by the renin-angiotensin system in vitro and in vivo. Free Radic Biol Med 31:1456–1464
Wu R, Millette E, Wu L, de Champlain J (2001) Enhanced superoxide anion formation in vascular tissues from spontaneously hypertensive and desoxycorticosterone acetate-salt hypertensive rats. J Hypertens 19:741–748
Wu R, Lamontagne D, de Champlain J (2002) Antioxidative properties of acetylsalicylic acid on vascular tissues from normotensive and spontaneously hypertensive rats. Circulation 105:387–392
Yada T, Shimokawa H, Hiramatsu O, Kajita T, Shigeto F, Goto M, Ogasawara Y, Kajiya F (2003) Hydrogen peroxide, an endogenous endothelium-derived hyperpolarizing factor, plays an important role in coronary autoregulation in vivo. Circulation 107:1040–1045
Yamawaki H, Haendeler J, Berk BC (2003) Thioredoxin: a key regulator of cardiovascular homeostasis. Circ Res 93:1029–1033
Yang S, Hardaway M, Sun G, Ries WL, Key L Jr (2000) Superoxide generation and tyrosine kinase. Biochem Cell Biol 78:11–17
Zafari AM, Ushio-Fukai M, Akers M, Griendling K (1998) Role of NADH/NADPH oxidase-derived H2O2 in angiotensin II-induced vascular hypertrophy. Hypertension 32:488–495
Zalba G, Beaumont FJ, San Jose G, Fortuno A, Fortuno MA, Etayo JC, Diez J (2000) Vascular NADH/NADPH oxidase is involved in enhanced superoxide production in spontaneously hypertensive rats. Hypertension 35:1055–1061
Zalba G, San Jose G, Moreno MU, Fortuno MA, Fortuno A, Beaumont FJ, Diez J (2001a) Oxidative stress in arterial hypertension: role of NAD(P)H oxidase. Hypertension 38:1395–1399
Zalba G, San Jose G, Beaumont FJ, Fortuno MA, Fortuno A, Diez J (2001b) Polymorphisms and promoter overactivity of the p22(phox) gene in vascular smooth muscle cells from spontaneously hypertensive rats. Circ Res 88:217–222
Zanzinger J (2002) Mechanisms of action of nitric oxide in the brain stem: role of oxidative stress. Auton Neurosci 98:24–27
Zhan CD, Sindhu RK, Vaziri ND (2004) Up-regulation of kidney NAD(P)H oxidase and calcineurin in SHR: reversal by lifelong antioxidant supplementation. Kidney Int 65:219–227
Zheng JS, Yang XQ, Lookingland KJ, et al (2003) Gene transfer of human guanosine 5′-triphosphate cyclohydrolase I restores vascular tetrahydrobiopterin level and endothelial function in low renin hypertension. Circulation 108:1238–1245
Acknowledgements
The work of the authors was supported by grants 13570, 37917, 44018 and a Group grant to the Multidisciplinary Research Group on Hypertension, all from the Canadian Institutes of Health Research (previously called the Medical Research Council of Canada). R.M.T. received a scholarship from the Fonds de la recherche en santé du Québec.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Touyz, R.M., Schiffrin, E.L. Reactive oxygen species in vascular biology: implications in hypertension. Histochem Cell Biol 122, 339–352 (2004). https://doi.org/10.1007/s00418-004-0696-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00418-004-0696-7