
Abstract
Oxidative stress is an imbalance between endogenous prooxidant and antioxidant systems leading to excessive production of reactive oxygen species (ROS), which potentially disrupt redox signalling and/or inflict damage to macromolecules. Numerous studies over the last two decades have suggested a central role for oxidative stress in the development of several cardiovascular diseases. This chapter aims to summarize the current experimental and clinical evidence about the major oxidant and antioxidant changes occurring in hypertension and heart failure, and to provide a critical overview of the relevance of oxidative stress in the pathophysiology of these prevalent diseases. Finally, the strategies known to prevent or ameliorate oxidative damage, both in animal models and in patients, will be discussed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
1.1 Reactive Oxygen Species (ROS) and Oxidative Stress: General Considerations
Oxygen is essential for cellular respiration and energy production by aerobic organisms. However, the partial reduction of oxygen by several metabolic pathways will inevitably result in the production of reactive oxygen species (ROS). In turn, these short-lived and highly reactive oxygen metabolites have the ability to attack cellular macromolecules, including lipids, proteins and nucleic acids, causing oxidative damage. Under normal conditions, intracellular ROS concentrations are maintained within a balanced, steady-state range, by integrated enzymatic and nonenzymatic antioxidant systems, which not only protect cells from the detrimental effects of ROS but also allow the activation of redox signalling pathways that regulate physiological functions. Disruption of redox equilibrium by persistently elevated ROS concentrations leads to oxidative stress and subsequent dysfunctional signalling and macromolecular damage [1,2,3]. Oxidative stress is widely recognized as an important contributor to ageing, which is characterized by a progressive decline in biological functions and in the organism’s ability to adapt to metabolic stress over time, being also aetiologically involved in the pathogenesis of a wide variety of disease processes, namely arterial hypertension, atherosclerosis, heart failure (HF), diabetic neuropathy, renal diseases, neurological diseases, as well as cancer [4, 5]. In contrast to the well documented role of oxidative stress in cardiac diseases, less is known regarding the role of reductive stress in these processes. Reductive stress is the counterpart of oxidative stress and is defined as an aberrant increase in reducing equivalents, leading to decreased ROS levels. Nevertheless, it is becoming increasingly clear that the biological extremes of the redox spectrum play critical roles in disease pathogenesis [6]. In addition to ROS, there is another class of chemically reactive molecules collectively designated as reactive nitrogen species (RNS), which include various nitric oxide (.NO)-derived compounds. RNS have been recognized as playing important functions in diverse physiological and pathological redox signalling processes [7, 8]. Similarly to ROS, excessive amounts of RNS have been implicated in cell injury and death by inducing nitrosative stress.
1.2 Main Types of ROS and RNS
ROS can be divided in two main groups: i) free radicals [e.g. superoxide anion (O2·−), hydroxyl radical (HO.), peroxyl radical (ROO•)], which are unstable and highly reactive species due to the presence of one or more unpaired electrons; and ii) non-radical oxidants [e.g. singlet oxygen (1O2), hydrogen peroxide (H2O2), hypochlorous acid (HOCl)], that have generally more specific reactivity and higher stability. RNS include .NO and nitrogen dioxide radicals (.NO2) and also non-radicals such as peroxynitrite (ONOO−), nitrous acid (HNO2), peroxynitrous acid (ONOOH) and alkyl peroxynitrites (ROONO) (Table 23.1). Among biological ROS and RNS, O2·−, H2O2, .NO and ONOO− appear to play a prominent role in vascular, cardiac, renal and neuronal regulation and dysregulation, thus representing important targets for strategies aiming to reduce redox dysfunction in cardiovascular diseases [9, 10].
1.3 Mechanisms of ROS Generation in Cardiovascular Diseases
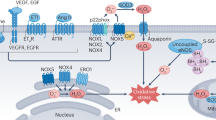
A key consideration for ROS/RNS chemistry and biology is the subcellular compartment where a particular species is generated, as discrete microenvironments can determine which targets will be preferentially attacked. ROS are derived from both endogenous and exogenous sources (Fig. 23.1). Intracellular compartments capable of ROS generation include mitochondria, the endoplasmic reticulum, peroxisomes, nuclei, the cytosol, and plasma membrane enzymatic systems. ROS can also be produced in response to external sources, including pollution, alcohol, tobacco smoke, heavy metals, UV radiation.

Major endogenous and exogenous sources leading to ROS production
1.3.1 Mitochondria
Mitochondria play a key role in energy metabolism in many tissues. More than 90% of the oxygen consumed by aerobic organisms is utilized by the mitochondrial electron transport chain (ETC), which generates ATP in a process coupled to the reduction of cellular oxygen to water. The mitochondrial respiratory chain complexes are also an important source of ROS within most mammalian cells [11,12,13]. In fact, about 1–4% of the oxygen used in these reactions is converted to O2·− and H2O2, which may have deleterious consequences to mitochondria if not adequately detoxified [14]. ROS formation in the mitochondria is regulated by the respiratory rate and by the antioxidant enzyme manganese superoxide dismutase (MnSOD) [12]. The mitochondrial respiratory chain appears to be a major source of oxidative stress in some experimental forms of arterial hypertension (e.g. mineralocorticoid hypertension, angiotensin II-induced hypertension) and the inhibition of mitochondrial ROS production has a significant blood pressure-lowering effect in these models [15, 16]. In HF there is also evidence of abnormal ROS production from mitochondrial respiratory chain. Furthermore, the scavenging of mitochondrial ROS has been shown to prevent or reverse HF and to eliminate sudden cardiac death in an animal model of non-ischemic HF that displays important features of human HF (e.g. prolonged QT interval, high incidence of spontaneous sudden cardiac death due to ventricular tachycardia/fibrillation) [17,18,19].
1.3.2 Other Prooxidant Enzymatic Systems
Besides mitochondrial oxidases, there are other important enzymatic sources of ROS, such as NADPH oxidases, myeloperoxidase, .NO synthases, xanthine oxidase and monoamine oxidases (Fig. 23.1).
1.3.2.1 Nicotinamide Adenine Nucleotide Phosphate (NADPH) Oxidases
NADPH oxidases (NOX) are multi-subunit transmembrane enzymes complexes that catalyze the one-electron reduction of molecular oxygen using NADPH as an electron donor. In general, the product of the electron transfer reaction is O2·−, but H2O2 is also rapidly formed from dismutation of NOX-derived O2·− due to the presence of superoxide dismutase in the cells or by spontaneous reaction. NOX-derived ROS play a role in host defence and also in various signalling pathways [20]. The NOX family contains seven members (NOX1-5 and Duox1-2) with distinct tissue distribution and roles [20]. NOX1, NOX2 and NOX4 isoforms enzymes appear to be particularly relevant in the pathophysiology of hypertension, being expressed in major sites of blood pressure regulation [20, 21]. For example, NOX1, NOX2 and NOX4 can be found in the central nervous system, where they contribute to sympathetic nerve activity control [21]. In the kidney, NOX2 and NOX4 appear to be the main isoforms regulating renal function and contributing to end-organ damage [21, 22]. These isoforms are also important determinants of vascular tone in several vascular beds, including the renal afferent arteriole, which is critical for the regulation of renal haemodynamics [23,24,25]. Endothelial function can be regulated by NOX2, which contributes to impaired vasodilation, or by NOX4, which improves endothelial-dependent vasodilation. NOX1 and NOX4 are also involved in vascular smooth muscle cell growth and migration [20, 23, 24]. Of note, recent studies suggest that NOX5, an isoform that is found in humans but absent in rodents, is also implicated in the pathogenesis of cardiovascular diseases, such as hypertension and atherosclerosis [26]. For example, renal proximal tubular cells from human hypertensive subjects appear to express NOX5 in a greater extent than the other isoforms [27]. Furthermore, in mice expressing human NOX5 in podocytes, the renal function becomes impaired and blood pressure increases [26]. NOX5 expression was also shown in human carotid artery atherosclerotic plaques and to be induced in macrophages exposed to a proinflammatory and prooxidant environment [28].
NOX2 and NOX4, the two isoforms expressed in the heart, appear to be especially relevant in HF [29, 30]. NOX2 contributes to angiotensin II-induced cardiac hypertrophy, atrial fibrillation, myocyte death under stress conditions and post-myocardial infarction remodelling. The inactivation of NOX2 was shown to attenuate ventricular dilatation and contractile dysfunction in experimental models of myocardial infarction. NOX2 deletion also abolished angiotensin II-induced cardiac hypertrophy but was not able to prevent the development of HF caused by severe pressure overload [29, 30]. The role of NOX4 in the heart is more controversial, with both protective and detrimental effects reported. For example, mice lacking cardiac NOX4 display either reduced or aggravated maladaptive remodelling in different models of pressure-overload-induced HF [29, 30]. In what concerns to ischemia-reperfusion injury, it appears that both NOX2 and NOX4 contribute to increased ROS production and damage, as evidenced by the reduced myocardial infarct size/area at risk and lower O2·− production in NOX2 knockout or NOX4 knockout mice subjected to ischemia-reperfusion injury. However, double knockout of NOX2 and NOX4 exacerbates ischemia-reperfusion injury, probably because low levels of ROS generated by these enzymes are necessary to activate adaptive mechanisms that protect the heart against ischemia-reperfusion injury [31].
1.3.2.2 Myeloperoxidase (MPO)
MPO, a haem-containing enzyme secreted by activated neutrophils and monocytes under inflammatory conditions, produces several oxidizing molecules that can affect lipids and proteins [32]. MPO uses H2O2 to produce other ROS/RNS, such as HOCl, chloramines, tyrosyl radicals and nitrogen dioxides [32]. Although MPO-derived ROS have a major role as bactericidal agents, they can also cause tissue damage in the heart, vessels, kidney and brain. Vascular tone and endothelial bioavailability of .NO appear to be significantly affected by MPO. Interestingly, the MPO G463A polymorphism was associated with an increased risk of hypertension [33]. MPO contributes to vascular and myocardial dysfunction, being significantly increased in acute coronary syndromes and HF [34,35,36]. Higher MPO values were reported to be associated with increasing likelihood of more advanced HF in chronic systolic HF patients and to predict future adverse clinical events [37].
1.3.2.3 NO Synthases
The .NO synthases (NOS) are a family composed of three enzyme isoforms (neuronal NOS, nNOS; inducible NOS, iNOS; endothelial NOS, eNOS) [38]. NOS are the endogenous sources of .NO in mammalian cells, in a reaction that converts L-arginine to L-citrulline [38]. .NO exerts a wide array of regulatory functions on the cardiovascular system, including regulation of vascular tone, blood pressure, cardiomyocyte contractility, sympathetic outflow, smooth muscle cell proliferation, renal renin release and natriuresis [39,40,41]. However, under conditions of limited bioavailability of the cofactor tetrahydrobiopterin (BH4) or the substrate L-arginine, NOS become unstable and reduces molecular oxygen to O2·− instead of producing .NO. This NOS uncoupling is more often described for eNOS and is triggered by oxidative/nitrosative stress [42]. There is evidence that eNOS dysregulation and consequent endothelial dysfunction occur both in hypertension and HF [43, 44]. Treatment with BH4, which contributes to eNOS recoupling, prevented or attenuated hypertension in spontaneously hypertensive rats [37]. It was also shown to reverse cardiac hypertrophy and fibrosis and to improve chamber and myocyte function in mice with heart disease induced by pressure overload [10, 45].
1.3.2.4 Xanthine Oxidase
The enzyme xanthine oxidoreductase displays two interchangeable forms, xanthine dehydrogenase (XDH) and xanthine oxidase (XO), that participate in the metabolism of purines by catalyzing the conversion of hypoxanthine to xanthine and xanthine to uric acid [38, 46]. XDH uses NAD+ as the preferential electron acceptor while XO reduces molecular oxygen in a reaction that generates O2·− and H2O2 [38, 46]. The XO form predominates in oxidative stress conditions and may contribute to endothelial dysfunction due to its localization in the luminal surface of vascular endothelium [38, 46]. Although XO is capable of generating ROS, both XDH and XO generate uric acid which has antioxidant properties, such as the ability to scavenge ONOO− and HO•, to prevent oxidative inactivation of endothelium enzymes and to stabilize vitamin C [47, 48]. In contrast, uric acid may also exhibit prooxidant and proinflammatory effects. Indeed, increased uric acid levels have been associated with cardiovascular disease [49, 50]. However, it is still unclear whether these effects reflect direct deleterious actions of uric acid or, alternatively, oxidative damage caused by XO-derived ROS.
XO appears to contribute to the pathophysiology of arterial hypertension in SHR, as evidenced by the significant reduction of blood pressure induced by the treatment with XO inhibitors [10, 51]. In humans, some studies have shown a blood pressure-lowering effect of XO inhibition in adolescents with newly diagnosed essential hypertension and an improvement of cardiovascular outcomes in adults with hypertension [52, 53].
In what concerns to heart diseases, XO inhibition was reported to improve left ventricle contractility and myocardial efficiency in an animal model of HF and to attenuate adverse left ventricular remodelling in experimental myocardial infarction [19]. XO expression and activity was also shown to be increased in coronary arteries from patients with coronary artery disease, contributing to the augmented production of O2·− [54]. The inhibition of XO with oxypurinol also improved myocardial contractility in patients with ischemic cardiomyopathy [55]. However, other studies failed to demonstrate clinical benefits of oxypurinol treatment in unselected patients with moderate-to-severe HF or in high-risk HF patients with reduced left ventricular ejection fraction and hyperuricemia [56, 57].
1.3.2.5 Monoamine Oxidases (MAO)
MAO-A and MAO-B are flavoenzymes predominantly located at the outer membrane of mitochondria, being responsible for the oxidative degradation of neurotransmitters (catecholamines, serotonin) and biogenic amines in a process that generates H2O2, ammonia and an aldehyde intermediate. All of these products are potentially deleterious, especially for mitochondria. Pathological stimuli such as neurohormonal and/or chronic hemodynamical stress, inflammation and ischemia-reperfusion can increase the availability of MAO substrates, thus augmenting H2O2-induced mitochondrial dysfunction in cardiovascular tissues/organs and leading to endothelial dysfunction and HF [9, 58, 59]. In experimental models of hypertension (induced by angiotensin II) and inflammation (induced by lipopolysaccharide), the expression of both MAO isoforms increased in endothelial cells and MAO inhibition attenuated ROS production and restored endothelial-dependent vasodilation [59]. MAO are also important sources of ROS in the heart. There are several important cardiac targets for MAO-derived ROS, besides mitochondria. These include sphingosine kinase-1, an enzyme involved in cell survival, whose inhibition may contribute to cardiomyocyte apoptosis, as well as the contractile proteins, actin and tropomyosin, whose oxidation correlates with ventricular dysfunction, and matrix metalloproteinases, whose activation induces extracellular matrix remodelling. The signalling pathways activated by MAO-derived H2O2 depend on the availability of MAO substrates and H2O2 concentration in tissues. Lower amounts of H2O2 trigger hypertrophy, cell proliferation and matrix remodelling, while higher concentrations lead to mitochondrial dysfunction, apoptosis or necrosis. MAO inhibition appears to be protective in ischemia-reperfusion injury and pressure overload-induced HF [59, 60].
1.4 Major Endogenous Antioxidant Systems
All living organisms have adapted and developed an endogenous antioxidant defence system, composed of enzymatic and nonenzymatic antioxidants, that is usually effective in neutralizing deleterious effects of ROS (Fig. 23.2). However, when the antioxidant systems are overwhelmed, as observed in most pathological conditions, oxidative stress ensues. Below we provide an overview of the major antioxidant systems with relevance to cardiovascular diseases.

Major enzymatic and nonenzymatic antioxidants
1.4.1 Enzymatic Antioxidants
1.4.1.1 Superoxide Dismutases
Superoxide dismutase (SOD) enzymes consist of three isoforms in mammals: the cytoplasmic Cu/ZnSOD (SOD1), the mitochondrial MnSOD (SOD2), and the extracellular Cu/ZnSOD (SOD3), all of which require catalytic metals (Cu or Mn) for their activity [61]. They are considered the major antioxidant defences against O2·−, being responsible for its dismutation to H2O2 and molecular oxygen, which limits the potentially harmful effects of this radical species [61].
1.4.1.2 Catalase and Glutathione Peroxidase
H2O2 produced by the action of SODs or oxidases, such as XO, can be further decomposed to water and oxygen. This is achieved primarily by catalase in the peroxisomes and by glutathione peroxidase (GPx) enzymes in the cytosol and mitochondria. Catalase exists as a tetramer composed of 4 identical monomers, each of which contains a haem group at the active site. Degradation of H2O2 is accomplished via the conversion between 2 conformations of catalase-ferricatalase and compound I. GPx are selenium-containing enzymes whose activity is dependent on the amount of reduced glutathione (GSH) available [62]. Besides neutralizing H2O2, GPx also degrades lipid hydroperoxides to lipid alcohols. These reactions lead to the oxidation of GSH to oxidized glutathione (GSSG). Catalase and GPx are differentially required for the clearance of high-levels or low-levels of H2O2, respectively [63].
1.4.1.3 Other Enzymatic Defences
In addition to the antioxidant enzymatic systems mentioned above, cells also express other specialized enzymes with direct and/or indirect antioxidant functions. Glutathione reductase (GR) regenerates GSH from GSSG in the presence of NADPH. Glutathione-S-transferase (GST) catalyzes the conjugation of GSH with reactive electrophiles and detoxifies some carbonyl-, peroxide- and epoxide-containing metabolites produced within the cell in oxidative stress conditions. Peroxiredoxins (Prx) are selenium-independent enzymes that decompose H2O2, organic hydroperoxides and peroxynitrite, and thioredoxin (Trx) and glutaredoxin (Grx) systems include various enzymes that regulate the thiol-disulphide state of proteins and modulate their structure and activity [10].
1.4.2 Nonenzymatic Antioxidants
Nonenzymatic antioxidants, such as GSH, ascorbic acid (vitamin C) and α-tocopherol (vitamin E) play a key role in protecting the cells from oxidative damage and are considered as the second line of defence against active radicals. GSH is termed the master antioxidant given its electron-donating capacity that renders GSH a potent antioxidant per se, besides acting as an important cofactor for GPx and other enzymes. Vitamins E and C are among the major dietary antioxidants. Vitamin E, found in lipoproteins, cell membranes and extracellular fluids, terminates lipid peroxidation processes and converts O2·− and HO• to less reactive forms. Vitamin C is a water-soluble antioxidant that can directly scavenge ROS and lipid hydroperoxides. Carotenoids, such as β-carotene, are lipid soluble antioxidants that function as efficient quenchers of 1O2 but may also scavenge ROO• radicals. Uric acid is a highly abundant aqueous antioxidant, considered to be the main contributor for the antioxidant capacity in the plasma. It has the ability to quench HO• and ONOO− and may prevent lipid peroxidation, but may also exert prooxidant effects once inside the cells. Bilirubin, the end-product of haem catabolism, has chainbreaking antioxidant properties. Plasma albumin, the predominant plasma protein, is also an antioxidant and can scavenge MPO-derived chlorinated reactive species and ROO• radicals [10].
1.5 The Dual “Faces” of ROS
It has long been accepted that elevated ROS levels can cause damage to macromolecules and have been implicated in a vast array of pathologies. More recently, it has become apparent that ROS also serve as signalling molecules to regulate biological and physiological processes and that dysregulated ROS signalling may contribute to a host of human diseases [3]. Downstream of ROS production, several signalling pathways are activated, including protein kinases [mitogen activated protein kinases (MAPKs), protein tyrosine kinases (PTKs), protein kinases B and C] and transcription factors (NF-κB, Nrf2) [64]. Nevertheless, our understanding of the signalling “face” of ROS is still in its infancy, as ROS can often act upstream and/or downstream within a given pathway and sometimes in opposing ways (i.e. inhibitory or stimulatory).
2 Evidence for Redox Changes in Experimental and Human Hypertension
2.1 Links Between Oxidative Stress and Hypertension
Arterial hypertension, currently defined as systolic blood pressure values ≥140 mmHg and/or diastolic blood pressure ≥90 mm Hg, is a multifactorial, complex disorder, involving many organ systems and constitutes a major risk factor for cardiovascular disease and premature mortality throughout the world [65]. Major pathophysiological mechanisms implicated in the development of hypertension include central nervous system dysregulation and increased activity of sympathetic nervous system, altered renal function with increased renal sodium and water retention and increased peripheral vascular resistance (Fig. 23.3) [51, 66]. The renin-angiotensin-aldosterone system (RAAS) also plays a central role in the regulation of arterial pressure by renal and extrarenal mechanisms (e.g. regulation of sodium homeostasis, autopotentiation of vasoconstrictor responses, vascular hypertrophy, regulation of sympathetic output, facilitation of sympathetic neurotransmitter release, promotion of oxidative stress and inflammation), being intimately involved in hypertension pathophysiology [67,68,69,70,71].

Organs and mechanisms involved in the development and maintenance of arterial hypertension
Oxidative stress has emerged as a unifying hypothesis for explaining these diverse mechanisms. Evidence gathered over the last two decades in both experimental models and humans suggests that hypertension arises from increased production of ROS and/or reduced antioxidant capacity in the cardiovascular, renal and central nervous systems [21, 42, 51].
By using animal models of genetic and drug-induced hypertension, we and others have demonstrated increased ROS levels and prooxidant activity, altered antioxidant defences and increased ROS-mediated damage, both at peripheral and central sites of cardiovascular regulation [72,73,74,75,76,77,78]. These studies have also underlined the importance of the kidney in the pathogenesis of hypertension and identified the renal medulla as a major target for angiotensin II-induced redox dysfunction in hypertension [72, 73]. Similarly to what happens in animals, there is also evidence of redox dysfunction in human hypertensive patients, although the association is less consistent and results vary depending on the biological marker of oxidative stress being investigated. The release of O2·− from peripheral polymorphonuclear leucocytes is increased in hypertensive patients in comparison with normotensive subjects [79]. Plasma H2O2 production is augmented in hypertensive patients and, among normotensive subjects, those with a family history of hypertension also exhibit a higher H2O2 production [80]. Increased levels of byproducts of protein, lipid and DNA oxidative damage, such as malondialdehyde, 8-isoprostanes, 8-oxo-2′-deoxyguanosine, oxidized low density lipoproteins, carbonyl groups and nitrotyrosine, have also been found in biofluids (i.e., plasma, serum and urine) and blood cells of hypertensive patients [81,82,83]. Furthermore, both enzymatic and nonenzymatic antioxidant defences appear to be reduced in human hypertension [81, 82, 84, 85]. Despite the vast number of studies reporting a close association between oxidative stress and hypertension, there is still an ongoing debate whether oxidative stress is a cause or a consequence of the disorder [86,87,88].
2.2 Oxidative Stress as Either a Cause or a Consequence of Hypertension
A large body of literature supports the hypothesis that oxidative stress is a major driver of arterial hypertension. In rats, the induction of oxidative stress through the administration of a common environmental heavy metal pollutant (lead), a glutathione synthesis inhibitor (buthionine sulfoximine-BSO) or a SOD inhibitor (sodium diethyldithiocarbamate-DETC), as well as the intrarenal or intrathecal infusion of H2O2, lead to increases in blood pressure [72, 89, 90]. The genetic manipulation of enzymes involved in ROS production or metabolism also modifies blood pressure in mice [91,92,93]. In addition, the exposure of cells and tissues to exogenous oxidants recapitulates molecular events implicated in the pathogenesis of hypertension [72, 94]. Also of importance are the facts that experimental hypertension can be prevented or attenuated by the administration of some antioxidants or inhibitors of ROS production [95,96,97,98] and that redox dysregulation, both at systemic and tissue level, precedes the rise in blood pressure [99, 100]. Collectively, these observations in preclinical models of hypertension suggest that oxidative stress plays a causal role in the development of hypertension.
Nevertheless, other authors have failed to demonstrate a direct involvement of oxidative stress in the pathogenesis of hypertension since the administration of antioxidants or inhibitors of ROS generation did not prevent or attenuate experimental hypertension [10]. Indeed, if oxidative stress is causally related to human hypertension, then antioxidants should be able to reduce blood pressure and oxidative damage. However, the majority of clinical trials did not find any blood pressure-lowering effects of antioxidants. One of the largest studies observed no improvement in blood pressure after a 5-year treatment with a combination of vitamin C, vitamin E, and β-carotene versus placebo in subjects thought to be at high risk of cardiovascular disease [101]. Likewise, a recent study found no beneficial effects against major cardiovascular events, including hypertension, after more than a decade of treatment with a multivitamin supplement versus placebo in a population of US male physicians [102]. Furthermore, a meta-analysis failed to reveal a clear benefit after antioxidant supplementation in cardiovascular mortality [103].
There is also evidence that lowering blood pressure per se leads to a reduction in oxidative stress and improvement in vascular function [10, 88]. Several antihypertensive drugs with different mechanisms of action, such as angiotensin-converting enzyme inhibitors, angiotensin II receptor antagonists, beta-blockers and calcium channel blockers, have been shown to attenuate oxidative stress markers in experimental and human hypertension [104, 105]. In light of these observations, some authors suggest that oxidative stress may be rather a consequence than a cause of hypertension. However, some of these antihypertensive agents have direct antioxidant properties and others block the RAAS, whose downstream effects are known to be mediated by ROS [10].
2.3 Pharmacological Interventions Aimed to Reduce Blood Pressure with Antioxidant Therapies
The rationale for reducing oxidative stress as a therapeutic strategy against hypertension stems from population-based observational studies showing an inverse correlation between plasma antioxidant concentrations, obtained by dietary intake, with blood pressure and cardiovascular risk factors [106]. However, in contrast with preclinical data, no significant improvement in blood pressure has been observed in the vast majority of studies after treatment with single or combination antioxidant therapy in subjects thought to be at high risk of cardiovascular disease (as discussed above in Sect. 23.2.2). A number of potential explanations for the failure of antioxidant supplementation in the chronic suppression of cardiovascular disease in humans have been put forward, including errors in trial design, choice of antioxidants, patient cohorts included in trials, the pathophysiological complexity of ROS/RNS signalling in humans with comorbidities, among others [107]. In what concerns the antioxidants, it is possible that the dose administered and duration of clinical trials were insufficient or agents examined were ineffective and nonspecific. Most antioxidant therapies that have been tested were not chosen because they were proved to be the best antioxidants, but rather because of their easy availability. It is also conceivable that the antioxidants administered failed to target the source of free radicals, particularly if ROS are generated in intracellular organelles and compartments, due to relatively poor uptake of antioxidants by target organs or the interference with other substances that, in some cases, reduce the antihypertensive effects. It is critical to remember that the lack of benefits seen in clinical trials to date does not rule out the essential role of oxidative stress in hypertension and other cardiovascular disorders. Rather, these results highlight the importance of evaluating optimal antioxidant therapies, the ideal cohort of patients to study, and the appropriate trial duration for the future improvement of antioxidant therapy.
3 Oxidative Stress in Heart Failure
3.1 The Heart, Metabolic Demand and ROS Production
The mammalian heart is the organ with the highest metabolic demand, consuming a large amount of cellular ATP to maintain the contraction-relaxation cycle. Under physiological conditions, this tremendous energy requirement is fulfilled by the high mitochondrial content of cardiomyocytes [19, 108,109,110]. Mitochondria ensure the production of more ATP through oxidative phosphorylation, whereby the mitochondrial ETC generates a proton gradient that drives ATP synthesis by ATP synthase. Since this process is sustained by O2, which functions as the final electron acceptor in the ETC, it is not surprising that the heart needs a continuous, as well as adjustable, high supply of O2 to maintain its function and viability [19, 108, 109]. Normally, most of the O2 consumed in oxidative phosphorylation is reduced to water. However, electron leakage from the ETC also occurs, thus resulting in the formation of a small amount of ROS, namely O2·− and H2O2, which can be detoxified by endogenous antioxidant enzymes [19, 108,109,110]. There are several other ROS-producing enzymes in the heart, namely NOXs, XO, uncoupled NOS, MAOs and MPO, that are present in several cell types such as cardiomyocytes, endothelial cells, vascular smooth muscle cells, fibroblasts, neutrophils, monocytes and macrophages [9, 19, 109,110,111].
Although large amounts of ROS are markedly detrimental, there is evidence that low-to-moderate ROS concentrations in the heart are involved in physiological processes and beneficial adaptive signalling in response to acute changes in workload or brief ischemic episodes [108, 110, 112]. For example, ROS contribute to cardiomyogenesis of embryonic stem cells and proliferation of neonatal cardiac cells [113, 114]. It has also been reported that H2O2 derived from dismutation of O2·− generated by myocardial ETC is involved in coronary dilation, thus linking myocardial oxygen consumption to coronary blood flow [115, 116]. In addition, an increase in mitochondrial-derived ROS appears to mediate the acute inotropic response of cardiomyocytes to β-adrenergic receptor stimulation, being part of the homeostatic physiological signalling in the heart [117]. Importantly, mitochondrial and NOX-derived ROS seem to participate in the protective adaptive responses to moderate hypoxia, through the redox regulation of cardiomyocyte hypoxia-inducible factor activation, and in myocardial ischemic preconditioning, a protective phenomenon triggered by transient ischemic episodes and responsible for enhanced heart resistance to prolonged ischemia-reperfusion scenarios [108, 112, 118, 119].
3.2 Role of Oxidative Stress in the Pathophysiology of Heart Failure
HF is a complex clinical syndrome derived from structural and/or functional abnormalities in the heart, leading to impaired ventricular filling or ejection [9, 120]. Cardiac dysfunction triggers compensatory haemodynamic and neurohormonal responses attempting to maintain proper tissue perfusion, but these ultimately become maladaptive and deleterious [121]. Typical symptoms of this syndrome include shortness of breath, ankle swelling, fatigue, tiredness and reduced tolerance to exercise [120]. HF is usually a chronic, progressive and terminal illness, associated to poor quality of life for the patient due to the increase in symptoms frequency, severity and distress along disease course. Its prevalence in developed countries ranges from 1–2% in adults but can increase to values equal or higher than 10% in people with more than 70 years old, posing an enormous economic burden on healthcare systems. Of note, HF is the most frequent diagnosis responsible for hospitalization among patients aged 65 years or older [122, 123]. HF aetiologies include those related with diseased myocardium (e.g. ischemic heart disease; toxic damage due to alcoholism, drugs of abuse, medications such as cytostatics, heavy metals or radiation; immune-mediated and inflammatory damage caused by infections or auto-immune conditions; metabolic derangements such as thyroid diseases and pheocromocytoma; infiltration related with malignancy or other diseases such as amyloidosis; genetic disorders), with abnormal loading conditions (e.g. arterial hypertension; valve and myocardial structural defects; pericardial and endomyocardial pathologies; high output states such as severe anaemia; volume overload caused by renal failure) or with arrhythmias [120].
Despite therapeutic advances, chronic HF often decompensates, leading to the rapid aggravation of symptoms and/or signs of HF and thus requiring hospitalization [120, 124]. The term acute HF frequently refers to this state of acute decompensation of chronic HF but may also represent new-onset HF (“de novo” HF) resulting, for example, from acute myocardial dysfunction due to ischemic, inflammatory or toxic insults, acute valve insufficiency or cardiac tamponade (a condition characterized by heart compression and dysfunction as a consequence of pericardial accumulation of fluid, pus, blood, clots or gas due to blunt or penetrating trauma, accidental cardiac perforation following catheterization, infection, cancer and aortic aneurysm rupture) [120, 125].
As mentioned previously, low-to-moderate amounts of ROS contribute to physiological and beneficial adaptive responses in the heart. However, when prooxidant and antioxidant systems are imbalanced, leading to a prevailing prooxidant status, macromolecular damage and harmful signalling may occur and contribute to the genesis and progression of HF [9, 19, 112]. In the heart, there are many processes or targets that can be adversely affected by ROS (Fig. 23.4), namely cardiac contractility, myocardial remodelling, cardiomyocyte apoptosis, mitochondria and endothelium [9, 19, 109, 112, 126]. ROS also contribute to ischemic cardiomyopathy by promoting the formation of oxidized low-density lipoprotein (oxLDL), which plays a central role in the pathogenesis of atherosclerosis [9, 109]. Furthermore, the redox sensitive alteration of apolipoprotein A-I, the major protein constituent in high-density lipoproteins (HDL), inhibits the efflux of cholesterol, contributing to atherosclerotic lesions formation and to a prooxidant and proinflammatory environment [127]. The activation of matrix metalloproteinases by ROS is also involved in coronary atheromatous plaque instability, rupture and subsequent coronary artery thrombosis [9, 109]. Of note, after a significant myocardial ischemic insult, the restoration of oxygen supply during the reperfusion phase is responsible for the generation of high amounts of ROS, which contribute to extensive damage and tissue necrosis in the heart [9, 109, 127].

Adverse effects of ROS in the heart. ADMA asymmetric dimethylarginine, eNOS endothelial nitric oxide synthase, DNA deoxyribonucleic acid, HDL high-density lipoprotein, MMPs matrix metalloproteinases, oxLDL oxidized low-density lipoprotein, ROS reactive oxygen species
Inflammation plays a central role in the development and progression of chronic HF, regardless of aetiologies [128, 129]. It is also considered an important precipitator and prognostic factor in acute HF [130]. Oxidative stress and inflammation are closely interconnected, contributing to the pathophysiology of HF [9, 109, 131]. Several transcription factors that regulate the expression of proinflammatory cytokines are activated under oxidative stress conditions [9, 109, 131]. In turn, proinflammatory cytokines induce the generation of ROS, thus creating a potential vicious cycle of oxidation and inflammation [9, 131, 132]. Moreover, the production of large amounts of ROS is a feature of activated inflammatory cells, and MPO, a major effector enzyme of neutrophils that is released into the extracellular space during leukocyte activation, also functions as a link between oxidative stress and inflammation [9, 34, 36]. This enzyme uses H2O2 as a substrate to produce HOCl, which is a potent prooxidant and proinflammatory molecule. Importantly, MPO has the ability to bind and infiltrate in the vascular wall and to utilize H2O2 derived not only from leukocyte oxidative burst but also from vascular NOX, thus amplifying vascular injury in conditions associated with higher than normal ROS production [9, 36, 133].
Our recent studies have demonstrated the interplay between oxidative stress and inflammatory processes in human HF. In a study involving patients with mild-to-moderate and severe chronic HF, we observed that severe patients had increased values of systemic MPO activity and lower concentrations of lipoxin A4 (LXA4), a specialized proresolving lipid mediator (SPM) that stimulates the resolution of inflammation and tissue regeneration [121, 134]. Furthermore, we found an inverse correlation between LXA4 with proinflammatory/prooxidant markers, such as C-reactive protein (CRP), uric acid and MPO activity, and with markers of heart dysfunction and/or injury, like B-type natriuretic peptide (BNP), troponin I and myoglobin [134]. In addition, in another study evaluating patients with acute HF, cardiogenic shock (the most severe form of HF) and healthy controls, we showed that patients with cardiogenic shock exhibited the highest values of endocan, a marker of endothelial dysfunction, which was significantly associated with inflammatory status [135, 136]. Among the controls and patients evaluated, serum nitrotyrosine, a marker of oxidative/nitrosative stress, was significantly and positively correlated with CRP and high-sensitivity-troponin I, which are markers of inflammation and myocardial damage, respectively [135]. We also observed that resolvin E1 (RvE1), another mediator of inflammation resolution, increased in line with acute HF severity and was significantly associated with inflammatory/oxidant status and endothelial dysfunction [136].
Noteworthy, LXs and Rvs, besides possessing proresolving and anti-inflammatory properties, have also been shown to exert several protective effects on redox status that may be particularly relevant in the context of HF. These include the blockade of NOX enzymes in endothelial cells and macrophages, inhibition of ROS generation by leukocytes and vascular smooth muscle cells, blockade of angiotensin II-, thrombin- or tumor necrosis factor-α (TNF-α)-induced ROS production in endothelial cells, increased SOD activity and reduced malondialdehyde (MDA) content in the heart, induction of haem oxygenase-1 in endothelial cells and cardiomyocytes and upregulation of nuclear factor erythroid-2 related factor 2 (Nrf2) in cardiomyocytes [121]. Thus, strategies targeting inflammation or promoting its resolution will likely attenuate oxidative stress, and vice-versa, in patients with HF.
3.3 Biomarkers of Oxidative Stress in Human Heart Failure
Human HF was recently divided into 3 categories according to left ventricular ejection fraction (LVEF): reduced (HFrEF), preserved (HFpEF) or mid-range (HFmrEF) [120]. This definition only comprises the clinical manifestation of an underlying structural and/or functional cardiac abnormality resulting from a myriad of insults, of which the ischemic is the most prevalent in HFrEF. Thus, in clinical trials it is difficult to understand oxidative stress as a cause or consequence of the disease because HF prevails in older ages and it remains underdiagnosed and untreated [137, 138]. It is well established that ageing is associated with increased ROS accumulation, lipid peroxidation and mitophagy as well as atherosclerosis, diabetes and obesity, major risk factors for ischemic heart disease and HF. Despite the association between oxidative stress with clinical outcome in patients with coronary artery disease, no redox biomarker is currently in routine clinical use, in part because they are not specific for individual disease processes [139, 140]. The question remains whether plasma oxidation products reflect systemic or vascular redox state or other biological processes, as well as what is their value for independent stratification and therapeutic management, critical issues to consider them as “biomarkers”. Nevertheless, and surmounting the difficulties associated with the short half-life, limited diffusion and requirement of invasive biopsies to quantify ROS in human tissues, indirect indexes of oxidative stress are gaining increasing acceptance among established biomarkers of HF [141]. These biomarkers can be grouped into three main categories, namely prooxidant enzymes, products of oxidized macromolecules and antioxidant defences.
3.3.1 Prooxidant Enzymes
Myeloperoxidase contributes to endothelial dysfunction and mediates dysregulation of vascular tone [10]. The plasma concentration of MPO is elevated in HF patients compared to controls and its systemic activity is also increased in severe chronic HF compared to mild to moderate HF [134, 142]. Treatment with the inodilator levosimendan seems to reduce the concentration of plasma MPO by decreasing its release from neutrophils in patients with acute decompensation of chronic HF [143]. MPO was selected among others as an incremental prognostic biomarker in a multimarker risk strategy of stratification for cardiovascular death or HF in patients with acute myocardial infarction [144] and it also seems to differentiate forms of acute HF with cardiorenal syndrome [145]. These results, along with its predictive value for cardiovascular morbidity and mortality observed in other relatively large prospective studies, its therapeutic implications and the feasibility of its commercial assays, make MPO one of the most promising redox biomarkers for clinical application [146, 147].
Although NOXs are important cardiovascular ROS sources, available data about the involvement of NOXs in human HF is scarce. One study has described increased NOX2 expression and raised NOX activity in myocardial tissue from human failing hearts compared with non-failing controls, but information is lacking regarding NOX2 association with prognosis or treatment, with the exception of a study reporting a downregulatory effect of mediterranean diet on soluble NOX2-derived peptide values in patients with atrial fibrillation [148]. Nevertheless, compelling evidence suggests that redox protective effects of RAAS inhibitors, which are part of HF pharmacological treatment, are due to the prevention of vascular and phagocytic NOX activation [24, 120, 137].
The .NO-generating enzyme eNOS has some limitations as a redox biomarker in humans, not only for its localization (in the vessel wall and cardiomyocytes) but also because the complex regulation of the biosynthesis of its cofactor, BH4, makes hard to estimate the ratio of reduced to oxidized forms (BH4/BH2) and consequently to calculate eNOS uncoupling, which is responsible for generating O2·− instead of NO. The administration of BH4 does not improve vascular oxidative stress in patients with coronary disease [149] but indirect strategies like folates [150], statins [9] or polyphenols [151] could do so, thus reinforcing the interest of this pathway for future research in HF. Of note, in high-risk diabetic patients, the cardioprotection and reduction of risk of re-infarction and all-cause mortality afforded by metformin, which is no longer contra-indicated in HF, seems to be related, at least in part, with increased .NO bioavailability [152]. Also, the superiority of ticagrelor vs. clopidogrel in reducing cardiovascular events can be explained by the higher .NO concentrations triggered by ticagrelor, compared to clopidogrel, through an adenosine-mediated pathway that activates eNOS [153, 154].
3.3.2 Products of Oxidized Macromolecules
Lipid peroxidation results from ROS attack to polyunsaturated fatty acids (PUFA) in cell membranes. End-products of lipid peroxidation, including isoprostanes and MDA, affect membrane fluidity, inactivate receptors and enzymes attached to it, and even threaten cell viability. This lipid susceptibility to ROS has attracted considerable attention to the evaluation of lipid peroxides as biomarkers of oxidative stress.
Isoprostanes are produced by ROS-induced peroxidation of arachidonic acid and then released by phospholipases [155]. The most stable and thus most commonly quantified are F2-isoprostanes, which can be assessed in tissues and biological fluids. In HF, isoprostane levels in plasma, urine and pericardial fluid correlate with disease severity and ventricular dilatation [156, 157]. Recent works are hypothesizing that they could be used in a precocious strategy to identify populations with sub-clinical increased cardiovascular risk. In addition, they could also be used to monitor the protective effect of diets (e.g. low-sodium diet), as well as dietary adequacy, in patients with HF [158, 159].
MDA, another product of lipid peroxidation, is routinely evaluated by the thiobarbituric acid-reactive substances (TBARS) assay. There is evidence of increased systemic and intraplatelet production of TBARS in patients with acute or chronic HF [160]. Furthermore, a reduction in TBARS levels was observed in HF patients after treatment with a beta-blocker, short-term inotropic support and vitamin C, but not with the addition of an angiotensin II receptor antagonist to angiotensin converting enzyme inhibitor therapy [161, 162]. MDA appears to contribute to the formation of OxLDL [163] which have been proposed to be an useful predictor of mortality in patients with CHF [164]. MDA or MDA-modified LDL are being evaluated in device studies in advanced HF to monitor oxidative stress in patients under device therapy (implantable cardioverter defibrillator, continuous-flow left ventricular assist device) [165, 166].
Oxidative posttranslational modifications of cellular proteins by means of tyrosine nitration, protein carbonylation, and S-glutathionylation, can accurately reflect oxidative stress in HF patients. One of the most emblematic examples of protein oxidation in HF is myocardial sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) inactivation by nitration, which may contribute to reduced contractility and progression of HF [167]. Furthermore, ceruloplasmin tyrosine nitration with consequent antioxidant reduced activity is associated with reduced survival in patients with HF [168]. Protein nitration by peroxynitrite and haem peroxidase can result in gain of function or inactivation of different proteins in plasma, vessel wall and myocardium that link nitrosative stress to cardiovascular disease and, for that reason, nitrotyrosine is also emerging as a good candidate for a marker of cardiovascular risk [169].
Protein carbonyls can result from oxidation of amino acid side chains, reaction with lipid peroxidation products and glycation/glycoxidation of Lys amino groups. They are very stable and represent a good mirror of protein oxidation Increased carbonyls were found in diaphragm biopsies from patients with end-stage HF, probably resulting from increased Nox2-derived ROS and imbalanced antioxidant enzymes [170]. The inodilator levosimendan prevented the increase in MDA, protein carbonyls and nitrotyrosine in hospitalized patients with worsening HF. These results point to a cardioprotective effect of this drug and thus its wider use in advanced CHF patients has been hypothesized [171]. Additionally, there is evidence that a polymorphism in angiotensin II type 1 receptor can predict the formation of carbonyls in HF patients, suggesting that angiotensin signalling contributes to oxidative stress in HF [172].
8-hydroxy-2′-deoxyguanosine (8-OHdG) results from oxidative DNA damage and its levels can be quantified in urine. In fact, 8-OHdG was demonstrated to be higher with increasing HF severity and correlated with left ventricular ejection fraction in patients with chronic HF [173]. It also seems to be a tool to evaluate beta-blocker responsiveness in chronic HF patients or even to diagnose subclinical left ventricular diastolic dysfunction in hypertensive patients [174, 175], but more data is needed and/or combination with other biomarkers in a multipanel strategy.
3.3.3 Antioxidant Defences
Antioxidant enzymes (e.g. catalase, GPx, SOD) can be measured in blood samples but their values are hard to interpret and these studies have low reproducibility or therapeutic/prognostic implications [176,177,178] On the other hand, there has been an enthusiastic exploration of non-enzymatic antioxidants, such as biopyrrins (oxidative metabolites of bilirubin) and albumin since urinary levels of biopyrrins have been shown to be associated with HF severity [179] and oxidative stress has been proposed as a cause for the development of hypoalbuminemia in ischemic HF [180]. Nevertheless, the disappointing results of studies evaluating the effects of antioxidant administration in HF patients, particularly the failure of vitamins C and E to improve prognosis and the deleterious effects observed in HOPE and HOPE TOO trials, restrained the enthusiasm in this area [181,182,183].
3.3.4 Other Oxidative Stress Markers
Uric acid is an end-product of purine metabolism in humans derived from XO that catalyses its conversion from hypoxanthine. Although it is one of the most abundant aqueous antioxidants in plasma, it can also exert prooxidant effects [10]. Uric acid is frequently accepted as a biomarker of HF [141] but remains a controversial issue because causality in its relationship with cardiovascular disease remains uncertain. Although affected by renal function and diuretic use, there is enough evidence demonstrating that it can work as an independent and simple, albeit nonspecific, predictor of excessive oxidative stress and of adverse prognosis in HF [184].
4 Concluding Remarks
A vast body of literature accumulated over the past decades has firmly implicated oxidative stress in the pathogenesis and progression of cardiovascular diseases, including hypertension and HF, as well as associated risk factors and comorbidities. Key molecular events in hypertension and HF, such as oxidative modification of lipids and proteins, endothelial cell activation and inflammation, are facilitated by oxidative stress. More recently, the role of redox signalling and specific molecular targets have also been appreciated. Despite the significant progress in understanding the pathophysiology of these conditions and the promising results in pre-clinical animal models, clinical trials of antioxidant approaches to prevent cardiovascular mortality and morbidity have been, so far, disappointing. Several hypotheses have been put forward, including the failure to appreciate the complexity of the effects of ROS or inappropriate antioxidant selection or dosage, which warrants future research on new compounds with improved properties. Finally, more human data is required to provide clinical relevance and determine the potential for clinical translation. Nevertheless, several studies indicate that oxidative stress biomarkers may be useful for risk stratification and to monitor the protective effects of pharmacological treatment, diets or devices in human HF.
References
Dickinson BC, Chang CJ (2011) Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat Chem Biol 7(8):504–511
Droge W (2002) Free radicals in the physiological control of cell function. Physiol Rev 82(1):47–95
Schieber M, Chandel NS (2014) ROS function in redox signaling and oxidative stress. Curr Biol 24(10):R453–R462
Alfadda AA, Sallam RM (2012) Reactive oxygen species in health and disease. J Biomed Biotechnol 2012:936486
Valko M et al (2007) Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol 39(1):44–84
Brewer AC et al (2013) Reductive stress linked to small HSPs, G6PD, and Nrf2 pathways in heart disease. Antioxid Redox Signal 18(9):1114–1127
Martinez MC, Andriantsitohaina R (2009) Reactive nitrogen species: molecular mechanisms and potential significance in health and disease. Antioxid Redox Signal 11(3):669–702
Turko IV, Murad F (2002) Protein nitration in cardiovascular diseases. Pharmacol Rev 54(4):619–634
Costa S et al (2016) Statins and oxidative stress in chronic heart failure. Rev Port Cardiol 35(1):41–57
Sousa T et al (2012) Lipid peroxidation and antioxidants in arterial hypertension. In: Catala A (ed) Lipid peroxidation. IntechOpen, Rijeka, pp 345–392
Chen YR, Zweier JL (2014) Cardiac mitochondria and reactive oxygen species generation. Circ Res 114(3):524–537
Murphy MP (2009) How mitochondria produce reactive oxygen species. Biochem J 417(1):1–13
Addabbo F, Montagnani M, Goligorsky MS (2009) Mitochondria and reactive oxygen species. Hypertension 53(6):885–892
Chance B, Sies H, Boveris A (1979) Hydroperoxide metabolism in mammalian organs. Physiol Rev 59(3):527–605
Nazarewicz RR et al (2013) Nox2 as a potential target of mitochondrial superoxide and its role in endothelial oxidative stress. Am J Physiol Heart Circ Physiol 305(8):H1131–H1140
Zhang A et al (2011) Relative contributions of mitochondria and NADPH oxidase to deoxycorticosterone acetate-salt hypertension in mice. Kidney Int 80(1):51–60
Dey S et al (2018) Mitochondrial ROS drive sudden cardiac death and chronic proteome remodeling in heart failure. Circ Res 123(3):356–371
Moris D et al (2017) The role of reactive oxygen species in myocardial redox signaling and regulation. Ann Transl Med 5(16):324
Tsutsui H, Kinugawa S, Matsushima S (2011) Oxidative stress and heart failure. Am J Physiol Heart Circ Physiol 301(6):H2181–H2190
Bedard K, Krause KH (2007) The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87(1):245–313
Datla SR, Griendling KK (2010) Reactive oxygen species, NADPH oxidases, and hypertension. Hypertension 56(3):325–330
Nistala R, Whaley-Connell A, Sowers JR (2008) Redox control of renal function and hypertension. Antioxid Redox Signal 10(12):2047–2089
Brandes RP, Takac I, Schroder K (2011) No superoxide--no stress?: Nox4, the good NADPH oxidase! Arterioscler Thromb Vasc Biol 31(6):1255–1257
Brown DI, Griendling KK (2009) Nox proteins in signal transduction. Free Radic Biol Med 47(9):1239–1253
Sedeek M et al (2013) NADPH oxidases, reactive oxygen species, and the kidney: friend and foe. J Am Soc Nephrol 24(10):1512–1518
Montezano AC et al (2015) Redox signaling, Nox5 and vascular remodeling in hypertension. Curr Opin Nephrol Hypertens 24(5):425–433
Yu P et al (2014) Unique role of NADPH oxidase 5 in oxidative stress in human renal proximal tubule cells. Redox Biol 2:570–579
Manea A et al (2015) Human monocytes and macrophages express NADPH oxidase 5; a potential source of reactive oxygen species in atherosclerosis. Biochem Biophys Res Commun 461(1):172–179
Munzel T et al (2017) Impact of oxidative stress on the heart and vasculature: part 2 of a 3-part series. J Am Coll Cardiol 70(2):212–229
Zhang M et al (2013) NADPH oxidases in heart failure: poachers or gamekeepers? Antioxid Redox Signal 18(9):1024–1041
Matsushima S et al (2013) Broad suppression of NADPH oxidase activity exacerbates ischemia/reperfusion injury through inadvertent downregulation of hypoxia-inducible factor-1alpha and upregulation of peroxisome proliferator-activated receptor-alpha. Circ Res 112(8):1135–1149
Davies MJ (2011) Myeloperoxidase-derived oxidation: mechanisms of biological damage and its prevention. J Clin Biochem Nutr 48(1):8–19
Liu YC et al (2013) Genetic polymorphisms of myeloperoxidase and their effect on hypertension. Blood Press 22(5):282–289
Anatoliotakis N et al (2013) Myeloperoxidase: expressing inflammation and oxidative stress in cardiovascular disease. Curr Top Med Chem 13(2):115–138
Baldus S et al (2006) Heparins increase endothelial nitric oxide bioavailability by liberating vessel-immobilized myeloperoxidase. Circulation 113(15):1871–1878
Nussbaum C et al (2013) Myeloperoxidase: a leukocyte-derived protagonist of inflammation and cardiovascular disease. Antioxid Redox Signal 18(6):692–713
Tang WH et al (2007) Prognostic value and echocardiographic determinants of plasma myeloperoxidase levels in chronic heart failure. J Am Coll Cardiol 49(24):2364–2370
Forstermann U, Sessa WC (2012) Nitric oxide synthases: regulation and function. Eur Heart J 33(7):829–837
Gewaltig MT, Kojda G (2002) Vasoprotection by nitric oxide: mechanisms and therapeutic potential. Cardiovasc Res 55(2):250–260
Horita S et al (2014) Regulatory roles of nitric oxide and angiotensin II on renal tubular transport. World J Nephrol 3(4):295–301
Massion PB et al (2003) Nitric oxide and cardiac function: ten years after, and continuing. Circ Res 93(5):388–398
Briones AM, Touyz RM (2010) Oxidative stress and hypertension: current concepts. Curr Hypertens Rep 12(2):135–142
Li H et al (2006) Reversal of endothelial nitric oxide synthase uncoupling and up-regulation of endothelial nitric oxide synthase expression lowers blood pressure in hypertensive rats. J Am Coll Cardiol 47(12):2536–2544
Yamamoto E et al (2015) The pivotal role of eNOS uncoupling in vascular endothelial dysfunction in patients with heart failure with preserved ejection fraction. Int J Cardiol 190:335–337
Moens AL et al (2008) Reversal of cardiac hypertrophy and fibrosis from pressure overload by tetrahydrobiopterin: efficacy of recoupling nitric oxide synthase as a therapeutic strategy. Circulation 117(20):2626–2636
Berry CE, Hare JM (2004) Xanthine oxidoreductase and cardiovascular disease: molecular mechanisms and pathophysiological implications. J Physiol 555(Pt 3):589–606
Kuzkaya N et al (2005) Interactions of peroxynitrite with uric acid in the presence of ascorbate and thiols: implications for uncoupling endothelial nitric oxide synthase. Biochem Pharmacol 70(3):343–354
Hooper DC et al (1998) Uric acid, a natural scavenger of peroxynitrite, in experimental allergic encephalomyelitis and multiple sclerosis. Proc Natl Acad Sci U S A 95(2):675–680
Johnson RJ et al (2003) Is there a pathogenetic role for uric acid in hypertension and cardiovascular and renal disease? Hypertension 41(6):1183–1190
Niskanen LK et al (2004) Uric acid level as a risk factor for cardiovascular and all-cause mortality in middle-aged men: a prospective cohort study. Arch Intern Med 164(14):1546–1551
Loperena R, Harrison DG (2017) Oxidative stress and hypertensive diseases. Med Clin North Am 101(1):169–193
Feig DI, Soletsky B, Johnson RJ (2008) Effect of allopurinol on blood pressure of adolescents with newly diagnosed essential hypertension: a randomized trial. JAMA 300(8):924–932
MacIsaac RL et al (2016) Allopurinol and cardiovascular outcomes in adults with hypertension. Hypertension 67(3):535–540
Guzik TJ et al (2006) Coronary artery superoxide production and NOX isoform expression in human coronary artery disease. Arterioscler Thromb Vasc Biol 26(2):333–339
Baldus S et al (2006) Inhibition of xanthine oxidase improves myocardial contractility in patients with ischemic cardiomyopathy. Free Radic Biol Med 41(8):1282–1288
Givertz MM et al (2015) Effects of xanthine oxidase inhibition in hyperuricemic heart failure patients: the xanthine oxidase inhibition for hyperuricemic heart failure patients (EXACT-HF) study. Circulation 131(20):1763–1771
Hare JM et al (2008) Impact of oxypurinol in patients with symptomatic heart failure. Results of the OPT-CHF study. J Am Coll Cardiol 51(24):2301–2309
Casas AI et al (2015) Reactive oxygen-related diseases: therapeutic targets and emerging clinical indications. Antioxid Redox Signal 23(14):1171–1185
Deshwal S et al (2017) Emerging role of monoamine oxidase as a therapeutic target for cardiovascular disease. Curr Opin Pharmacol 33:64–69
Kaludercic N et al (2011) Monoamine oxidases (MAO) in the pathogenesis of heart failure and ischemia/reperfusion injury. Biochim Biophys Acta 1813(7):1323–1332
Fukai T, Ushio-Fukai M (2011) Superoxide dismutases: role in redox signaling, vascular function, and diseases. Antioxid Redox Signal 15(6):1583–1606
Lubos E, Loscalzo J, Handy DE (2011) Glutathione peroxidase-1 in health and disease: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal 15(7):1957–1997
Wassmann S, Wassmann K, Nickenig G (2004) Modulation of oxidant and antioxidant enzyme expression and function in vascular cells. Hypertension 44(4):381–386
Morgan MJ, Liu ZG (2011) Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res 21(1):103–115
Williams B et al (2018) ESC/ESH guidelines for the management of arterial hypertension. Eur Heart J 39(33):3021–3104
Coffman TM (2011) Under pressure: the search for the essential mechanisms of hypertension. Nat Med 17(11):1402–1409
Davisson RL, Zimmerman MC (2010) Angiotensin II, oxidant signaling, and hypertension: down to a T? Hypertension 55(2):228–230
Kobori H et al (2007) The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev 59(3):251–287
Reckelhoff JF, Romero JC (2003) Role of oxidative stress in angiotensin-induced hypertension. Am J Phys Regul Integr Comp Phys 284(4):R893–R912
Weir MR, Dzau VJ (1999) The renin-angiotensin-aldosterone system: a specific target for hypertension management. Am J Hypertens 12(12 Pt 3):205S–213S
Zimmerman MC et al (2004) Hypertension caused by angiotensin II infusion involves increased superoxide production in the central nervous system. Circ Res 95(2):210–216
Makino A et al (2003) Increased renal medullary H2O2 leads to hypertension. Hypertension 42(1):25–30
Sousa T et al (2012) Role of H(2)O(2) in hypertension, renin-angiotensin system activation and renal medullary disfunction caused by angiotensin II. Br J Pharmacol 166(8):2386–2401
Gomes P et al (2009) Aging increases oxidative stress and renal expression of oxidant and antioxidant enzymes that are associated with an increased trend in systolic blood pressure. Oxidative Med Cell Longev 2(3):138–145
Simao S et al (2011) Age-related changes in renal expression of oxidant and antioxidant enzymes and oxidative stress markers in male SHR and WKY rats. Exp Gerontol 46(6):468–474
Gomes P et al (2013) Loss of oxidative stress tolerance in hypertension is linked to reduced catalase activity and increased c-Jun NH2-terminal kinase activation. Free Radic Biol Med 56:112–122
Ulker S et al (2003) Impaired activities of antioxidant enzymes elicit endothelial dysfunction in spontaneous hypertensive rats despite enhanced vascular nitric oxide generation. Cardiovasc Res 59(2):488–500
Chan SH et al (2009) Oxidative impairment of mitochondrial electron transport chain complexes in rostral ventrolateral medulla contributes to neurogenic hypertension. Hypertension 53(2):217–227
Kristal B et al (1998) Participation of peripheral polymorphonuclear leukocytes in the oxidative stress and inflammation in patients with essential hypertension. Am J Hypertens 11(8 Pt 1):921–928
Lacy F et al (2000) Plasma hydrogen peroxide production in human essential hypertension: role of heredity, gender, and ethnicity. Hypertension 36(5):878–884
Zhou L et al (2006) Reduction in extracellular superoxide dismutase activity in African-American patients with hypertension. Free Radic Biol Med 41(9):1384–1391
Redon J et al (2003) Antioxidant activities and oxidative stress byproducts in human hypertension. Hypertension 41(5):1096–1101
Kedziora-Kornatowska K et al (2004) The markers of oxidative stress and activity of the antioxidant system in the blood of elderly patients with essential arterial hypertension. Cell Mol Biol Lett 9(4A):635–641
Wen Y et al (1996) Lipid peroxidation and antioxidant vitamins C and E in hypertensive patients. Ir J Med Sci 165(3):210–212
Pedro-Botet J et al (2000) Decreased endogenous antioxidant enzymatic status in essential hypertension. J Hum Hypertens 14(6):343–345
Ward NC, Croft KD (2006) Hypertension and oxidative stress. Clin Exp Pharmacol Physiol 33(9):872–876
Ceriello A (2008) Possible role of oxidative stress in the pathogenesis of hypertension. Diabetes Care 31(Suppl 2):S181–S184
Grossman E (2008) Does increased oxidative stress cause hypertension? Diabetes Care 31(Suppl 2):S185–S189
Lin HH et al (2003) Hydrogen peroxide increases the activity of rat sympathetic preganglionic neurons in vivo and in vitro. Neuroscience 121(3):641–647
Vaziri ND et al (2000) Induction of oxidative stress by glutathione depletion causes severe hypertension in normal rats. Hypertension 36(1):142–146
Dikalova A et al (2005) Nox1 overexpression potentiates angiotensin II-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation 112(17):2668–2676
Godin N et al (2010) Catalase overexpression prevents hypertension and tubular apoptosis in angiotensinogen transgenic mice. Kidney Int 77(12):1086–1097
Gavazzi G et al (2006) Decreased blood pressure in NOX1-deficient mice. FEBS Lett 580(2):497–504
Simao S et al (2011) H2 O2 stimulates Cl- /HCO 3- exchanger activity through oxidation of thiol groups in immortalized SHR renal proximal tubular epithelial cells. J Cell Biochem 112(12):3660–3665
Baumer AT et al (2007) The NAD(P)H oxidase inhibitor apocynin improves endothelial NO/superoxide balance and lowers effectively blood pressure in spontaneously hypertensive rats: comparison to calcium channel blockade. Clin Exp Hypertens 29(5):287–299
Beswick RA et al (2001) NADH/NADPH oxidase and enhanced superoxide production in the mineralocorticoid hypertensive rat. Hypertension 38(5):1107–1111
Sousa T et al (2008) Role of superoxide and hydrogen peroxide in hypertension induced by an antagonist of adenosine receptors. Eur J Pharmacol 588(2–3):267–276
Zhang Y et al (2004) The antioxidant tempol prevents and partially reverses dexamethasone-induced hypertension in the rat. Am J Hypertens 17(3):260–265
Nabha L et al (2005) Vascular oxidative stress precedes high blood pressure in spontaneously hypertensive rats. Clin Exp Hypertens 27(1):71–82
Wilcox CS (2005) Oxidative stress and nitric oxide deficiency in the kidney: a critical link to hypertension? Am J Phys Regul Integr Comp Phys 289(4):R913–R935
Heart Protection Study Collaborative, G (2002) MRC/BHF heart protection study of antioxidant vitamin supplementation in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet 360(9326):23–33
Sesso HD et al (2012) Multivitamins in the prevention of cardiovascular disease in men: the physicians’ health study II randomized controlled trial. JAMA 308(17):1751–1760
Vivekananthan DP et al (2003) Use of antioxidant vitamins for the prevention of cardiovascular disease: meta-analysis of randomised trials. Lancet 361(9374):2017–2023
Baykal Y et al (2003) Effects of antihypertensive agents, alpha receptor blockers, beta blockers, angiotensin-converting enzyme inhibitors, angiotensin receptor blockers and calcium channel blockers, on oxidative stress. J Hypertens 21(6):1207–1211
de Cavanagh EM et al (2010) Vascular structure and oxidative stress in salt-loaded spontaneously hypertensive rats: effects of losartan and atenolol. Am J Hypertens 23(12):1318–1325
Dauchet L et al (2006) Fruit and vegetable consumption and risk of coronary heart disease: a meta-analysis of cohort studies. J Nutr 136(10):2588–2593
Steinhubl SR (2008) Why have antioxidants failed in clinical trials? Am J Cardiol 101(10A):14D–19D
Brown DA et al (2017) Expert consensus document: mitochondrial function as a therapeutic target in heart failure. Nat Rev Cardiol 14(4):238–250
Giordano FJ (2005) Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest 115(3):500–508
Kalogeris T, Bao Y, Korthuis RJ (2014) Mitochondrial reactive oxygen species: a double edged sword in ischemia/reperfusion vs preconditioning. Redox Biol 2:702–714
Haworth RA, Potter KT, Russell DC (2010) Role of arachidonic acid, lipoxygenase, and mitochondrial depolarization in reperfusion arrhythmias. Am J Physiol Heart Circ Physiol 299(1):H165–H174
Santos CX et al (2011) Redox signaling in cardiac myocytes. Free Radic Biol Med 50(7):777–793
Buggisch M et al (2007) Stimulation of ES-cell-derived cardiomyogenesis and neonatal cardiac cell proliferation by reactive oxygen species and NADPH oxidase. J Cell Sci 120(Pt 5):885–894
Sauer H et al (2000) Role of reactive oxygen species and phosphatidylinositol 3-kinase in cardiomyocyte differentiation of embryonic stem cells. FEBS Lett 476(3):218–223
Saitoh S et al (2007) Redox-dependent coronary metabolic dilation. Am J Physiol Heart Circ Physiol 293(6):H3720–H3725
Saitoh S et al (2006) Hydrogen peroxide: a feed-forward dilator that couples myocardial metabolism to coronary blood flow. Arterioscler Thromb Vasc Biol 26(12):2614–2621
Andersson DC et al (2011) Mitochondrial production of reactive oxygen species contributes to the beta-adrenergic stimulation of mouse cardiomycytes. J Physiol 589(Pt 7):1791–1801
Loor G, Schumacker PT (2008) Role of hypoxia-inducible factor in cell survival during myocardial ischemia-reperfusion. Cell Death Differ 15(4):686–690
Zhang M et al (2010) NADPH oxidase-4 mediates protection against chronic load-induced stress in mouse hearts by enhancing angiogenesis. Proc Natl Acad Sci U S A 107(42):18121–18126
Ponikowski P et al (2016) ESC guidelines for the diagnosis and treatment of acute and chronic heart failure: the task force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail 18(8):891–975
Reina-Couto M et al (2016) Resolving inflammation in heart failure: novel protective lipid mediators. Curr Drug Targets 17(10):1206–1223
Braunwald E (2015) The war against heart failure: the lancet lecture. Lancet 385(9970):812–824
Guha K, McDonagh T (2013) Heart failure epidemiology: European perspective. Curr Cardiol Rev 9(2):123–127
Ramani GV, Uber PA, Mehra MR (2010) Chronic heart failure: contemporary diagnosis and management. Mayo Clin Proc 85(2):180–195
Spodick DH (2003) Acute cardiac tamponade. N Engl J Med 349(7):684–690
von Haehling S et al (2010) Elevated levels of asymmetric dimethylarginine in chronic heart failure: a pathophysiologic link between oxygen radical load and impaired vasodilator capacity and the therapeutic effect of allopurinol. Clin Pharmacol Ther 88(4):506–512
Kanaan GN, Harper ME (2017) Cellular redox dysfunction in the development of cardiovascular diseases. Biochim Biophys Acta Gen Subj 1861(11 Pt A):2822–2829
Heymans S et al (2009) Inflammation as a therapeutic target in heart failure? A scientific statement from the translational research Committee of the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail 11(2):119–129
Hofmann U, Frantz S (2013) How can we cure a heart “in flame”? A translational view on inflammation in heart failure. Basic Res Cardiol 108(4):356
Mueller C et al (2006) Inflammation and long-term mortality in acute congestive heart failure. Am Heart J 151(4):845–850
Khaper N et al (2010) Targeting the vicious inflammation-oxidative stress cycle for the management of heart failure. Antioxid Redox Signal 13(7):1033–1049
Chen X et al (2008) Role of reactive oxygen species in tumor necrosis factor-alpha induced endothelial dysfunction. Curr Hypertens Rev 4(4):245–255
Zhang C et al (2003) Interaction of myeloperoxidase with vascular NAD(P)H oxidase-derived reactive oxygen species in vasculature: implications for vascular diseases. Am J Physiol Heart Circ Physiol 285(6):H2563–H2572
Reina-Couto M et al (2014) Impaired resolution of inflammation in human chronic heart failure. Eur J Clin Investig 44(6):527–538
Reina-Couto M et al (2018) Endocan as a new biomarker of severity in acute heart failure. Eur J Heart Fail 20:P459
Reina-Couto M et al (2018) Inflammation resolution mediators in acute heart failure. J Hypertens 36(e-Supplement 1):e211
Munzel T et al (2015) Pathophysiological role of oxidative stress in systolic and diastolic heart failure and its therapeutic implications. Eur Heart J 36(38):2555–2564
van Riet EE et al (2014) Prevalence of unrecognized heart failure in older persons with shortness of breath on exertion. Eur J Heart Fail 16(7):772–777
Karimi Galougahi K et al (2015) Redox biomarkers in cardiovascular medicine. Eur Heart J 36(25):1576–1582. 1582a-b
Patel RS et al (2016) Novel biomarker of oxidative stress is associated with risk of death in patients with coronary artery disease. Circulation 133(4):361–369
Braunwald E (2008) Biomarkers in heart failure. N Engl J Med 358(20):2148–2159
Tang WH et al (2006) Plasma myeloperoxidase levels in patients with chronic heart failure. Am J Cardiol 98(6):796–799
Adam M et al (2015) Levosimendan displays anti-inflammatory effects and decreases MPO bioavailability in patients with severe heart failure. Sci Rep 5:9704
O’Donoghue ML et al (2016) Multimarker risk stratification in patients with acute myocardial infarction. J Am Heart Assoc 5(5):e002586
Virzi GM et al (2018) Levels of proinflammatory cytokines, oxidative stress, and tissue damage markers in patients with acute heart failure with and without cardiorenal syndrome type 1. Cardiorenal Med 8(4):321–331
Kataoka Y et al (2014) Myeloperoxidase levels predict accelerated progression of coronary atherosclerosis in diabetic patients: insights from intravascular ultrasound. Atherosclerosis 232(2):377–383
Meuwese MC et al (2007) Serum myeloperoxidase levels are associated with the future risk of coronary artery disease in apparently healthy individuals: the EPIC-norfolk prospective population study. J Am Coll Cardiol 50(2):159–165
Pastori D et al (2015) Does mediterranean diet reduce cardiovascular events and oxidative stress in atrial fibrillation? Antioxid Redox Signal 23(8):682–687
Cunnington C et al (2012) Systemic and vascular oxidation limits the efficacy of oral tetrahydrobiopterin treatment in patients with coronary artery disease. Circulation 125(11):1356–1366
Shirodaria C et al (2007) Global improvement of vascular function and redox state with low-dose folic acid: implications for folate therapy in patients with coronary artery disease. Circulation 115(17):2262–2270
Santos CN et al (2018) Pure polyphenols applications for cardiac health and disease. Curr Pharm Des 24(19):2137–2156
Driver C et al (2018) Cardioprotective effects of metformin. J Cardiovasc Pharmacol 72(2):121–127
Alemayehu M et al (2017) Effect of ticagrelor versus clopidogrel on vascular reactivity. J Am Coll Cardiol 69(17):2246–2248
Nanhwan MK et al (2014) Chronic treatment with ticagrelor limits myocardial infarct size: an adenosine and cyclooxygenase-2-dependent effect. Arterioscler Thromb Vasc Biol 34(9):2078–2085
Montuschi P, Barnes PJ, Roberts LJ 2nd (2004) Isoprostanes: markers and mediators of oxidative stress. FASEB J 18(15):1791–1800
Mallat Z et al (1998) Elevated levels of 8-iso-prostaglandin F2alpha in pericardial fluid of patients with heart failure: a potential role for in vivo oxidant stress in ventricular dilatation and progression to heart failure. Circulation 97(16):1536–1539
Polidori MC et al (2004) Increased F2 isoprostane plasma levels in patients with congestive heart failure are correlated with antioxidant status and disease severity. J Card Fail 10(4):334–338
Davies SS, Roberts LJ 2nd (2011) F2-isoprostanes as an indicator and risk factor for coronary heart disease. Free Radic Biol Med 50(5):559–566
Hummel SL et al (2012) Low-sodium dietary approaches to stop hypertension diet reduces blood pressure, arterial stiffness, and oxidative stress in hypertensive heart failure with preserved ejection fraction. Hypertension 60(5):1200–1206
de Meirelles LR et al (2011) Platelet activation, oxidative stress and overexpression of inducible nitric oxide synthase in moderate heart failure. Clin Exp Pharmacol Physiol 38(10):705–710
Ellis GR et al (2002) Addition of candesartan to angiotensin converting enzyme inhibitor therapy in patients with chronic heart failure does not reduce levels of oxidative stress. Eur J Heart Fail 4(2):193–199
White M et al (2006) Increased systemic inflammation and oxidative stress in patients with worsening congestive heart failure: improvement after short-term inotropic support. Clin Sci (Lond) 110(4):483–489
Amaki T et al (2004) Circulating malondialdehyde modified LDL is a biochemical risk marker for coronary artery disease. Heart 90(10):1211–1213
Charach G et al (2015) Usefulness of antibodies to oxidized low-density lipoproteins as predictors of morbidity and prognosis in heart failure patients aged >/=65 years. Am J Cardiol 116(9):1379–1384
Kato M et al (2017) Stretching exercises improve vascular endothelial dysfunction through attenuation of oxidative stress in chronic heart failure patients with an implantable cardioverter defibrillator. J Cardiopulm Rehabil Prev 37(2):130–138
Mondal NK et al (2016) Systemic inflammatory response syndrome in end-stage heart failure patients following continuous-flow left ventricular assist device implantation: differences in plasma redox status and leukocyte activation. Artif Organs 40(5):434–443
Lokuta AJ et al (2005) Increased nitration of sarcoplasmic reticulum Ca2+-ATPase in human heart failure. Circulation 111(8):988–995
Cabassi A et al (2014) Low serum ferroxidase I activity is associated with mortality in heart failure and related to both peroxynitrite-induced cysteine oxidation and tyrosine nitration of ceruloplasmin. Circ Res 114(11):1723–1732
Peluffo G, Radi R (2007) Biochemistry of protein tyrosine nitration in cardiovascular pathology. Cardiovasc Res 75(2):291–302
Ahn B et al (2016) Diaphragm abnormalities in patients with end-stage heart failure: NADPH oxidase upregulation and protein oxidation. Front Physiol 7:686
Parissis JT et al (2007) Effects of Levosimendan on circulating markers of oxidative and nitrosative stress in patients with advanced heart failure. Atherosclerosis 195(2):e210–e215
Cameron VA et al (2006) Angiotensin type-1 receptor A1166C gene polymorphism correlates with oxidative stress levels in human heart failure. Hypertension 47(6):1155–1161
Kobayashi S et al (2011) Urinary 8-hydroxy-2′-deoxyguanosine reflects symptomatic status and severity of systolic dysfunction in patients with chronic heart failure. Eur J Heart Fail 13(1):29–36
Masugata H et al (2013) Association between oxidative stress assessed by urinary 8-hydroxydeoxyguanosine and the cardiac function in hypertensive patients without overt heart disease. Clin Exp Hypertens 35(5):308–312
Susa T et al (2012) Urinary 8-hydroxy-2′-deoxyguanosine as a novel biomarker for predicting cardiac events and evaluating the effectiveness of carvedilol treatment in patients with chronic systolic heart failure. Circ J 76(1):117–126
Blankenberg S et al (2003) Glutathione peroxidase 1 activity and cardiovascular events in patients with coronary artery disease. N Engl J Med 349(17):1605–1613
Caruso R et al (2007) Pre-operative redox state affects 1-month survival in patients with advanced heart failure undergoing left ventricular assist device implantation. J Heart Lung Transplant 26(11):1177–1181
Sam F et al (2005) Increased reactive oxygen species production and functional alterations in antioxidant enzymes in human failing myocardium. J Card Fail 11(6):473–480
Hokamaki J et al (2004) Urinary biopyrrins levels are elevated in relation to severity of heart failure. J Am Coll Cardiol 43(10):1880–1885
Ellidag HY et al (2014) Oxidative stress and ischemia-modified albumin in chronic ischemic heart failure. Redox Rep 19(3):118–123
Lonn E et al (2005) Effects of long-term vitamin E supplementation on cardiovascular events and cancer: a randomized controlled trial. JAMA 293(11):1338–1347
Thomson MJ, Frenneaux MP, Kaski JC (2009) Antioxidant treatment for heart failure: friend or foe? QJM 102(5):305–310
Yamauchi Y et al (2017) Is serum uric acid independently associated with left ventricular mass index, ejection fraction, and B-type natriuretic peptide among female and male cardiac patients? Int Heart J 58(4):562–569
Wannamethee SG et al (2018) Serum uric acid as a potential marker for heart failure risk in men on antihypertensive treatment: the British regional heart study. Int J Cardiol 252:187–192
Acknowledgements
We apologize to all colleagues whose work has not been discussed or cited owing to space limitations. TS is currently supported by FEDER funds via COMPETE (Portugal 2020) and by national funds through the Portuguese Foundation for Science and Technology (FCT) (project grant PTDC/MEC-CAR/32188/2017; SFRH/BPD/112005/2015). PG is funded by FEDER, Centro2020 Regional Operational Programme: CENTRO-01-0145-FEDER-000012-HealthyAging2020, COMPETE 2020-Operational Programme for Competitiveness & Internationalization; and FCT (strategic project UID/NEU/04539/2013; SFRH/BPD/111815/2015).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Sousa, T., Reina-Couto, M., Gomes, P. (2019). Role of Oxidative Stress in the Pathophysiology of Arterial Hypertension and Heart Failure. In: Chakraborti, S., Dhalla, N., Ganguly, N., Dikshit, M. (eds) Oxidative Stress in Heart Diseases. Springer, Singapore. https://doi.org/10.1007/978-981-13-8273-4_23
Download citation
DOI: https://doi.org/10.1007/978-981-13-8273-4_23
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-8272-7
Online ISBN: 978-981-13-8273-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)