
Abstract
ATP8A2-related disorders are autosomal recessive conditions that associate encephalopathy with or without hypotonia, psychomotor delay, abnormal movements, chorea, tremor, optic atrophy and cerebellar atrophy (CARMQ4). Through a multi-centric collaboration, we identified six point mutations (one splice site and five missense mutations) involving ATP8A2 in six individuals from five families. Two patients from one family with the homozygous p.Gly585Val mutation had a milder presentation without encephalopathy. Expression and functional studies of the missense mutations demonstrated that protein levels of four of the five missense variants were very low and lacked phosphatidylserine-activated ATPase activity. One variant p.Ile215Leu, however, expressed at normal levels and displayed phospholipid-activated ATPase activity similar to the non-mutated protein. We therefore expand for the first time the phenotype related to ATP8A2 mutations to less severe forms characterized by cerebellar ataxia without encephalopathy and suggest that ATP8A2 should be analyzed for all cases of syndromic or non-syndromic recessive or sporadic ataxia.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The cerebellar ataxia, mental retardation, and disequilibrium syndrome (CAMRQ) is a heterogeneous group of genetic disorders of autosomal recessive inheritance [1]. Several genes responsible for the condition have been identified to date, namely VLDLR (MIM: 224050) [2], WDR81 (MIM: 610185) [3] and CA8 (MIM: 613227) [4] causing, respectively, CAMRQ1, CAMRQ2, and CAMRQ3.
More recently, disease-causing mutations in ATP8A2 have been identified in CAMRQ4 patients. The initial CAMRQ4 report identified a single ATP8A2 missense mutation segregating in four patients of a large, multigenerational consanguineous family [5]. Since then, a total of 26 patients with CAMRQ4 have been described, confirming the involvement of the ATP8A2 in severe early-onset hypotonia with psychomotor delay, abnormal movements, tremor, mental retardation, optic and cerebellar atrophy [6,7,8].
We report five additional patients from four families and we describe for the first time two patients presenting with a novel ATP8A2 phenotype characterized by mild cerebellar ataxia.
Subjects and methods
Genetic studies and ethics statement
Human genetic studies conducted in research laboratories were approved by local ethics committees from participating centers (Montpellier, France; Baltimore, USA; Padova-Bergamo, Italy; Ankara, Turkey). Written informed consent was obtained from all study participants. All five affected individuals underwent extensive clinical examination by at least one expert in the ataxia field. Either whole-exome (individuals 1-A, 1-B, 3 and 4), mini-exome (individual 2) or neuromuscular gene panel (individual 5) sequencing and data analysis were performed according to previously published protocols [5, 7, 9,10,11].
Generation of human ATP8A2 mutant constructs
Human ATP8A2 constructs (NCBI NM_016529.6) containing encoding a C-terminal 9 amino acid 1D4 tag in pcDNA 3.1 have been described previously [12]. Disease-associated missense mutations were generated using the Q5 site-directed mutagenesis kit (NEB, #E0552S—New England Biolabs, Whitby, ON) with primers specific to each point mutation (Supplementary Table S2). The mutant plasmids were verified by Sanger sequencing of the entire coding and promoter region.
Expression of ATP8A2 constructs
HEK293T cells (American type culture collection, Manassas, VA) were transfected in 10 cm plates at 80% confluency with 5 µg of human ATP8A2-1D4 and 5 µg of CDC50A plasmids using 30 µg of the transfection agent polyethylenimine (PEI). Cells were harvested 24 h post-transfection and lysed in 4% SDS with stirring. Samples were centrifuged at 40,000 rpm for 10 min and supernatant was quantified for total protein. Protein expression was analyzed on western blots labeled for ATP8A2 and tubulin as a loading control. Briefly, SDS-PAGE gels were transferred onto immobilon FL membranes (millipore) and blocked for 30 min in 1% milk/PBS. ATP8A2 expression was determined using an in-house Rho-1D4 antibody (1/500 dilution, 1 h labeling) and goat anti-mouse Ig secondary antibody coupled to IR dye 680 (1/40,000, 40 min labeling). Anti-β-tubulin antibody (Abcam, ab15568) was used to detect β-tubulin together with donkey anti-rabbit Ig secondary antibody coupled to IR dye 800. Membranes were washed in between antibody incubations with PBS containing 0.5% Tween 20. Imaging of blots was carried out on the LI-COR Odyssey infrared imaging system. Band intensities of both Rho1D4 and β-tubulin labeling were quantified and the ratio of 1D4/β-tubulin was calculated and plotted for each ATP8A2 variant. All experiments were done at least three times.
The ATP8A2-CDC50A variants were purified on a Rho 1D4-Sepharose immunoaffinity matrix as described previously [13]. For more details, see the supplementary method in Appendix A.
ATPase activity assay
1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC) and 1,2-dioleoyl-sn-glycero-3-phosphoserine (DOPS) (Avanti Polar Lipids, Alabaster, AL) were dried at a concentration of 50 mg/ml under N2 gas and resuspended in ATPase Assay buffer (50 mM HEPES–NaOH pH 7.5, 150 mM NaCl, 12.5 mM MgCl2, 1 mM DTT, and 10 mM CHAPS) to make a final concentration of 5 mg/ml. Each protein eluate was diluted in ATPase Assay buffer at 0.4 ng/µl. Resuspended lipids contained either 100% DOPC or 84% DOPC and 16% DOPS. Each reaction contained 12.5 µl of 5 mg/ml lipids, 10 µl of 1.25 mM ATP in assay buffer, and 2.5 µl of diluted protein. Controls contained 25 µl of 12% SDS and each sample was done in triplicate. All samples were incubated at 37 C for 30 min to stimulate the ATPase reaction. Twenty-five microliter of 12% SDS was added to each sample (except controls) to terminate the reaction. The amount of hydrolyzed phosphate was measured using a colorimetric assay previously described [14]. Absorbance measurements were compared to those of known phosphate standards carried out in parallel. The specific activity (µmol Pi released per min per mg protein) was calculated. Data were analyzed for n = 2 for 100% PC and n = 3 for 84%PC/16%PS with error bars (SD) or as indicated.
Results
Identification of point mutation variants disrupting ATP8A2
Through a multi-centric collaboration, we identified six point mutations (one splice site and five missense mutations) involving ATP8A2 in six individuals from five families.
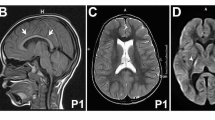
Because of parental consanguinity, we investigated all homozygous variants found by whole exome (individuals 1-A, 1-B, 3), mini-exome (individual 2) or neuromuscular panel (individual 5) sequencing analysis. We identified three homozygous non-conservative missense changes: c.1754G > T, p.Gly585Val (G585V) in individuals 1-A and 1-B, c.1762C > T, p.Arg588Trp (R588W) in individual 2 and c.1312A > G, p.Met438Val (M438V) in individual 5, all located in the catalytic cytoplasmic domain [amino acids 364–877] of ATP8A2 (NM_016529.4) and predicted to be pathogenic (Fig. 1b, c).

a On the left: T1-weighted sagittal brain MRI of individual 1A at age 4 years, showing mild atrophy of the anterior superior cerebellar vermis. On the right: T2-weighted MRI brain showing early bilateral frontal atrophy at age 2 years. Full MRI sequence analysis revealed no sulcation abnormality for individual 5. b Schematic representation of P4-ATPase ATP8A2 protein with ten transmembrane helixes (1–10), A (actuator), P (phosphorylation), N (nucleotide binding) domains, adapted from Vestergaard et al. [18], and showing localizations of the mutations previously reported (italics) and described in this study (bold and underlined). Compound heterozygous mutations are indicated by dotted boxes. Compound heterozygous A544P and R625W were found in a patient described by Martin-Hernandez [6]. c Amino-acid sequence comparison of orthologous P4-ATPase ATP8A2. Amino acid changes described in our study (bold and underlined) are predicted to be pathogenic (isoleucine 215, methionine 438, glycine 585 and arginine 588 are highly conserved up to invertebrates and fungi)
In individual 3, WES revealed a homozygous point mutation, c.1057 + 5G > C, affecting the splice donor site of intron 11. Human splicing finder 3.1 (https://www.umd.be/HSF/index.html) predicts a reduction of the splice site score from 92 (wild type) to 65 (mutant), most probably affecting splicing.
In individual 4 who was born to unrelated parents from Italian origin, WES revealed two segregating variants predicted to be pathogenic: c.643A > T, p.Ile215Leu (I215L) located in the actuator domain of ATP8A2 and c.1916A > G, p.Tyr639Cys (Y639C) located in the phosphorylation domain (Fig. 1b, c, Table 1).
Clinical features of individuals with ATP8A2 variants
The five affected individuals in our cohort display developmental delay of differing severity ranging from delayed walking with ataxia to severe encephalopathy with no ambulation and severe intellectual disability (Table 1).
Two siblings (individual 1-A and 1-B) born from first-degree consanguineous parents of Turkish origin were affected by 2 years of age with head titubation, ataxic gait and tremor. Both siblings have borderline intellectual functioning with IQ ranging from 70 to 80. Cerebral magnetic resonance imaging (MRI) revealed very mild vermian atrophy in the brother at 4 years (Fig. 1a). Both patients were still ambulant, with unilateral aid, by ages ranging from 8 to 11 years.
In the second family from Algeria, a girl (individual 2) was affected by a transient encephalopathy with brutal post measles coma at around 15 months of age. She experienced delayed walking, disequilibrium, severe hypotonia, dysmetria, multidirectional nystagmus and dysarthria. During a short stay in France, this child progressed very significantly: she became able to stand alone with support and to do three consecutive walking steps with aid.
Individual 3 is an 8 year old female with Caucasian origin who was adopted at 4 years of age. She has encephalopathy, developmental delay, hypotonia, muscle weakness, seizures, chorea, dystonia, mild/moderate intellectual disability, microcephaly, optic atrophy and no ambulation. She is able to say 20–30 words with dysarthria and is G-tube-dependent. She had several EEGs around the ages of 5–6 which revealed nonspecific background slowing, and subsequently right occipital spike wave discharges and occasional right central spikes.
Individual 4 is a 28 year old female with Italian origin who is wheelchair-bound. She presents with a similar phenotype to that of individual 3 but with severe intellectual disability, anarthria, strabismus and without optic atrophy. EEGs were normal up to the age of 17 years, and then revealed focal paroxysms and slow wave activity. MRI at the age of 5 years and 11 years showed microcephaly, oligogyria with few shallow sulci, bilateral moderate thinning of white matter, mild thinning of the corpus callosum and normal cerebellum.
Individual 5 is a 2 year old boy from Turkey who presents with developmental delay, intellectual disability, severe hypotonia, muscle weakness, chorea, dystonia, facial dyskinesia, strabismus, severe ptosis, ophthalmoplegia, hearing impairment and bilateral frontal atrophy on brain MRI. At 24 months, he experienced generalized febrile seizures. Two routine EEGs obtained at different timepoints showed no epileptiform abnormality. He also has feeding difficulties.
Expression and functional analysis
To determine the effect of the missense mutations on ATP8A2, HEK293T cells were co-transfected with plasmids containing the ATP8A2 variant and CDC50A (also known as TMEM30A). The expression of the ATP8A2 variants relative to the non-mutated protein was examined on Western blots (Fig. 2). The I215L variant expressed at levels comparable to control ATP8A2, whereas the four other variants expressed at levels less than 15% of control ATP8A2.

Effect of disease-causing mutations on ATP8A2 protein expression. HEK293T cells co-expressing ATP8A2 variants and CDC50A were solubilized in SDS and analyzed on western blots labeled for the ATP8A2 variants with the Rho 1D4 antibody. Left: example of a western blot of SDS-solubilized non-mutated ATP8A2, and the I215L, M438V, G585V, R588W, and Y639C variants and anti-tubulin as a loading control. Right: quantification of expression levels normalized to non-mutated ATP8A2. Values are the mean ± SD for n = 3 independent experiments
The ATPase activity of immunoaffinity purified ATP8A2 and the I215L variant was measured in the presence of 100% phosphatidylcholine and 84% phosphatidylcholine-16% phosphatidylserine. As shown in Fig. 3, the ATPase activity of the I215L variant, like control ATP8A2, was strongly activated by increasing concentrations of phosphatidylserine. The M438V, G585V, R588W, and Y639C variants expressed at very low levels making ATPase activity measurements difficult. However, by increasing the number of transfected cells, we were able to obtain sufficient protein to assess the ATPase activity of the G585V and M438V variants. As shown in Fig. 3, neither variant displayed significant phosphatidylserine-activated ATPase.

The ATPase activity of ATP8A2 disease-associated variants. a Effect of increasing phosphatidylserine concentration on the ATPase activity of non-mutated ATP8A2 and the I215L variant. b ATPase activity of ATP8A2 variants in the presence of 100% phosphatidylcholine (PC) and 84% phosphatidylcholine-16% phosphatidylserine (PS). Data were normalized to the phosphatidylserine-activated ATPase activity of non-mutated ATP8A2. n = 3 for ATP8A2 and the I215L variant. n = 1 for M438V and G585V variants
Discussion
Through a multi-centric collaboration, we identified five patients from four unrelated families who presented in childhood with neurological deficits distinguished by ataxia and/or developmental delay of differing severities that were caused by mutations in ATP8A2.
To date, 26 patients from thirteen families have been described in the literature: six families have homozygous or compound heterozygous truncating mutations, one sporadic case has a presumed dominant de novo balanced translocation of chromosomes 10 and 13 disrupting the ATP8A2 coding sequence, while the six remaining families have homozygous or compound heterozygous missense mutations, almost all located in the catalytic cytoplasmic domain and adjacent transmembrane segment VI [amino acids 364–877] of the phospholipid-transporting ATP8A2 (6/7 mutations).
Mutations in genes coding for flippases are globally rare and are often responsible for severe early onset encephalopathy. In the initial report of CAMRQ4, p.Ile376Met homozygous mutation was predicted to change the secondary structure of the ATP8A2 protein. The patients with p.Ile376Met presented with encephalopathy, developmental delay, hypotonia, quadrupedal gait, truncal ataxia and dysarthric speech.
Since then, 22 additional cases have been described and presented with an even more severe phenotype with absence of ambulation, non-verbal or absent language and feeding difficulties. Among them, nine individuals also experienced optic atrophy [6,7,8, 15] (see Supplementary clinical data Table S1).
In this report we expand the phenotype of ATP8A2 mutations describing for the first time two patients with a less severe form characterized by cerebellar ataxia without encephalopathy (individuals 1-A and 1-B).
Remarkably, individual 2 who presented at birth with encephalopathy, clearly improved with physiotherapy and had a relatively mild presentation at 10 years. As individuals 1-A and 1-B, she was able to walk with unilateral aid and could speak with dysarthria.
In our study, the pathogenicity of the missense mutations was evaluated by analysis of the ATP8A2 variants expressed in culture cells. Four variants (M438V, G585V, R588W, and Y639C) expressed at exceedingly low levels compared to control ATP8A2. The low expression of these variants harboring missense mutations in the catalytic domains is likely caused by significant misfolding of the protein together with proteasomal degradation. Interestingly, the I215L variant harboring a relatively conserved isoleucine to leucine substitution displayed a level of expression and phosphatidylserine (PS)-activated ATPase activity comparable to non-mutated control ATP8A2. It is unclear why this variant is associated with the severe disease phenotype in patient 4 also harboring the severe p.Y639C mutation. It is possible that there is an additional mutation in the introns or promoter regions, or a gene rearrangement (which could not be ruled out from exome analysis) of the allele encoding the I215L variant or alternatively this mutation affects a property of ATP8A2 not reproduced in the heterologous expression system used here. Likewise, it is unclear why patients 1 and 2 homozygous for the G585V mutation display a mild disease phenotype despite the finding that this variant expresses at low levels and is devoid of ATPase activity. It is possible that other genetic modifiers or P4-ATPases may compensate for the loss in expression/activity of this variant.
In the central nervous system, apoptosis plays an important role during development and is a primary pathogenic mechanism in several adult neurodegenerative diseases. Among apoptotic signaling pathways, the PS pathway appears to have a crucial and unique role [16]. P4-ATPase ATP8A2 is a 1188-amino-acid protein involved in the maintenance of transbilayer lipid asymmetry by actively transporting specific phospholipids such as PS across cell membranes [17]. ATP8A2-encoded flippase is strongly expressed in the brain, cerebellum, retina and testis [5, 15]. ATP8A2 partial loss of function contributes to PS exposure and possible initiation of the early phase of apoptosis. On the surface of cells, PS is recognized by macrophages through PtdSerR, a phosphatidylserine receptor used for specific induction of phagocytosis. The lack of genotype/phenotype correlation in ATP8A2-related disorders suggests that variability of macrophage activation may also be an important contributor to clinical severity.
On the basis of amino acid sequence alignment, the P4-ATPase ATP8A2 is predicted to possess a transmembrane domain with 10 helices and three cytoplasmic domains: P (phosphorylation) that contains the phosphorylated canonical aspartic acid residue, N (nucleotide binding) that contains the ATP-binding pocket, and A (actuator) that serves to dephosphorylate the phosphorylated P domain as part of the reaction cycle of the P4-ATPase (Fig. 1b) [18]. P and N belong to the haloacid dehalogenase domain shared by a superfamily of enzymes that include phosphatases, phosphonatases, P-type ATPases, beta-phosphoglucomutases, phosphomannomutases, and dehalogenases. Interestingly, both missense mutations associated with the mildest phenotypes (G585V, individuals 1-A, 1-B and R588W, individual 2) are located in the N domain (Fig. 1b). The three other missense mutations identified in individuals with the classic severe phenotype (individual 4 and 5) were located in the A (I215L) and P (M438V, Y639C) domains.
Mild cerebellar ataxia without encephalopathy has never been reported in ATP8A2 disorders. The present report underscores the strikingly variable clinical presentations resulting from ATP8A2 mutations, ranging from early-onset severe epileptic encephalopathy with cerebello-ocular syndrome to isolated ataxia. Since the detection of these milder and new phenotypes is now possible by next generation sequencing techniques (NGS), ATP8A2 should be included in NGS screening panels for the diagnosis of syndromic and non-syndromic inherited ataxias.
Several classifications of inherited ataxias have been proposed. Only the latest classifications, resulting from consensus among panels of international experts, attempt to grasp the complexity and phenotypic and genotypic heterogeneity of ataxias that result from the explosion of gene identification. In these classifications, ATP8A2-related disorders are classified in the group of “other metabolic or complex autosomal recessive disorders that have ataxia as an associated feature [19] or that have only occasional ataxia presentation [20]. Our report of novel patients is in agreement with this classification since, for most ATP8A2 patients, ataxia remains an associated feature.
The huge functional diversity of affected proteins in autosomal recessive ataxia impedes their exhaustive classification according to physiopathological mechanisms. On the contrary, current knowledge about autosomal recessive ataxias indicates that no particular pathophysiological pathway explains the occurrence of this symptom, which results rather from an extreme sensitivity of cerebellar, spinocerebellar and sensory neurons to mild metabolic disturbances [21] often by partial loss of function [22,23,24,25].
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Tan U (2006) A new syndrome with quadrupedal gait, primitive speech, and severe mental retardation as a live model for human evolution. Int J Neurosci 116:361–369. https://doi.org/10.1080/00207450500455330
Schlotawa L, Hotz A, Zeschnigk C et al (2013) Cerebellar ataxia, mental retardation and dysequilibrium syndrome 1 (CAMRQ1) caused by an unusual constellation of VLDLR mutation. J Neurol 260:1678–1680. https://doi.org/10.1007/s00415-013-6941-z
Doldur-Balli F, Ozel MN, Gulsuner S et al (2015) Characterization of a novel zebrafish (Danio rerio) gene, wdr81, associated with cerebellar ataxia, mental retardation and dysequilibrium syndrome (CAMRQ). BMC Neurosci 16:96. https://doi.org/10.1186/s12868-015-0229-4
Türkmen S, Guo G, Garshasbi M et al (2009) CA8 mutations cause a novel syndrome characterized by ataxia and mild mental retardation with predisposition to quadrupedal gait. PLoS Genet 5:e1000487. https://doi.org/10.1371/journal.pgen.1000487
Onat OE, Gulsuner S, Bilguvar K et al (2013) Missense mutation in the ATPase, aminophospholipid transporter protein ATP8A2 is associated with cerebellar atrophy and quadrupedal locomotion. Eur J Hum Genet 21:281–285. https://doi.org/10.1038/ejhg.2012.170
Martín-Hernández E, Rodríguez-García ME, Camacho A et al (2016) New ATP8A2 gene mutations associated with a novel syndrome: encephalopathy, intellectual disability, severe hypotonia, chorea and optic atrophy. Neurogenetics 17:259–263. https://doi.org/10.1007/s10048-016-0496-y
McMillan HJ, Telegrafi A, Singleton A et al (2018) Recessive mutations in ATP8A2 cause severe hypotonia, cognitive impairment, hyperkinetic movement disorders and progressive optic atrophy. Orphanet J Rare Dis 13:86. https://doi.org/10.1186/s13023-018-0825-3
Alsahli S, Alrifai MT, Al Tala S et al (2018) Further delineation of the clinical phenotype of cerebellar ataxia, mental retardation, and disequilibrium syndrome type 4. J Cent Nerv Syst Dis 10:1179573518759682. https://doi.org/10.1177/1179573518759682
Guissart C, Li X, Leheup B et al (2015) Mutation of SLC9A1, encoding the major Na+/H+ exchanger, causes ataxia-deafness Lichtenstein–Knorr syndrome. Hum Mol Genet 24:463–470. https://doi.org/10.1093/hmg/ddu461
Marelli C, Guissart C, Hubsch C et al (2016) Mini-exome coupled to read-depth based copy number variation analysis in patients with inherited ataxias. Hum Mutat 37:1340–1353. https://doi.org/10.1002/humu.23063
Platzer K, Sticht H, Edwards SL et al (2019) De novo variants in MAPK8IP3 cause intellectual disability with variable brain anomalies. Am J Hum Genet 104:203–212. https://doi.org/10.1016/j.ajhg.2018.12.008
Lee S, Uchida Y, Wang J et al (2015) Transport through recycling endosomes requires EHD1 recruitment by a phosphatidylserine translocase. EMBO J 34:669–688. https://doi.org/10.15252/embj.201489703
Coleman JA, Molday RS (2011) Critical role of the β-subunit CDC50A in the stable expression, assembly, subcellular localization, and lipid transport activity of the P 4-ATPase ATP8A2. J Biol Chem 286:17205–17216. https://doi.org/10.1074/jbc.M111.229419
Coleman JA, Kwok MCM, Molday RS (2009) Localization, purification, and functional reconstitution of the P4-ATPase Atp8a2, a phosphatidylserine flippase in photoreceptor disc membranes. J Biol Chem 284:32670–32679. https://doi.org/10.1074/jbc.M109.047415
Cacciagli P, Haddad M-R, Mignon-Ravix C et al (2010) Disruption of the ATP8A2 gene in a patient with a t(10;13) de novo balanced translocation and a severe neurological phenotype. Eur J Hum Genet 18:1360–1363. https://doi.org/10.1038/ejhg.2010.126
De Simone R, Ajmone-Cat MA, Minghetti L (2004) Atypical antiinflammatory activation of microglia induced by apoptotic neurons: possible role of phosphatidylserine-phosphatidylserine receptor interaction. Mol Neurobiol 29:197–212. https://doi.org/10.1385/MN:29:2:197
Wang J, Molday LL, Hii T et al (2018) Proteomic analysis and functional characterization of P4-ATPase phospholipid flippases from murine tissues. Sci Rep 8:10795. https://doi.org/10.1038/s41598-018-29108-z
Vestergaard AL, Coleman JA, Lemmin T et al (2014) Critical roles of isoleucine-364 and adjacent residues in a hydrophobic gate control of phospholipid transport by the mammalian P4-ATPase ATP8A2. Proc Natl Acad Sci U S A 111:E1334–E1343. https://doi.org/10.1073/pnas.1321165111
Beaudin M, Matilla-Dueñas A, Soong B-W et al (2019) The classification of autosomal recessive cerebellar ataxias: a consensus statement from the society for research on the cerebellum and ataxias task force. Cerebellum. https://doi.org/10.1007/s12311-019-01052-2
Rossi M, Anheim M, Durr A et al (2018) The genetic nomenclature of recessive cerebellar ataxias. Mov Disord 33:1056–1076. https://doi.org/10.1002/mds.27415
Anheim M, Tranchant C, Koenig M (2012) The autosomal recessive cerebellar ataxias. N Engl J Med 366:636–646. https://doi.org/10.1056/NEJMra1006610
Renaud M, Guissart C, Mallaret M et al (2016) Expanding the spectrum of PEX10-related peroxisomal biogenesis disorders: slowly progressive recessive ataxia. J Neurol 263:1552–1558. https://doi.org/10.1007/s00415-016-8167-3
Guissart C, Drouot N, Oncel I et al (2016) Genes for spinocerebellar ataxia with blindness and deafness (SCABD/SCAR3, MIM# 271250 and SCABD2). Eur J Hum Genet 24:1154–1159. https://doi.org/10.1038/ejhg.2015.259
Carré G, Marelli C, Anheim M et al (2017) Xeroderma pigmentosum complementation group F: a rare cause of cerebellar ataxia with chorea. J Neurol Sci 376:198–201. https://doi.org/10.1016/j.jns.2017.03.021
Marelli C, Hamel C, Quiles M et al (2018) ACO2 mutations: a novel phenotype associating severe optic atrophy and spastic paraplegia. Neurol Genet 4:e225. https://doi.org/10.1212/NXG.0000000000000225
Acknowledgements
The authors thank the families and patients described herein for their participation in this study. The corresponding author had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. This study was supported by funds from the Agence Nationale pour la Recherche (ANR)/E-rare Joint Trans-national Call (JTC) 2011 ‘Euro-SCAR’ (2011-RARE-004–01 to M.K.) and the Canadian Institutes of Health Research grant (PJT 148649 to RSM).
Author information
Authors and Affiliations
Contributions
CG and MK: conception and design of the study; AH, AC, CG, EAA, HT, IO, KB, LL, MB, MI, MK, NEE, PC, RSM, RT: contributed to acquisition, analysis, and interpretation of data; CG, MB, MK and RSM: contributed to drafting the manuscript or figures; AC, CG, EAA, HT, IO, KB, LL, MB, MC, MI, MK, NEE, PC, RSM, RT: contributed to critical revision of the manuscript, gave final approval of the version to be published, and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no conflicts of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Guissart, C., Harrison, A.N., Benkirane, M. et al. ATP8A2-related disorders as recessive cerebellar ataxia. J Neurol 267, 203–213 (2020). https://doi.org/10.1007/s00415-019-09579-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-019-09579-4