
Abstract
Peroxisomal biogenesis disorders (PBDs) consist of a heterogeneous group of autosomal recessive diseases, in which peroxisome assembly and proliferation are impaired leading to severe multisystem disease and early death. PBDs include Zellweger spectrum disorders (ZSDs) with a relatively mild clinical phenotype caused by PEX1, (MIM# 602136), PEX2 (MIM# 170993), PEX6 (MIM# 601498), PEX10 (MIM# 602859), PEX12 (MIM# 601758), and PEX16 (MIM# 603360) mutations. Three adult patients are reported belonging to a non-consanguineous French family affected with slowly progressive cerebellar ataxia, axonal neuropathy, and pyramidal signs. Mental retardation and diabetes mellitus were optional. The age at onset was in childhood or in adolescence (3–15 years). Brain MRI showed marked cerebellar atrophy. Biochemical blood analyses suggested a mild peroxisomal defect. With whole exome sequencing, two mutations in PEX10 were found in the three patients: c.827G>T (novel) causing the missense change p.Cys276Phe and c.932G>A causing the missense change p.Arg311Gln. The phenotypic spectrum related to PEX10 mutations includes slowly progressive, syndromic recessive ataxia.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Autosomal recessive cerebellar ataxias (ARCAs) consist of a heterogeneous group of inherited neurodegenerative disorders of the cerebellum often associated with peripheral nervous system involvement and systemic abnormalities, including ophthalmologic disturbances [1]. Peroxisomal biogenesis disorders (PBDs) associated with the loss of multiple peroxisomal metabolic functions are a rare cause of ARCAs. PBDs are due to mutations in PEX genes encoding peroxisome biogenesis factors involved in the import of peroxisomal membrane and matrix proteins [2]. PBDs include the Zellweger spectrum disorders (ZSDs), a clinical and biochemical spectrum of recessively inherited diseases caused by mutations in one of 13 different PEX genes [3]. The clinical spectrum ranges from patients presenting in the neonatal period who die within the first year of life to patients presenting in adulthood with only minor symptoms. ZSDs with a relatively mild clinical phenotype caused by PEX1, (MIM# 602136), PEX2 (MIM# 170993), PEX6 (MIM# 601498), PEX10 (MIM# 602859), PEX12 (MIM# 601758), and PEX16 (MIM# 603360) mutations have been reported [4–7]. Here, we report three patients with ataxia and unusually prolonged survival caused by missense mutations in the RING Zinc finger domain of PEX10.
Patients and methods
Clinical and biological analysis
Age at onset of the disease, disease duration, and age at last examination were noted. Spinocerebellar degeneration functional score (SDFS) was used to evaluate the disability stage from 1 to 7 (0: no functional handicap; 1: no functional handicap but signs at examination; 2: mild, able to run; 3: moderate, unable to run; 4: severe, walking with one stick, walking unlimited; 5: walking with two sticks; 6: unable to walk, requiring wheelchair, limited walking without aid; and 7: confined to bed) [8].
Patients underwent neurological examination, motor and sensory nerve conduction studies, and brain MRI. Biochemical determinations of plasma amino acids, urinary organic acids, serum alpha-fetoprotein, vitamin E, lactate, very long-chain fatty acids (VLCFA), as well as pristanic, phytanic, and pipecolic acids have been performed. Several peroxisomal parameters were studied in cultured skin fibroblasts, including catalase immunofluorescence microscopy [9], VLCFA profile [10], and dihydroxyacetonephosphate-acyltransferase (DHAPAT) activity [11].
Written informed consent has been obtained from all individuals providing the biological samples for molecular and biochemical investigations and local ethics committee approved the study.
Genetic studies
DNA was extracted by the standard procedures. Whole exome sequencing was performed by exon capture with the Agilent SureSelect kit and high throughput sequencing with an Illumina HiSeq 2500 sequencer [IGBMC Microarray and Sequencing platform, a member of the “France Génomique” consortium (ANR-10-INBS-0009)]. Reads were mapped to the human reference genome (hg19). The total number of mapped reads was 106 242 472, the median read depth over exons was 82, total exons covered by 10 reads or more was 91 %, and exon coverage by at least one sequence 96 %. Variations were annotated, ranked, and analyzed with an in-house pipeline which combine splice site prediction and protein coding changes [12] and the VaRank program [13]. In particular, based on dbSNP (137), the 1000 Genomes project [14], and the Exome Variant Server (NHLBI GO Exome Sequencing Project), single nucleotide polymorphisms with a frequency of 1 % or above were filtered out. Pathogenicity of the variations were assessed for the several type of effects, including splice site alterations (thanks to the Alamut Batch annotations included in VaRank), and missenses using SIFT and PolyPhen-2 [15, 16]. Protein domain annotation was retrieved from the Pfam database (version 29.0) [17].
Results
Clinical data
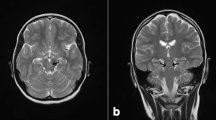
Two sisters and one brother from a French non-consanguineous family were affected by a slowly progressive cerebellar ataxia. Age at onset was between 3 and 15 years, but disease progression was slow despite SDFS was above 5 at last examination in at least two patients. Neurological examination showed sensorimotor axonal polyneuropathy, pyramidal signs with no spasticity, and mental retardation (in 2 out of 3 patients), in addition to cerebellar signs. Neuropsychological tests revealed intellectual deficiency with difficulties for reasoning, judgment, and structural constructive skills for both patients II.2 and II.4. Patient II.4 had verbal IQ of 56, performance IQ of 52, and limited memory abilities. Intellectual deficiency was marked and stable. Brain MRI showed marked cerebellar atrophy and cerebral white matter hyperintensities (Fig. 1). No motor evoked potentials were available. Diabetes mellitus was diagnosed in two patients. The main clinical data are summarized in Table 1.
Biological data
In patients II.2 and II.4, plasma analysis revealed normal C26:0 concentration but increased C26/C22 and C24/C22 ratios and increased phytanic, pristanic, and pipecolic acid levels (Table 2). Almost no biochemical abnormalities could be observed in cultured fibroblasts of patients II2 and II4 (Table 2): fibroblasts had normal C26/C22 and C24/C22 ratios, normal DHAPAT activity (Dihydroxyacetonephosphate-acyltransferase, the first enzyme in the plasmalogen biosynthesis pathway and located in the peroxisome), normal very long-chain fatty acid profile, and normal immunoblot profile both for peroxisomal Acyl-CoA oxidase 1 (ACOX1) and peroxisomal 3-ketoacyl-CoA thiolase. Only a very small number of cells had absent peroxisomal staining when performing immunofluorescence microscopy analyses with antibodies against catalase (a peroxisomal matrix protein) in patient II4 (Table 2) both at 37 and 40 °C.
Mutation analysis
DNA sample of patient II.4 was analyzed by the whole exome sequencing. Because the reported family was non-consanguineous, we analyzed both homozygous and compound heterozygous mutations on autosomes. After selection of unique variants out of a set of 12 independent exomes and exclusion of variants found in more than 1 % of the population as well as of variants with a VaRank score lower than 55, we identified 19 homozygous variants and 44 compound heterozygous variants. Among them, two different mutations were found in PEX10 (NM_002617): c.827G>T causing the missense change p.Cys276Phe and c.932G>A causing the missense change p.Arg311Gln.
Both missenses are part of the RING Zinc finger domain of PEX10 (PFAM: PF13920 positions 272–311). Looking at the multiple sequence alignment of the 8919 sequences available in the PFAM database, both amino acids are highly conserved amino acids that help defining the motif of this domain. In addition, SIFT and PolyPhen-2 predict both changes to be very probably pathogenic (SIFT score: 0.00 and Polyphen2 score: 1.00 for both mutations) [15, 16].
We confirmed the presence of the two heterozygous PEX10 mutations, p.Cys276Phe and p.Arg311Gln, in all three patients by direct sequencing. Only one heterozygous mutation, p.Arg311Gln, was found in the youngest healthy sister (Fig. 2), demonstrating that the two mutations are allelic and that the three patients are compound heterozygous for the mutations. DNA samples from the parents and the older healthy sister were not available.

Brain MRI abnormalities in patient II.4. a Cerebellar atrophy (sagittal T1). b Periventricular white matter hyperintensities (axial FLAIR)
Discussion
Herein, we report the identification of an unusual cause of recessive ataxia, achieved by the exome sequencing analysis. In our family, affected members were clinically characterized by early-onset, slowly progressive cerebellar ataxia associated with axonal polyneuropathy, pyramidal signs, mental retardation, and diabetes mellitus. In the three siblings, phytanic, pristanic, and pipecolic acid plasma levels were increased which was consistent with PBD. Interestingly, C26:0/C22:0 and C24:0/C22:0 ratios were increased in plasma but normal in fibroblasts. Immunoblot analysis of ACOX1 and 3-ketoacyl-CoA thiolase revealed normal peroxisomal processing of these proteins. The only abnormality observed in cultured skin fibroblasts of patient II.3 was the absence of peroxisomal staining in a very small number of cells with the catalase immunofluorescence microscopy analysis both at 37 and 40 °C. In this family, the mutations were not truncating, but missense mutations located in the RING Zinc finger domain of PEX10 which is a crucial domain for its function.
PEX10 is a 326 amino acid peroxisomal membrane protein, part of the PEX2–PEX10–PEX12 ubiquitin ligase complex [18, 19]. More than 30 different mutations have been identified in the PEX10 gene, including missense, nonsense, deletion, insertion, splice site mutations, and disruption of the start codon [20]. As recently reported [15], we used the shorter (most abundant) isoform of PEX10 (NM_002617) as reference and relabeled previously published mutations accordingly. The most common PEX10 mutation, c.814_815delCT, is found mainly in the Japanese population [21], and the second most common mutation is c.704_705insA [22]. Both mutations cause the occurrence of a truncated protein missing the RING Zinc finger domain. The phenotype is characterized clinically by severe global neurological involvement, dysmorphy, retinitis pigmentosa, sensorineural deafness, and other systemic features (Fig. 1).

Identification of PEX10 mutations in the 3 patients. a Localization of PEX10 on the map of chromosome 1 is indicated by an arrow. b Pedigree showing segregation of the disease with heterozygous compound mutation: c.827G>T causing the p.Cys276Phe missense change (C276F) and c.932G>A causing the p.Arg311Gln missense change (R311Q). Sanger sequencing results are indicated on the right of the pedigree: the three affected are compound heterozygous for C276F and R311Q (for the sake of clarity, electropherograms of only one patient are shown), and the healthy sister is heterozygous for C276F only. c Sequence comparison of amino acids in PEX10 and of orthologous proteins from different species. Amino acids that are identical to the human PEX10 sequence are shown in bold. The compound heterozygous mutations (on the top of the mutated amino acid cysteine, C, and arginine, R, highlighted in yellow) are located in a region coding for the RING Zinc finger domain (273–311, boxed). The mutated amino acids are conserved in all eukaryotes, including plants. Positions defining the RING motif are shown with a “*”
Three patients with milder presentations of PBDs due to PEX10 mutations have been previously reported [5]. Two of them had cerebellar ataxia, axonal motor neuropathy, and posterior column dysfunction, but neither had mental retardation nor pyramidal signs or diabetes mellitus. The first one had an age of onset at 5 years and was compound heterozygous for the c.704_705insA and p.Arg311Gln mutations. The second had an age of onset at 6 years and was compound heterozygous for a c.730C>T mutation, causing the nonsense change p.Arg244* (again resulting in a truncation before the RING Zinc finger domain) and a c.2>T mutation which abolishes the initiation codon [5]. This latter mutation is assumed to cause re-initiation at the next in-frame ATG codon and production of an N-terminally truncated PEX10 protein, but still containing the RING Zinc finger domain. Similar to our cases, no biochemical abnormalities could be observed in the cultured fibroblasts of both patients. Brain MRI showed severe cerebellar atrophy with normal white matter.
The third previously reported patient [23] developed unsteady gait around 3 years of age, then obvious isolated ataxia with dysarthria, severe cerebellar atrophy, and cerebral white matter changes on brain MRI. Again, he was compound heterozygous for a frame-shift mutation (c.337delC) and a de novo missense mutation in a highly conserved amino acid of the RING Zinc finger domain (p.Leu277Pro).
Our three additional cases expand the phenotypic spectrum of patients with PEX10 mutations with cerebellar ataxia, peripheral neuropathy, pyramidal signs with no spasticity, and mental retardation. Age at onset is in childhood or in adolescence and the course is very slowly progressive with disease duration of up to 56 years [1]. Whether diabetes mellitus is part of the phenotype or is incidental finding remains unclear. Brain MRI shows obvious cerebellar atrophy without cerebellar white matter changes. All PEX10 cerebellar ataxia cases have at least one missense mutation in the RING Zinc finger domain or one mutation that produces a truncated protein, but that preserves the RING Zinc finger domain. All these mutations are predicted to be hypomorphic, and therefore, presumably explain the mild presentation of the patients compared to patients at the other end of the PBD spectrum with, for example, homozygous or compound heterozygous mutations that completely abolish the RING Zinc finger domain [2]. These results support the growing concept of spinocerebellar ataxia by mildly affected metabolic pathway, due to the exquisite sensitivity of cerebellar/spinocerebellar neurons to even modest metabolic insult [1].
In conclusion, our data suggest searching for PBDs mutations in patients with unexplained early-onset, slowly progressive ARCA with peripheral neuropathy and pyramidal signs, particularly, since early diagnosis should prompt evaluation of appropriate treatments, such as bile acid supplements or dietary restriction of phytanic acid.
References
Anheim M, Tranchant C, Koenig M (2012) The autosomal recessive cerebellar ataxias. N Engl J Med 366(7):636–646
Steinberg SJ, Dodt G, Raymond GV, Braverman NE, Moser AB, Moser HW (2006) Peroxisome biogenesis disorders. Biochim Biophys Acta 1763(12):1733–1748
Waterham HR, Ebberink MS (2012) Genetics and molecular basis of human peroxisome biogenesis disorders. Biochim Biophys Acta 1822(9):1430–1441
Sevin C, Ferdinandusse S, Waterham HR, Wanders RJ, Aubourg P (2011) Autosomal recessive cerebellar ataxia caused by mutations in the PEX2 gene. Orphanet J Rare Dis. BioMed Central Ltd 6(1):8
Régal L, Ebberink MS, Goemans N, Wanders RJA, de Meirleir L, Jaeken J et al (2010) Mutations in PEX10 are a cause of autosomal recessive ataxia. Ann Neurol 68(2):259–263
Zeharia A, Ebberink MS, Wanders RJA, Waterham HR, Gutman A, Nissenkorn A et al (2007) A novel PEX12 mutation identified as the cause of a peroxisomal biogenesis disorder with mild clinical phenotype, mild biochemical abnormalities in fibroblasts and a mosaic catalase immunofluorescence pattern, even at 40 °C. J Hum Genet 52(7):599–606
Guissart C, Drouot N, Oncel I, Leheup B, Gershoni-Barush R, Muller J et al (2015) Genes for spinocerebellar ataxia with blindness and deafness (SCABD/SCAR3, MIM# 271250 and SCABD2). Eur J Hum Genet EJHG. Nature Publishing Group 1–6
Anheim M, Fleury M, Monga B, Laugel V, Chaigne D, Rodier G et al (2010) Epidemiological, clinical, paraclinical and molecular study of a cohort of 102 patients affected with autosomal recessive progressive cerebellar ataxia from Alsace, Eastern France: implications for clinical management. Neurogenetics 11(1):1–12
Wanders RJ, Wiemer EA, Brul S, Schutgens RB, van den Bosch H, Tager JM (1989) Prenatal diagnosis of Zellweger syndrome by direct visualization of peroxisomes in chorionic villus fibroblasts by immunofluorescence microscopy. J Inherit Metab Dis 12(Suppl 2):301–304
Dacremont G, Vincent G (1995) Assay of plasmalogens and polyunsaturated fatty acids (PUFA) in erythrocytes and fibroblasts. J Inherit Metab Dis 18(Suppl 1):84–89
Wanders RJ, Ofman R, Romeijn GJ, Schutgens RB, Mooijer PA, Dekker C et al (1995) Measurement of dihydroxyacetone-phosphate acyltransferase (DHAPAT) in chorionic villous samples, blood cells and cultured cells. J Inherit Metab Dis 18(Suppl 1):90–100
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C et al (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43(5):491–498
Geoffroy V, Pizot C, Redin C, Piton A, Vasli N, Stoetzel C et al (2015) VaRank: a simple and powerful tool for ranking genetic variants. Peer J. 3:e796
Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA et al (2010) A map of human genome variation from population-scale sequencing. Nature 467(7319):1061–1073
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P et al (2010) A method and server for predicting damaging missense mutations. Nat Methods 7(4):248–249
Kumar P, Henikoff S, Ng PC (2009) Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4(7):1073–1081
Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR et al (2014) Pfam: the protein families database. Nucleic Acids Res 42(Database issue):D222–D230
Okumoto K, Itoh R, Shimozawa N, Suzuki Y, Tamura S, Kondo N et al (1998) Mutations in PEX10 is the cause of Zellweger peroxisome deficiency syndrome of complementation group B. Hum Mol Genet 7(9):1399–1405
Chang CC, Warren DS, Sacksteder KA, Gould SJ (1999) PEX12 interacts with PEX5 and PEX10 and acts downstream of receptor docking in peroxisomal matrix protein import. J Cell Biol 147(4):761–774
Ebberink MS, Mooijer PAW, Gootjes J, Koster J, Wanders RJA, Waterham HR (2011) Genetic classification and mutational spectrum of more than 600 patients with a Zellweger syndrome spectrum disorder. Hum Mutat 32(1):59–69
Shimozawa N, Nagase T, Takemoto Y, Suzuki Y, Kondo N (2003) Genetic heterogeneity in Japanese patients with peroxisome biogenesis disorders and evidence for a founder haplotype for the most common mutation in PEX10 gene. Adv Exp Med Biol 544:71
Warren DS, Wolfe BD, Gould SJ (2000) Phenotype-genotype relationships in PEX10-deficient peroxisome biogenesis disorder patients. Hum Mutat 15(6):509–521
Steinberg SJ, Snowden A, Braverman NE, Chen L, Watkins PA, Clayton PT et al (2009) A PEX10 defect in a patient with no detectable defect in peroxisome assembly or metabolism in cultured fibroblasts. J Inherit Metab Dis 32(1):109–119
Acknowledgments
We wish to acknowledge the help of Bernard Jost, Serge Vicaire, Stéphanie Legras, Michael Dumas, and Véronique Geoffroy for the next-generation sequencing and analysis.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The final manuscript has been read and approved by all authors who accepted full responsibility for the design and undertaking of the original article, had access to the data and controlled the decision to publish.
Ethical standards
The study was approved by the local ethics committee.
Funding
This study was supported by funds from the Agence Nationale pour la Recherche-Maladies Rares and Maladies Neurologiques et Psychiatriques (ANR-09-MNPS-001-01 to M.K.), the ANR/E-rare JTC 2011 “Euro-SCAR” (2011-RARE-004-01 to M.K.).
Web resources
UCSC Genome Browser: http://genome.ucsc.edu/index.html [April, 2015].
Ensembl Genome Browser: http://www.ensembl.org/index.html [April, 2015].
Exome Variant Server (EVS), NHLBI GO Exome Sequencing Project (ESP), Seattle, WA: http://evs.gs.washington.edu/EVS [June, 2013].
Human Splicing Finder version 2.4.1: http://www.umd.be/HSF/ [June, 2013].
VaRank: http://www.lbgi.fr/VaRank/ [April, 2015].
PEX database: http://www.dbpex.org/ [April, 2015].
Additional information
Mathilde Renaud and Claire Guissart have equally contributed to this work.
Rights and permissions
About this article

Cite this article
Renaud, M., Guissart, C., Mallaret, M. et al. Expanding the spectrum of PEX10-related peroxisomal biogenesis disorders: slowly progressive recessive ataxia. J Neurol 263, 1552–1558 (2016). https://doi.org/10.1007/s00415-016-8167-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-016-8167-3