
Abstract
Accumulation of the DNA/RNA binding protein fused in sarcoma (FUS) as inclusions in neurons and glia is the pathological hallmark of amyotrophic lateral sclerosis patients with mutations in FUS (ALS-FUS) as well as in several subtypes of frontotemporal lobar degeneration (FTLD-FUS), which are not associated with FUS mutations. Despite some overlap in the phenotype and neuropathology of FTLD-FUS and ALS-FUS, significant differences of potential pathomechanistic relevance were recently identified in the protein composition of inclusions in these conditions. While ALS-FUS showed only accumulation of FUS, inclusions in FTLD-FUS revealed co-accumulation of all members of the FET protein family, that include FUS, Ewing’s sarcoma (EWS) and TATA-binding protein-associated factor 15 (TAF15) suggesting a more complex disturbance of transportin-mediated nuclear import of proteins in FTLD-FUS compared to ALS-FUS. To gain more insight into the mechanisms of inclusion body formation, we investigated the role of Transportin 1 (Trn1) as well as 13 additional cargo proteins of Transportin in the spectrum of FUS-opathies by immunohistochemistry and biochemically. FUS-positive inclusions in six ALS-FUS cases including four different mutations did not label for Trn1. In sharp contrast, the FET-positive pathology in all FTLD-FUS subtypes was also strongly labeled for Trn1 and often associated with a reduction in the normal nuclear staining of Trn1 in inclusion bearing cells, while no biochemical changes of Trn1 were detectable in FTLD-FUS. Notably, despite the dramatic changes in the subcellular distribution of Trn1 in FTLD-FUS, alterations of its cargo proteins were restricted to FET proteins and no changes in the normal physiological staining of 13 additional Trn1 targets, such as hnRNPA1, PAPBN1 and Sam68, were observed in FTLD-FUS. These data imply a specific dysfunction in the interaction between Trn1 and FET proteins in the inclusion body formation in FTLD-FUS. Moreover, the absence of Trn1 in ALS-FUS provides further evidence that ALS-FUS and FTLD-FUS have different underlying pathomechanisms.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD) are clinically, genetically and neuropathologically heterogeneous groups of neurodegenerative diseases. The neuropathology is characterized by the abnormal intracellular accumulation of specific proteins with the accumulation of the fused in sarcoma protein (FUS, also known as translocated in liposarcoma, TLS) being the characteristic lesion in ~3 % of ALS cases and ~10 % of FTLD cases, subsumed as FTLD-FUS [20].
FUS is a multifunctional DNA/RNA binding protein and belongs to the FET family of proteins that also includes Ewing’s sarcoma (EWS) protein, TATA-binding protein-associated factor 15 (TAF15) and the drosophila orthologue Cabeza [14, 17]. The FET proteins are predominantly nuclear, ubiquitously expressed, highly conserved proteins with predicted roles in RNA transcription, processing, transport, microRNA processing and DNA repair [14, 17, 34]. All of the FET proteins are shuttling proteins [12, 39, 40] and their nuclear import is mediated by a non-classical nuclear localization signal, called PY-NLS, recognized by the nuclear import protein Transportin 1 (Trn1) [7, 18].
ALS cases with FUS pathology are almost always associated with a mutation in the FUS gene (ALS-FUS). Mutations in the gene FUS were first identified in 2009 as the cause of familial ALS type 6 [15, 37] and then rapidly confirmed in large ALS cohorts accounting for ~3 % of familial and <1 % of sporadic ALS [20]. The neuropathology of ALS-FUS is characterized by abnormal cytoplasmic inclusions in neurons and glia that are immunoreactive for FUS [3, 9, 10, 15, 29, 37]. Significant pathological heterogeneity with respect to morphological and regional distribution of FUS pathology has been described in ALS-FUS allowing delineation of two distinct patterns correlating with disease severity and specific mutations [23]. The majority of FUS mutations cluster in the C terminus of the protein which includes its PY-NLS. They have been shown to disrupt the PY-NLS motif leading to impaired Trn1-mediated nuclear import of FUS and subsequent increase of cytoplasmic FUS levels [7, 11, 13]. Notably, the level of nuclear import impairment that results from specific FUS mutations in cultured cells directly correlates with the clinical phenotype and the neuropathological patterns observed in ALS-FUS, thereby strongly suggesting that altered nuclear import of FUS is a key event in the pathogenesis of ALS-FUS.
FTLD-FUS includes three distinct clinico-pathological entities; atypical FTLD-U (aFTLD-U), basophilic inclusion body disease (BIBD) and neuronal intermediate filament inclusion disease (NIFID) [19, 24–26]. FTLD-FUS is mostly associated with sporadic early onset of behavioral variant of frontotemporal dementia with the exception of single cases with positive family history [16, 25, 26, 31, 32, 36]. While possibly other genetic factors might be involved in FTLD-FUS, in contrast to pure ALS with FUS pathology which is usually associated with FUS mutations, no genetic abnormalities of FUS have been identified in FTLD-FUS, and the mechanisms underlying inclusion body formation in FTLD-FUS remain unknown.
Although there is some clinical and pathological overlap between ALS-FUS and FTLD-FUS, the presence of significant differences in the genetics, phenotypes and the morphological patterns of FUS pathology [21] raised the question as to whether these conditions represent closely related conditions with a shared pathomechanism or whether the pathogenic pathways triggered by FUS mutations may be different from those involved in FTLD-FUS. Indeed, the analysis of the FUS homologues TAF15 and EWS has recently identified striking differences in the protein composition of inclusions in these conditions, with co-accumulation of all FET proteins in FTLD-FUS and restricted accumulation of only FUS in ALS-FUS [27]. These data provided strong evidence for different pathological processes underlying inclusion body formation and cell death between ALS-FUS and FTLD-FUS, with ALS-FUS being restricted to dysfunction of FUS, while FTLD-FUS seems to be associated with a broader and more complex dysregulation of Trn1-mediated nuclear import affecting all FET proteins and perhaps other Trn1 cargos.
In the present study, we extend our immunohistochemical analysis of the protein composition of inclusion bodies in the spectrum of FUS-opathies to Trn1 and 13 additional known Trn1 cargo proteins with a similar PY-NLS motif as FET proteins.
Methods
Case selection
The FUS-opathy cases included aFTLD-U (n = 17), BIBD (n = 8), NIFID (n = 4) and six ALS cases with four different FUS mutations. Most of these cases have been described in detail in previous studies [21, 23–27] and brief clinical and demographic features are summarized in Supplementary Table 1.
The original set of neurological control cases included FTLD-TDP (n = 6; two with subtype A (one with GRN mutation), two with subtype B (one with C9ORF72 mutation) and two with subtype C [22]), FTLD-tau (n = 6; two Pick’s disease, two corticobasal degeneration, and two progressive supranuclear palsy), FTLD with CHMP2B mutations (n = 2), FTLD-ni (n = 3), sporadic ALS with TDP pathology (n = 2), familial ALS with SOD1 mutations (n = 2), Alzheimer’s disease (n = 2), dementia with Lewy bodies (n = 2), Parkinson’s disease (n = 2), multiple system atrophy (n = 2), Huntington’s disease (n = 2), spinocerebellar ataxia (n = 3; one SCA-1 and two SCA-3) and neuronal intranuclear inclusion body disease (n = 1). When it was found that rare classical Lewy bodies in the substantia nigra of the cases of Parkinson’s disease showed some Trn1 immunoreactivity, the following additional controls were added: sections of midbrain from six additional cases of Parkinson’s disease, sections of hippocampus from two additional cases of dementia with Lewy bodies (to check if immunoreactivity was restricted to Lewy bodies in the substantia nigra) and sections of midbrain from three additional cases of PSP (to check if immunoreactivity was generalized to all types of cytoplasmic inclusions in nigral neurons). Normal control tissue (n = 4) was from elderly patients with no history of neurological disease.
Staining protocols for the 13 antibodies against other PY-NLS proteins (Table 3) were established using a tissue microarray including biopsy material from a glioblastoma and a brain metastasis of a colon carcinoma as well as postmortem tissue from the dentate granule cell layer and temporal cortex of three controls with no history of neurological disease.
Antibodies
A mouse monoclonal antibody was used for Trn1 immunohistochemistry (IHC) (clone 45, Abcam, 1:200). All FUS-opathy cases were stained for FUS (using either the polyclonal anti-FUS HPA008784, Sigma-Aldrich, 1:2,000 or the monoclonal anti-FUS, ProteintechGroup, 1:1,000), TAF15 using the polyclonal anti-TAF15 IHC-00094-1 (Bethyl Laboratories, 1:200), and EWS (using the monoclonal anti-EWS clone G5, Santa Cruz, 1:200, or the polyclonal anti-EWS IHC-00086, Bethyl Laboratories, 1:200). The list of antibodies against 13 other Trn1 cargo proteins with a PY-NLS investigated in this study is provided in Table 3.
Immunohistochemistry and double-label immunofluorescence
Trn1 IHC was performed on 5-μm thick sections of formalin-fixed paraffin-embedded tissue for all FUS-opathy and control cases on selected neuroanatomical regions known to have robust pathology using the Ventana BenchMark XT automated staining system (Ventana, Tuscon, AZ) following microwave antigen retrieval and developed with aminoethylcarbizole (AEC) or using the NovoLink™ Polymer Detection Kit and developed with DAB. Staining with antibodies against other PY-NLS proteins was performed on selected cases with severe FUS pathology (BIBD n = 1, aFTLD-U n = 4).
Incubation with primary antibodies was 32 min using the automated staining system or 1 h for non-automated staining procedure.
Double-label immunofluorescence (IF) was performed on selected cases for FUS, TAF15, EWS and Trn1 or FUS and other Trn1 substrates (Table 3). The secondary antibodies were Alexa Fluor 594 and Alexa Fluor 488-conjugated anti-mouse and anti-rabbit IgG (Invitrogen, 1:500). 4′-6-Diamidino-2-phenylindol (DAPI) was used for nuclear counterstaining. IF images were obtained by wide-field fluorescence microscopy (BX61 Olympus with digital camera F-view, Olympus).
Biochemical analysis
Fresh-frozen postmortem frontal grey matter from aFTLD-U (n = 5), BIBD (n = 1), NIFID (n = 1), FTLD with TDP-43 pathology (n = 5), and normal controls (n = 4) was used for the sequential extraction of proteins with buffers of increasing stringency, using an established protocol [25, 27]. Briefly, gray matter was extracted at 2 ml/g (v/w) by repeated homogenization and centrifugation steps (120,000×g, 30 min, 4 °C) with high-salt buffer (50 mM Tris–HCl, 750 mM NaCl, 10 mM NaF, 5 mM EDTA, pH 7.4), 1 % Triton-X 100 in HS buffer, radioimmunoprecipitation assay buffer [50 mM Tris–HCl, 150 mM NaCl, 5 mM EDTA, 1 % NP-40, 0.5 % sodium deoxycholate, 0.1 % sodium dodecyl sulfate (SDS)] and 2 % SDS buffer. To prevent carry over, each extraction step was performed twice. Only supernatants from the first extraction steps were analyzed while supernatants from the second wash steps were discarded. The 2 %-SDS insoluble pellet was extracted in 70 % formic acid at 0.5 ml/g (v/w), evaporated in a SpeedVac system and the dried pellet was resuspended in sample buffer and the pH was adjusted with NaOH. Protease inhibitors were added to all buffers prior to use. For immunoblot analysis, equal volumes of fractions from different samples (10 μl of high-salt and TX, 20 μl of RIPA and SDS, 25 μl of formic acid) were resolved by 7.5 % SDS-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes (Millipore, Billerica, MA, USA). Following transfer, membranes were blocked with Tris buffered saline containing 3 % powdered milk and probed with the mouse monoclonal anti-Trn1 antibody (clone 45, Abcam, 1:500) or polyclonal anti-FUS antibody (300-302A, Bethyl Laboratories, 1:10,000). Primary antibodies were detected with horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG (Jackson ImmunoResearch Europe, UK), signals were visualized by a chemiluminescent reaction (Millipore) and the Chemiluminescence Imager Stella 3200 (Raytest, Switzerland).
Results
Trn1 co-aggregates with FET proteins in all FTLD-FUS subtypes
Although there was some variation among cases and anatomical regions, the normal physiological staining pattern for Trn1 usually consisted of moderate to strong immunoreactivity of nuclei and weak diffuse cytoplasmic immunoreactivity in neurons (Fig. 1a).

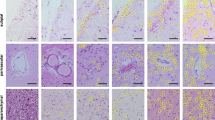
Trn1 immunohistochemistry in controls, FTLD-FUS and ALS-FUS. The physiological staining pattern of Trn1 consists of moderate to strong immunoreactivity of neuronal nuclei and weak diffuse cytoplasmic staining as shown in control cortex (a). Abundant Trn1-positive pathology is present in all affected brain regions in FTLD-FUS cases (b–g), including neuronal cytoplasmic inclusions (NCI) in the hippocampus in aFTLD-U (b, c), the neocortex (d) and spinal cord (g) in NIFID and the pons in BIBD (e). In addition to NCI, round and vermiform neuronal intranuclear inclusions (NII) were labeled in cases of aFTLD-U and NIFID (arrows in c) and glial cytoplasmic inclusions were present in the cerebral white matter (f). In striking contrast to FTLD-FUS, no Trn1 positive pathology was seen in ALS-FUS cases as shown for the spinal cord of the p.[R514S/;E516V] mutation (h); note the absence of Trn1 immunoreactivity of the basophilic inclusion (arrowhead in h) while nuclear staining is retained (arrow in h). Scale bar 125 μm (a), 63 μm (b, e, h), 20 μm (c), 90 μm (d, g), 45 μm (f)
In all subtypes of FTLD-FUS, Trn1 IHC revealed strong labeling of neuronal and glial inclusions (Table 1; Fig. 1b–g) with the frequency and morphology of inclusions being comparable to those demonstrated with FUS and TAF15 IHC. Antibodies against EWS labeled fewer inclusions, particularly in aFTLD-U, as previously described [27]. Specifically, aFTLD-U cases showed strong Trn1 staining of neuronal cytoplasmic inclusions (NCI) and neuronal intranuclear inclusions (NII) in the hippocampal dentate fascia (Fig. 1b, c) and the frontotemporal cortex. The numerous NCI in cortical, subcortical, brainstem and spinal cord regions in NIFID (Fig. 1d, g) and BIBD (Fig. 1e) cases were also strongly immunoreactive for Trn1. In addition to NCI, FTLD-FUS cases showed variable numbers of Trn1-positive glial cytoplasmic inclusions (GCI; Fig. 1f). The variability of physiological Trn1 immunoreactivity among cases mainly influenced by the time of tissue fixation did not allow a detailed quantification, but in cases with robust staining, inclusion bearing cells sometimes showed a clear reduction in nuclear Trn1 staining compared to cells without inclusions (Supplementary Fig. 1).
To confirm co-accumulation of Trn1 in the characteristic inclusions in FTLD-FUS with all FET proteins, we performed double-label immunohistochemistry for Trn1 and FUS, TAF15 or EWS of selected cases. This demonstrated consistent co-labeling of all Trn1-positive NCI and NII in cortical and spinal cord sections in aFTLD-U, NIFID and BIBD for FUS (Fig. 2a–c), and TAF15 (Supplementary Fig. 2). EWS and Trn1 co-localized in all inclusions in BIBD and NIFID cases, while in aFTLD-U only a subset of Trn1-positive inclusions was EWS immunoreactive (Supplementary Fig. 3).

Co-localization of Trn1 and FUS in all FTLD-FUS subtypes. Double-label immunofluorescence for FUS (red) and Trn1 (green), with DAPI staining of nuclei in the merged images. FUS-positive inclusions in all FTLD-FUS subtypes consistently showed co-localization of FUS and Trn1, as shown for neuronal cytoplasmic inclusions (NCI) and neuronal intranuclear inclusions (NII, arrow in a) in the dentate granule cells in aFTLD-U (a), NCI in the temporal cortex of NIFID (b) and NCI and glial cytoplasmic inclusions in the spinal cord of BIBD (c). Scale bar 10 μm
Absence of Trn1 in inclusions in ALS with FUS mutations
Trn1 staining was investigated in six ALS-FUS cases with four different FUS mutations. All had previously been shown to have robust FUS-positive (but TAF15- and EWS-negative) pathology, particularly in the spinal cord and motor cortex, that included NCI and variable numbers of GCI [23]. In striking contrast to FTLD-FUS, no Trn1 immunoreactive neuronal or glial inclusions were detectable in cortical, subcortical, or spinal cord regions in any of the ALS-FUS cases, while the physiological nuclear staining pattern for Trn1 was retained (Fig. 1h). The absence of Trn1 accumulation in FUS-positive NCI and GCI in ALS-FUS was further confirmed by double-label IF (Fig. 3a–c).

Absence of Trn1 pathology in ALS-FUS. Double-label immunofluorescence for FUS (red) and Trn1 (green), with DAPI staining of nuclei in the merged images. FUS-positive inclusions in ALS-FUS were not labeled for Trn1 as shown for neuronal cytoplasmic inclusions in the spinal cord for three different FUS mutations (a–c). Note the physiological nuclear staining for Trn1 in inclusion bearing cells. FUS-positive glial cytoplasmic inclusions present in a subset of ALS-FUS cases also showed no co-labeling for Trn1 (arrow in a). Scale bar 10 μm
Trn1 staining in neurological controls
The normal and neurological control cases did not reveal Trn1 immunoreactive pathology with one exception (Table 2). Specifically, there was no labeling of the characteristic inclusions in AD, FTLD-tau, FTLD-TDP, ALS-TDP, ALS-SOD1, and MSA. The exception was weak labeling of a subset of classical Lewy bodies in the substantia nigra in 7 of 8 PD cases with a frequency ranging from 2 to 14 %. Notably, NII in SCA, HD and NIIBD, which have previously been shown to be consistently FUS positive and more variably EWS positive [6, 27, 38], were negative for Trn1; a finding which further underlines the specificity of FET and Trn1 immunoreactive inclusions in FTLD-FUS.
Biochemical analysis of Trn1 in FTLD-FUS
To investigate potential biochemical alterations of Trn1 which might explain its co-accumulation in FTLD-FUS, proteins were sequentially extracted from frozen brain tissue from FTLD-FUS, as well as normal and neurological controls, using the same extraction protocol previously used to demonstrate changes in the solubility of FET protein as consistent biochemical alteration in FTLD-FUS [25, 27, 28].
Trn1 could be detected as major band at the expected molecular mass of ~97 kDa, in the HS, Triton-X, RIPA and SDS fractions in FTLD-FUS, as well as controls but not in the formic acid fraction enriched for highly insoluble proteins (Fig. 4). There was some variability in the signal intensities between the different fractions among the various cases and controls; however, no consistent changes such as a shift in Trn1 solubility or additional Trn1 isoforms were observed in FTLD-FUS compared to controls. As positive control and validation of the extraction protocol, the same protein fractions were analyzed by immunoblot for FUS (Fig. 4), demonstrating a clear shift of FUS from the soluble HS fraction towards the insoluble SDS fraction in all FTLD-FUS cases compared to control cases, in accordance with previous reports.

Biochemical analysis of Trn1 and FUS in FTLD-FUS. Proteins were sequentially extracted from frontal cortex of aFTLD-U, NIFID, BIBD, controls without neurodegenerative disease as well as FTLD-TDP as neurological controls. High-salt (fraction 1), Triton-X-100 (fraction 2), RIPA (fraction 3), 2 % SDS (fraction 4) and formic acid (fraction 5) protein fractions were separated by SDS-PAGE and immunoblotted. Trn1 was present as one major band at the expected molecular size for the full-length protein (~97 kDa) in fractions 1–4 in FTLD-FUS and controls and was absent in fraction 5 enriched for highly insoluble proteins. While there was some variability among signal intensities in the different fractions among cases, no clear correlation of a distinct pattern was detectable in FTLD-FUS compared to controls. For validation of protein extractions, samples were analyzed by anti-FUS immunoblot, demonstrating full-length FUS at a molecular size of ~73 kDa with the amount of FUS present in the SDS fraction (fraction 4) being much higher in FTLD-FUS cases compared to controls, in accordance with previous reports
Absence of other PY-NLS proteins in inclusions in FTLD-FUS
The co-accumulation of all FET proteins together with Trn1 in FTLD-FUS suggests a complex impairment of Trn1-mediated nuclear import in FTLD-FUS pathogenesis, raising the possibility that besides FET proteins other Trn1 cargo proteins might also be affected in FTLD-FUS. In order to test this hypothesis, we performed an immunohistochemical analysis of FTLD-FUS cases for 13 additional PY-NLS proteins (Table 3) described as validated cargo proteins of Trn1 [8, 18, 33]. IHC for all showed a predominantly nuclear staining pattern that was robust in neurons and weaker in glial cells in controls as well as FTLD-FUS cases. However, none of the antibodies against the additional PY-NLS proteins labeled inclusions in FTLD-FUS and no change in their normal physiological nuclear staining was observed in inclusion bearing cells. The absence of labeling of inclusions was confirmed by double-label immunofluorescence as illustrated for hnRNP A1, the best-characterized substrate of Trn1 (Fig. 5a), SAM68 (Fig. 5b), and PABPN1 (Fig. 5c).

Absence of selected other Trn1 cargos (hnRNP A1, SAM68 and PABPN1) in FTLD-FUS. Double-label immunofluorescence for FUS (red) and other Trn1 cargos with PY-NLS (hnRNP A1, SAM68 and PABPN1, respectively, green) with DAPI staining of nuclei in the merged images in FTLD-FUS. FUS-positive inclusions in FTLD-FUS as shown here in the dentate gyrus of aFTLD-U were not labeled for hnRNP A1 (a), SAM68 (b) and PABPN1 (c). Scale bar 10 μm
Discussion
FUS accumulation characterizes all cases of ALS with FUS mutations and a variety of FTLD subtypes, collectively referred to as FTLD-FUS [15, 19, 24–26, 37]; however, the mechanisms of neurodegeneration in FUS-opathies are only poorly understood.
Based on our recent findings on differences in the protein composition of inclusions between ALS-FUS and FTLD-FUS with respect to the co-accumulation of the FUS homologues EWS and TAF15 [27], the hypothesis has emerged that these conditions might have different underlying pathomechanisms. The abnormality in ALS-FUS is restricted to alterations and dysfunction of FUS that result from FUS mutations that disrupt its PY-NLS and thereby interfere with proper binding to its nuclear transport receptor Trn1 and consecutive cytoplasmic accumulation of FUS, but not TAF15 and EWS [7, 27]. In contrast, FTLD-FUS is associated with accumulation of all FET proteins, thereby suggesting a broader defect of nuclear import pathways [5, 27], although the mechanism(s) of FET accumulation in the absence of mutations in their PY-NLS remain unclear.
Data presented in this study provide further strong evidence for different underlying mechanisms of inclusion body formation in ALS-FUS and FTLD-FUS by expanding the differences in the protein composition of inclusions between ALS-FUS and FTLD-FUS to Trn1.
None of the six ALS-FUS cases investigated (including three different FUS missense and one truncation mutations) showed alterations in the physiological staining pattern of Trn1 or evidence for co-accumulation of Trn1 in the FUS-positive neuronal and glial inclusions. Thus, the impaired binding capability of FUS with a mutated PY-NLS to Trn1 [7] is not associated with detectable changes in the subcellular distribution of Trn1 by IHC, which is also reflected by the unaffected nuclear import of other Trn1 cargo proteins such as TAF15 and EWS in ALS-FUS [27]. Thus, the absence of Trn1 pathology in ALS-FUS is consistent with the hypothesis that the pathological processes underlying ALS-FUS are restricted to dysfunctions of FUS.
In striking contrast to ALS-FUS, we demonstrate robust and consistent co-accumulation of Trn1 with all FET proteins in the various types of inclusions (NCI, GCI and NII) in our large collection of FTLD-FUS cases, including 17 aFTLD-U, 8 BIBD and 4 NIFID cases, in accordance with previous reports in smaller series of FTLD-FUS [4, 5]. Accumulation of Trn1 into cytoplasmic inclusions was often associated with a reduction in the physiological nuclear staining of Trn1 in FTLD-FUS. Notably, the co-labeling of inclusions for Trn1 and all FET proteins was found to be a highly specific and unique feature of all subtypes of FTLD-FUS that was not present in any of our neurological controls, including those (HD, SCA, NIIBD) characterized by FUS and variable EWS immunoreactive NII [6, 27, 38]. These findings support the concept that aFTLD-U, NIFID and BIBD are closely related disease entities sharing the same pathomechanism [21]. Interestingly, a small subset of LBs in the midbrain of PD cases was found to be weakly positive for Trn1; however, the significance of this finding needs further investigation.
The dramatic changes in the subcellular distribution of Trn1 together with the accumulation of all FET proteins in FTLD-FUS support the idea of a complex dysregulation of Trn1-associated nuclear import in FTLD-FUS. This could result from a primary defect of Trn1 itself, either by posttranslational modifications, genetic variations or altered expression levels. In this scenario, one might expect to see changes of other Trn1 substrates and/or biochemical alterations Trn1 itself.
However, our immunohistochemical analysis of a large series of additional Trn1 cargo proteins failed to demonstrate any abnormal accumulation or alterations in their subcellular distribution. The Trn1 cargo proteins with PY-NLS motifs similar to FET proteins included in this study have all been validated in vitro as Trn1 binding proteins and included hnRNP A1, the best studied Trn1 cargo [8, 18, 33]. While we cannot exclude that some other PY-NLS proteins, in addition to FET proteins, might be affected in FTLD-FUS, our data strongly argue against a general dysfunction of Trn1 as a primary event in FTLD-FUS inclusion body formation. In line with that, we did not detect any specific biochemical alterations of Trn1 such as solubility changes or presence of abnormal Trn1 species in FTLD-FUS using a well-established sequential protein extraction protocol, able to reveal clear solubility changes for FUS, TAF15 and EWS in FTLD-FUS (this study, [25, 27]). However, our biochemical data are in disagreement with a previous publication describing a shift towards insolubility for Trn1 in aFTLD-U and NIFID [4]. This discrepancy might be due to different extraction protocols used, an issue which needs to be further addressed in future studies.
Based on our data we hypothesize an alternate scenario in which some abnormal posttranslational modification(s) that specifically affect FET proteins interferes with their normal Trn1 binding and dissociation and subsequently the proper nucleocytoplasmic transport of these shuttling proteins and their subcompartmental distribution in FTLD-FUS. Described posttranslational modifications of FET proteins include phosphorylation and arginine methylation and these have been shown to affect the cellular distribution, RNA/DNA-binding ability, protein–protein interaction, and protein stability of FET proteins in vitro [1, 2, 12, 30, 34, 35]. Beside changes in solubility of FET proteins [25, 27, 28], no disease-associated posttranslational changes have yet been reported in FTLD-FUS; however, this requires more detailed characterization. Notably, arginine methylation of FUS has been recently reported to weaken its interaction with Trn1 [8]. Together with the findings of co-accumulation of Trn1 in inclusions in FTLD-FUS, it is tempting to speculate that a reduced methylation status of FET proteins might be present in FTLD-FUS which would lead to a very tight binding to Trn1, thus hampering the normal dissociation of the transport complex resulting in co-accumulation of Trn1 and FET proteins in cytoplasmic and nuclear inclusions.
In summary, we have demonstrated striking differences in the accumulation of Trn1 in the spectrum of FUS-opathies with consistent Trn1 accumulation in FTLD-FUS but not in ALS-FUS, thereby providing further strong evidence for different mechanisms underlying inclusion body formation in FTLD-FUS and ALS-FUS. Notably, despite the dramatic changes in the subcellular distribution of Trn1 in FTLD-FUS, alterations of its cargo proteins were found to be restricted to FET proteins in FTLD-FUS, implying a specific dysfunction in the interaction between Trn1 and all FET proteins, most likely by posttranslational modifications of FET proteins, in the pathogenesis of FTLD-FUS.
References
Belyanskaya LL, Gehrig PM, Gehring H (2001) Exposure on cell surface and extensive arginine methylation of Ewing sarcoma (EWS) protein. J Biol Chem 276:18681–18687
Belyanskaya LL, Delattre O, Gehring H (2003) Expression and subcellular localization of Ewing sarcoma (EWS) protein is affected by the methylation process. Exp Cell Res 288:374–381
Blair IP, Williams KL, Warraich ST et al (2010) FUS mutations in amyotrophic lateral sclerosis: clinical, pathological, neurophysiological and genetic analysis. J Neurol Neurosurg Psychiatry 81:639–645
Brelstaff J, Lashley T, Holton JL et al (2011) Transportin1: a marker of FTLD-FUS. Acta Neuropathol 122:591–600
Davidson YS, Robinson AC, Hu Q et al (2012) Nuclear carrier and RNA binding proteins in frontotemporal lobar degeneration associated with fused in sarcoma (FUS) pathological changes. Neuropathol Appl Neurobiol. doi:10.1111/j.1365-2990.2012.01274.x
Doi H, Koyano S, Suzuki Y, Nukina N, Kuroiwa Y (2010) The RNA-binding protein FUS/TLS is a common aggregate-interacting protein in polyglutamine diseases. Neurosci Res 66:131–133
Dormann D, Rodde R, Edbauer D et al (2010) ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J 29:2841–2857
Fronz K, Guttinger S, Burkert K et al (2011) Arginine methylation of the nuclear poly(a) binding protein weakens the interaction with its nuclear import receptor, transportin. J Biol Chem 286:32986–32994
Groen EJ, van Es MA, van Vught PW et al (2010) FUS mutations in familial amyotrophic lateral sclerosis in the Netherlands. Arch Neurol 67:224–230
Hewitt C, Kirby J, Highley JR et al (2010) Novel FUS/TLS mutations and pathology in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol 67:455–461
Ito D, Seki M, Tsunoda Y, Uchiyama H, Suzuki N (2010) Nuclear transport impairment of amyotrophic lateral sclerosis-linked mutations in FUS/TLS. Ann Neurol 69(1):152–162
Jobert L, Argentini M, Tora L (2009) PRMT1 mediated methylation of TAF15 is required for its positive gene regulatory function. Exp Cell Res 315:1273–1286
Kino Y, Washizu C, Aquilanti E et al (2011) Intracellular localization and splicing regulation of FUS/TLS are variably affected by amyotrophic lateral sclerosis-linked mutations. Nucleic Acids Res 39:2781–2798
Kovar H (2011) Dr. Jekyll and Mr. Hyde: the two faces of the FUS/EWS/TAF15 Protein Family. Sarcoma 837474. doi:10.1155/2011/837474
Kwiatkowski TJ Jr, Bosco DA, Leclerc AL et al (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323:1205–1208
Lashley T, Rohrer JD, Bandopadhyay R et al (2011) A comparative clinical, pathological, biochemical and genetic study of fused in sarcoma proteinopathies. Brain 134:2548–2564
Law WJ, Cann KL, Hicks GG (2006) TLS, EWS and TAF15: a model for transcriptional integration of gene expression. Brief Funct Genomic Proteomic 5:8–14
Lee BJ, Cansizoglu AE, Suel KE et al (2006) Rules for nuclear localization sequence recognition by karyopherin beta 2. Cell 126:543–558
Mackenzie IR, Neumann M, Bigio EH et al (2010) Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol 119:1–4
Mackenzie IR, Rademakers R, Neumann M (2010) TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol 9:995–1007
Mackenzie IR, Munoz DG, Kusaka H et al (2011) Distinct pathological subtypes of FTLD-FUS. Acta Neuropathol 121:207–218
Mackenzie IR, Neumann M, Baborie A et al (2011) A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol 122:111–113
Mackenzie IRA, Ansorge O, Strong M et al (2011) Pathological heterogeneity in amyotrophic lateral sclerosis with FUS mutations: two distinct patterns correlating with disease severity and mutation. Acta Neuropathol 122:87–98
Munoz DG, Neumann M, Kusaka H et al (2009) FUS pathology in basophilic inclusion body disease. Acta Neuropathol 118:617–627
Neumann M, Rademakers R, Roeber S et al (2009) A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain 132:2922–2931
Neumann M, Roeber S, Kretzschmar HA et al (2009) Abundant FUS-immunoreactive pathology in neuronal intermediate filament inclusion disease. Acta Neuropathol 118:605–616
Neumann M, Bentmann E, Dormann D et al (2011) FET proteins TAF15 and EWS are selective markers that distinguish FTLD with FUS pathology from amyotrophic lateral sclerosis with FUS mutations. Brain 134:2595–2609
Page T, Gitcho MA, Mosaheb S et al (2011) FUS immunogold labeling TEM analysis of the neuronal cytoplasmic inclusions of neuronal intermediate filament inclusion disease: a frontotemporal lobar degeneration with FUS proteinopathy. J Mol Neurosci 45:409–421
Rademakers R, Stewart H, DeJesus-Hernandez M et al (2010) FUS gene mutations in familial and sporadic amyotrophic lateral sclerosis. Muscle Nerve 42:170–176
Rappsilber J, Friesen WJ, Paushkin S, Dreyfuss G, Mann M (2003) Detection of arginine dimethylated peptides by parallel precursor ion scanning mass spectrometry in positive ion mode. Anal Chem 75:3107–3114
Rohrer JD, Lashley T, Holton J et al (2011) The clinical and neuroanatomical phenotype of FUS associated frontotemporal lobar degeneration. J Neurol Neurosurg Psychiatry 82:1405–1407
Snowden JS, Hu Q, Rollinson S et al (2011) The most common type of FTLD-FUS (aFTLD-U) is associated with a distinct clinical form of frontotemporal dementia but is not related to mutations in the FUS gene. Acta Neuropathol 122:99–110
Suel KE, Gu H, Chook YM (2008) Modular organization and combinatorial energetics of proline-tyrosine nuclear localization signals. PLoS Biol 6:e137
Tan AY, Manley JL (2009) The TET family of proteins: functions and roles in disease. J Mol Cell Biol 1:82–92
Tradewell ML, Yu Z, Tibshirani M et al (2012) Arginine methylation by PRMT1 regulates nuclear-cytoplasmic localization and toxicity of FUS/TLS harbouring ALS-linked mutations. Hum Mol Genet 21:136–149
Urwin H, Josephs KA, Rohrer JD et al (2010) FUS pathology defines the majority of tau- and TDP-43-negative frontotemporal lobar degeneration. Acta Neuropathol 120:33–41
Vance C, Rogelj B, Hortobagyi T et al (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323:1208–1211
Woulfe J, Gray DA, Mackenzie IR (2010) FUS-immunoreactive intranuclear inclusions in neurodegenerative disease. Brain Pathol 20:589–597
Zakaryan RP, Gehring H (2006) Identification and characterization of the nuclear localization/retention signal in the EWS proto-oncoprotein. J Mol Biol 363:27–38
Zinszner H, Sok J, Immanuel D, Yin Y, Ron D (1997) TLS (FUS) binds RNA in vivo and engages in nucleo-cytoplasmic shuttling. J Cell Sci 110:1741–1750
Acknowledgments
We thank Margaret Luk and Jay Tracy for their excellent technical assistance. This work was supported by grants from the Swiss National Science Foundation (31003A-132864, MN); the Synapsis Foundation (MN); the Canadian Institutes of Health Research (74580 and 179009, IM), the Pacific Alzheimer’s Research Foundation (C06-01, IM); and the NIHR Oxford Biomedical Research Centre (OA).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Neumann, M., Valori, C.F., Ansorge, O. et al. Transportin 1 accumulates specifically with FET proteins but no other transportin cargos in FTLD-FUS and is absent in FUS inclusions in ALS with FUS mutations. Acta Neuropathol 124, 705–716 (2012). https://doi.org/10.1007/s00401-012-1020-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-012-1020-6