
Abstract
The term frontotemporal lobar degeneration (FTLD) describes a group of disorders that are subdivided by the presence of one of a number of pathological proteins identified in the inclusion bodies observed post-mortem. The FUS variant is defined by the presence of the fused in sarcoma protein (FUS) in the pathological inclusions. However, similar to other FTLDs, the disease pathogenesis of FTLD-FUS remains largely poorly understood. Here we present data that the protein transportin1 (TRN1) is abundant in the FUS-positive inclusions. TRN1, the protein product of the TNP01 gene, is responsible for shuttling proteins containing an M9 nuclear localisation signal between the nuclear and cytoplasmic compartments. RNA interacting proteins, including FUS, have been implicated as targets of TRN1. Using TRN1 immunohistochemistry and Western blotting in this study, we investigated 13 cases of FTLD-FUS including 6 cases with neuronal intermediate filament inclusion disease (NIFID) and 7 atypical frontotemporal lobar degeneration with ubiquitinated inclusion (aFTLD-U) cases. The data from our immunohistochemical studies show that FUS-immunoreactive inclusions are also strongly labelled with the anti-TRN1 antibody and double-label immunofluorescence studies indicate good co-localisation between the FUS and TRN1 pathologies. Our biochemical investigations demonstrate that urea-soluble TRN1 is present in aFTLD-U and NIFID, but not in normal control brains. These findings implicate abnormalities of FUS transport in the pathogenesis of FTLD-FUS.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The term frontotemporal lobar degeneration (FTLD) defines a heterogeneous group of neurodegenerative diseases with distinct yet overlapping clinical presentations with behavioural variant frontotemporal dementia, progressive non-fluent aphasia and semantic dementia being the three best-characterised clinical syndromes. Although there is some correlation between the clinical and pathological subtypes, the clinical presentation does not always predict the underlying pathology. Disease classification of FTLDs relies on the identification of the main protein component of neuronal inclusions. The proteins responsible for the majority of the cases are tau, the TAR DNA-binding protein-43 (TDP-43) and the ‘fused in sarcoma’ (FUS) protein [16, 17]. The significance of FUS in neurodegeneration was recognised with the discovery that mutations in the FUS gene are responsible for familial amyotrophic lateral sclerosis (ALS) type 6 (ALS-FUS) [10, 27]. Subsequently FUS has also emerged as a significant disease protein in a subgroup of FTLDs, previously characterized by immunoreactivity of the neuronal inclusions for ubiquitin, but not for TDP-43 or tau with a proportion of the inclusions also containing α-internexin in a further subgroup known as neuronal intermediate filament inclusion disease (NIFID). The disease entities which are now considered subtypes of FTLD-FUS are atypical frontotemporal lobar degeneration with ubiquitinated inclusions (aFTLD-U), NIFID (otherwise known as neurofilament inclusion body disease [8]) and basophilic inclusion body disease (BIBD), which together with ALS-FUS comprise the FUS-opathies [18–21].
The cause and pathomechanism of inclusion formation in the FUS-opathies is only partially understood. FUS is a 526-amino acid long protein with a predicted molecular mass of 53 kDa, which has diverse cellular functions [1, 4, 31]. The C-terminal region of FUS is involved in RNA–protein interactions while its N-terminus has a role in transcription activation [22]. FUS is ubiquitously expressed [1] and is able to bind both RNA and DNA [4]. In keeping with its important functions in transcription regulation, FUS protein is present in considerably larger amounts in the nucleus than in the cytoplasm of neurons while it is restricted to the nuclei and not found in the cytoplasm of glial cells [2]. Under normal physiological conditions, the FUS protein shuttles between the nucleus and cytoplasm through the nuclear pore [11, 32]. Transport of FUS from the cytoplasm to the nucleus takes place with the aid of transportin1 (TRN1), also known as M9-interacting protein or karyopherinβ2 (Karβ2), which is an 890-amino acid long protein (OMIM; http://www.ncbi.nlm.nih.gov/omim/602901). Karyopherinβ-s (Karβs), also known as importins and exportins are responsible for the majority of the cellular nucleocytoplasmic transport. In humans, ten Karβs have been shown to carry diverse macromolecular substrates into the nucleus and in one of these import pathways Karβ2 is responsible for the import of a significant group of RNA processing proteins, including FUS [3, 5, 13, 15, 29, 30]. Binding of substrates to import and export Karβs is dependent on the nuclear localisation signal (NLS) and nuclear export signal, which are predicted to be located at the C-terminal end and the beginning of the RNA recognition motif of FUS, respectively [11]. The NLS of FUS can be described as a PY motif very similar to previously described M9 signals and a likely target of the TRN1 nuclear import system [5, 13]. During nuclear import of proteins, the Karβ-protein cargo complex translocates into the nucleus through its association with the nuclear pore complex. Once inside the nucleus the Karβ-protein complex dissociates, which is dependent on RanGTP binding to the import Karβs. The free Karβs are then recycled into the cytoplasm to be available for a new cycle of cargo import [29].
In this study, we wished to investigate whether TRN1 is also incorporated into the FUS-positive inclusions in FTLD-FUS, as TRN1 is a key player of the finely tuned cellular machinery responsible for the nuclear import of FUS [5] and several other proteins involved in RNA processing [13]. For this we performed a detailed immunohistochemical study and have found strong TRN1 immunoreactivity in neuronal cytoplasmic inclusions (NCIs) and neuronal intranuclear inclusions (NIIs) with an overall good co-localisation between TRN1 and FUS immunoreactivity in both the NIFID and aFLTD-U subgroups of FTLD-FUS. Furthermore, our biochemical investigations have demonstrated that highly insoluble (urea-soluble) TRN1 is present in FTLD-FUS, but not in normal controls, indicating that aggregation of TRN1 could be a significant event of pathomechanism in FTLD-FUS.
Materials and methods
Cases
Brains were donated to the Queen Square Brain Bank for Neurological Disorders, UCL Institute of Neurology, University College London; the MRC London Brain Bank for Neurodegenerative Diseases, Institute of Psychiatry, King’s College, London, UK; Neuropathology Department, Århus Kommunehospital, Århus, Denmark and NeuroResource, UCL Institute of Neurology, University College London. All cases had previously been diagnosed as NIFID (6 cases) or aFTLD-U (7 cases) (Table 1). Two aFTLD-U (case 13 was the mother of case 9) cases are from the same family, although no mutations in the FUS gene have been found [12]. In addition, three normal control cases and three cases each with multiple system atrophy, corticobasal degeneration, motor neuron disease, Alzheimer’s disease, Parkinson’s disease, progressive supranuclear palsy, Pick’s disease and FTLD-TDP types 1 and 3 were selected from the archives of the Queen Square Brain Bank. Three cases with FTLD-TDP type 2 pathology, which included one of the motor neuron disease cases with extensive cortical TDP-43 pathology and also used as motor neuron disease control, were also used for this study. In addition to frontal and hippocampal regions, the cervical or thoracic spinal cord from each motor neurone disease case was also stained for TRN1. No BIBD cases were available for this study in our archives.
TRN1 antibodies and western blotting
Two commercially available anti-TRN1 antibodies were used in this study (Abcam ab10303, monoclonal; Abcam ab67352, polyclonal).
Tissue samples from frontal cortex (grey matter) from four controls, three NIFID and four aFTLD-U cases were homogenised at a ratio of 1:2 (wt/vol) in high-salt (HS) buffer (50 mM Tris–HCl, 750 mM NaCl, 10 mM NaF, 5 mM EDTA) containing 1% Triton-X and protease and phosphatase inhibitors (Roche). Tissue homogenate was spun at 1,000g to remove nuclear and membrane debris. The resulting supernatant was subjected to ultracentrifugation at 120,000g for 30 min at 4°C, following which the supernatant was retained (HS fraction). Using this HS fraction, we performed immunoblotting with both the monoclonal (Abcam ab10303, 1:500) and the polyclonal antibodies (Abcam ab67352, 1:500) to test antibody specificity. Antibody specificity was confirmed by omission of the primary antibody. The results of these preliminary experiments indicated that the monoclonal antibody identified a strong band at ~100 kDa representing the expected molecular weight of TRN1. In addition to this band, the polyclonal antibody also labelled additional low molecular weight bands. Therefore, the monoclonal antibody was regarded as more specific and was chosen for the full biochemical analysis.
The pellet, retained after harvesting the HS fraction, was subjected to further extractions with RIPA buffer (50 mM Tris–HCl, 150 mM NaCl, 1% NP-40, 0.5% deoxycholate) containing 2% SDS and protease and phosphatase inhibitors as before, which was subjected to ultracentrifugation at 120,000g for 30 min at 15°C to avoid SDS precipitation, with the resulting supernatant being termed RIPA-SDS fraction. The final pellet was resuspended in 8 M urea containing 8% SDS (urea-soluble) fraction. Protein concentration was determined by the BCA protein assay (Pierce) and 20 μg of protein from the HS and RIPA-SDS fraction, and 5 μg of protein from the urea fraction, of each case was loaded onto 10% Bis–Tris polyacrylamide gels (Invitrogen) and run at 200 V with MES buffer (Invitrogen) under reducing conditions. Following electrophoresis, the proteins were transferred onto Hybond P membrane (GE Healthcare), blocked with 5% non-fat dried milk in PBS containing 0.1% Tween (PBS-T) and probed overnight with the monoclonal anti-TRN1 (Abcam ab10303, 1:500) antibody at 4°C. Following washes in PBS-T, the blot was treated with HRP-conjugated secondary antibody (Santa Cruz). Blots were visualised by enhanced chemiluminescence (Pierce) and the image captured onto Kodak, X-Omat (Sigma) films.
Immunohistochemistry
For TRN1 immunohistochemistry, both the monoclonal (Abcam ab10303, 1:200) and the polyclonal antibody (Abcam ab67352, 1:200) were tested. Although both antibodies stained normal nuclear protein, NCIs and NIIs, the monoclonal antibody was used for full immunohistochemical analysis as this antibody was shown to be more specific with western blotting. Furthermore, the polyclonal antibody only stained TRN1 when frozen tissue was used while the monoclonal antibody gave positive staining on paraffin sections.
For detailed immunohistochemical studies, 8-μm thick tissue sections from frontal cortex, hippocampus, medulla and spinal cord were cut from paraffin embedded tissue. Endogenous peroxidase activity was blocked with 0.3% H2O2 in methanol followed by pressure cooker pre-treatment in citrate buffer pH 6.0. Sections were treated with 10% dried milk solution to block non-specific binding. Tissue sections were incubated with the primary anti-TRN1 (Abcam, ab10303, 1:200) antibody for 1 h at room temperature, followed by biotinylated anti-mouse (Dako, 1:200) and ABC complex (Dako). Colour was developed with di-aminobenzidine/H2O2.
Double-label immunofluorescence and morphometry
The frontal cortex and hippocampus from all FTLD-FUS cases were stained using a polyclonal anti-FUS antibody (aa1-50, Novus, 1:200) in combination with a monoclonal anti-TRN1 (Abcam, ab10303, 1:200). After appropriate pre-treatment, tissue sections were incubated with the primary antibodies overnight, followed by the secondary antibody anti-mouse Alexa Fluor 488 (Invitrogen, Paisley, UK, 1:300) for 1 h at room temperature. Sections were next incubated with swine anti-rabbit secondary (Dako, 1:200), followed by ABC and visualized using TSA Rhodamine kit (Perkin-Elmer) and 4′-6-diamidino-2-phenylindole (DAPI) was used for nuclear counterstaining. Sections were viewed with a Leica DM5500B fluorescence microscope using 3D deconvolution post-processing.
To establish the proportion of FUS-positive inclusions also labelled with the anti-TRN1 antibody, double-labelled sections of the hippocampus were chosen from three NIFID and three aFTLD-U cases. The granule cell layer was identified using the Leica DM5500B fluorescence microscope and ten sequential visual fields of this structure were captured using a 60× objective. Subsequently z-stack of images through the full depth of the tissue section was taken and a non-blind deconvolution algorithm was applied. A maximum projection of the z-stack provided the final image for analysis. TRN1 or FUS-positive NCIs and NIIs were visually identified on the appropriate channels and the co-localisation was confirmed on the combined images.
Results
TRN1 immunoblot analysis
To establish the biochemical characteristics of TRN1 in FTLD-FUS and normal control brains, protein was sequentially extracted from flash frozen frontal cortex. Buffers containing increasing detergent strength were used to investigate the different biochemical fractions with different solubility characteristics. Our immunoblot data demonstrate two key points: (1) The two antibodies tested were found to recognise an ~100 kDa band that corresponds to full-length TRN1 protein and (2) analysis of the immunoblots using the monoclonal antibody (Abcam ab10303) has demonstrated that, although a significant amount of TRN1 is present in the HS and RIPA-SDS fractions in both FTLD-FUS and controls, urea-soluble TRN1 (highly insoluble fraction of TRN1) is present in FTLD-FUS (Fig. 1).

Representative immunoblots demonstrating TRN1 in fractions of varying solubility in FTLD-FUS and normal control brains. Proteins were sequentially extracted into high salt (lane 1), RIPA-SDS (lane 2), and urea (lane 3) fractions from 4 control, 3 NIFID, and 4 aFTLD-U cases. 20 μg of protein was loaded from high salt and RIPA-SDS fractions, while 5 μg of protein was loaded from the urea fractions. A single band at approximately 100 kDa corresponding to full-length TRN1 appears in high salt and RIPA-SDS fractions in both control and disease cases, but the highly insoluble urea fraction is seen in the FTLD-FUS cases
Normal localization of TRN1
The cellular localization of TRN1, examined in normal control cases using formalin fixed tissue and TRN1 immunohistochemistry showed that immunoreactivity was localized to the nuclei of both neurons and glial cells (Fig. 2a). A similar staining pattern was seen in unaffected neurons in the FTLD-FUS cases and cases with other neurodegenerative diseases.

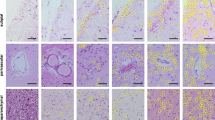
In normal controls transportin 1 (TRN1) immunoreactivity is nuclear in both neurons (double arrow) and glial cells (arrow) (a). TRN1-positive neuronal cytoplasmic inclusions in the hippocampal granule cells in a NIFID case (b arrow) and both neuronal cytoplasmic inclusions and neuronal intranuclear inclusions in granule cells in an aFTLD-U case (c double arrow). Note the lack of nuclear staining on b due to prolonged fixation in formaldehyde. Different inclusion types in motor neurons in the XIIth cranial nerve nucleus (d, f) and spinal cord (e, g) in NIFID (d, e) and aFTLD-U (f, g). The inclusion types included dot-like/granular (d), large globular inclusions without nuclear positivity (e), large globular inclusions with nuclear positivity (f), inclusions formed in a neuronal process (g arrow). Bar in a represents 5 μm on d–g, 20 μm on a and 40 μm on b and c
Localization of TRN1 in FTLD-FUS
Tissue sections of the frontal cortex, hippocampus with the entorhinal cortex, and whenever available the medulla with the XIIth cranial nerve nucleus and/or the spinal cord (available in 4 NIFID and 3 aFTLD-U cases) were investigated in the 12 post-mortem cases, and the frontal cortex alone in the case diagnosed using a frontal cortical biopsy. The tissue sections from all areas showed TRN1-immunoreactive NCIs (Figs. 2b–g, 3a–d, f, g), NIIs (Figs. 2c, 3c, e) and threads/neurites in all 13 FTLD-FUS cases. The intensity of the immunolabelling of inclusions varied between and within individual cases, but this together with the variability of the morphological appearances of the inclusions, the overall severity and distribution of the TRN1 pathology were comparable to those seen with FUS immunohistochemistry.

TRN1 immunohistochemistry highlighted different inclusion types in the frontal cortex in both NIFID (a–c, f, g) and aFTLD-U (d, e). In some neurons with neuronal cytoplasmic inclusions the normal nuclear TRN1 staining was retained (a, d), but this was lost in the majority (a, b, c, f). A number of inclusion types were seen in cortical neurons including crescent/annular shaped inclusions in both NIFID and aFTLD-U (a, d), Pick body-like inclusions in NIFID (b) and bean-shaped inclusions in aFTLD-U (c). Vermiform neuronal intranuclear inclusions were seen in both subtypes (c, e). Granular, dot-like cytoplasmic positivity often extending into dendrites (arrows) was found in NIFID (f). TRN1-positive grains were observed in the entorhinal cortex in NIFID (f double arrow). Skein-like inclusions were seen in motor neurons (g). Bar on a represents 20 μm on a; 5 μm on b–e, g; 10 μm on f
Although the preservation of the normal nuclear staining appeared to be dependent on the length of the fixation time (Fig. 2b), variability in the strength of the TRN1 nuclear staining could be ascertained in cases with otherwise good preservation of nuclear TRN1 immunoreactivity. In such cases, although strong TRN1 nuclear immunoreactivity was present in a significant proportion of the nuclei of neurons with NCIs (Figs. 2f, 3a, d, g), the TRN1 staining was decreased or was entirely absent in others (Figs. 2c, 3a). In the NIFID cases, variable numbers of small, 1–2 μm large, dot-like TRN1-positive structures were frequently found in the neuropil of the frontal cortex, but these structures were generally more numerous in the entorhinal cortex. In one of the most severely affected NIFID cases (case 2), larger TRN1-positive spindle or comma-shaped grain-like structures with a diameter of up to 4 μm were found to intermingle with TRN1-positive dots in the entorhinal cortex with TRN1-positive grain-like structures being particularly frequent in the cellular islands of the pre-α layer of the entorhinal cortex. A characteristic, rather coarse, dot-like immunoreactivity was also commonly seen in the cytoplasm and dendrites of the large neurons of the pre-α clusters. Albeit less abundant, TRN1 dot-like positivity was also present in the cortical neuropil in the aFTLD-U cases (Fig. 3f). Fine TRN1-positive threads or delicate neurites and often large and rather thick neurites were evident in most cases. Similar to the findings seen with FUS immunohistochemistry, the motor neurons of the XIIth nerve nucleus and spinal cord contained morphologically variable NCIs, including granular, globular or filamentous skein-like inclusions (Fig. 3g).
TRN1 staining in other neurodegenerative diseases
The frontal cortex and hippocampus from examples of different neurodegenerative diseases (multiple system atrophy, corticobasal degeneration, motor neuron disease, Alzheimer’s disease, Parkinson’s disease, progressive supranuclear palsy, Pick’s disease and FTLD-TDP types 1, 2 and 3 were stained with the anti-TRN1 antibody. In addition, sections of the cervical or thoracic spinal cord were stained in the motor neuron disease cases. In all cases, TRN1 immunoreactivity was seen in the nuclei of neurons and glial cells, but, apart from a single cortical Lewy body, the characteristic inclusions seen in these diseases showed no immunoreactivity for TRN1 (data not shown).
TRN1 and FUS double-label immunofluorescence and morphometry
In all cases, histological sections of the frontal cortex and hippocampal formation including the granule cell layer of the dentate fascia were double stained for TRN1 and FUS. Qualitative assessment of the fluorescence images showed that there is an overall very good co-localisation of TRN1 with FUS in NCIs and NIIs in both NIFID and aFTLD-U cases (Fig. 4). Furthermore, quantitative analysis of three NIFID and three FTLD-U cases also demonstrated that the overwhelming majority of the FUS-positive inclusions also had TRN1 immunolabelling in the granule cells of the dentate fascia (99.4% in NIFID and 100% in aFTLD-U). It was also noted that the TRN1 labelling of NII was often disproportionate in that a proportion of such inclusions were only weakly immunoreactive for TRN1.

Double-label immunofluorescence in aFTLD-U (a–c) and NIFID (d–f) demonstrating co-localisation of TRN1 (green) and FUS (red) in neuronal cytoplasmic inclusions (single arrow) and in neuronal intranuclear inclusions (double arrow) in the granule cell layer of the hippocampus (nuclear DAPI in blue). Note that the neuronal nuclei are largely negative for TRN1 (a, d), but they show FUS immunoreactivity (b, e). Bar on a represents 5 μm on all panels
Discussion
In this study of seven aFTLD-U cases, which included two members of the same family and of six NIFID cases, we have shown by immunohistochemistry that TRN1 is abundant in neuronal inclusions in the two main subtypes of FTLD-FUS, NIFID and aFTLD-U. Our western blot studies indicate that the antibody (Abcam, ab10303) used for these immunohistochemical investigations recognises an ~100 kDa band, corresponding to full-length TRN1, and does not cross-react with FUS, which runs as two distinct bands at 53 and 75 kDa in our FUS-opathy cases [12]. Furthermore, our detailed biochemical studies have also demonstrated that highly insoluble TRN1 is present in FTLD-FUS, but not in normal brains. These findings indicate that TRN1 is a reliable marker for inclusions in at least these subtypes of FTLD-FUS, and moreover, it provides a new molecular target for future investigations. Results of our qualitative assessment of tissue sections of the frontal cortex, hippocampus, entorhinal cortex, and wherever available, brainstem and/or spinal cord motor neurons, indicate that TRN1 immunoreactive inclusions occur in all neuron types affected by FUS inclusions and that the TRN1-positive inclusions conform to the morphologies, which have previously been described in aFTLD-U and NIFID using FUS immunohistochemistry [12, 20, 21]. Furthermore, our double-label fluorescence experiments supplemented with morphometry confirm a close co-localisation of TRN1 and FUS in both NCIs and NIIs. In addition to NCIs and NIIs, the TRN1 antibody also strongly labelled fine neuropil threads, coarse neurites, dots and occasional grain-like structures, which are also labelled with the anti-FUS antibody. A dendritic origin of the dots and “grains” is likely as they were readily recognised in the dendritic processes of larger neurons such as those of the cellular islands of the pre-α layer of the entorhinal cortex. This is interesting in view of the observation that FUS has a role in mRNA export and mRNA transport to dendritic spines [6, 7].
The protocol used in this study for the biochemical investigation of TRN1 is based on increasingly insoluble sequential protein extraction and is identical to that previously used for FUS [12, 20]. This approach allowed us to separate HS-soluble, RIPA-SDS-soluble and urea-soluble fractions of TRN1. While previous studies, including our own have showed that FUS is present in all three biochemical fractions in both FTLD-FUS and normal controls [12, 20], the data of our current study indicate that highly insoluble (urea-soluble) TRN1 is only present in FTLD-FUS cases. This finding indicates that the presence of highly insoluble TRN1 may be a robust biochemical marker of both NIFID and aFTLD-U and this notion is further underpinned by the absence of TRN1 immunoreactivity in inclusions of a number of neurodegenerative diseases such as Alzheimer’s disease, multiple system atrophy, Pick’s disease, progressive supranuclear palsy, MND and FTLD-TDP.
Interference with the transport mechanism of nuclear proteins can result in their redistribution into the cytoplasm, which has been strongly implicated in the pathogenesis of both TDP-43 and FUS proteinopathies [11, 23, 28] and there are data indicating that classical NLS-mediated nuclear import may also be disrupted in Alzheimer’s disease [14]. The hypothesis that cytoplasmic redistribution of FUS is central to the pathogenesis of FUS-opathies is supported by a number of observations. Cellular stress disrupts the nuclear transport of FUS by its recruitment into stress granules, markers of which have recently been shown to co-deposit with FUS-positive inclusions in both ALS-FUS and FTLD-FUS [5]. Cell culture experiments, utilizing mutations associated with familial ALS-FUS, have shown that modification of the C-terminal tail of FUS, which contains the NLS, is able to disrupt the normal binding of TRN1. This results in the failure of the cellular machinery responsible for the import of FUS into the cell nucleus with its concomitant cytoplasmic accumulation [5]. The cellular and molecular events triggering aggregation of FUS are not known in sporadic FTLD-FUS, but further detailed biochemical studies could answer whether abnormalities of TRN1 and FUS interactions or involvement of the nuclear pore and RanGTPase could be factors contributing to the disease mechanism. So far no biochemical modifications of the disease-associated FUS have been reported [10, 20, 21, 27], although studies indicate that in vitro, post-translational modifications of FUS such as tyrosine phosphorylation or dimethylation of arginine residues may alter its subcellular localisation [9, 11, 23, 26].
In this immunohistochemical study, we consistently observed strong TRN1 immunoreactivity of the FUS-positive inclusions with co-localisation of these two, functionally closely associated proteins, indicating that it is likely that both FUS and TRN1 are major components of the inclusions in FTLD-FUS. These findings also indicate that a significant amount of TRN1 protein may be sequestered in the neuronal inclusions with a decrease or absence of normal nuclear staining of TRN1 in some nerve cells. This event could, in turn, further impact on the nuclear import of FUS, which may still be available for transport, and compromise the nuclear import of a host of other RNA binding proteins, which are known to be mediated by TRN1 [13]. Therefore, investigation of whether involvement of TRN1 transport mechanism, found in FTLD-FUS, could influence the transport of other relevant proteins should be undertaken. However, recent studies have indicated the possibility of a degree of redundancy in the transportin system. It has been suggested that a splicing variant of transportin2 has a M9 recognition motif with a high degree of similarity to that of TRN1 [24]. In our current study, we were unable to comment on the biology of TRN2b as no suitable antibodies were available. However, we recognise that this avenue of research requires further investigation in order to elucidate the role TRN1 plays in the pathogenesis of FTLD-FUS.
The observation that TRN1 protein becomes insoluble and is incorporated into the inclusions (both nuclear and cytoplasmic) in FTLD-FUS is consistent with a role in inclusion formation. The study by Dormann et al. [5] and the findings of our current investigations implicate the TRN1 transport pathway in the pathogenesis of FTLD-FUS, which presents an interesting window of opportunity for research. Whilst FTLD-FUS represents a relatively small proportion of the total FTLD disease spectrum [25], a better understanding of the pathogenic events resulting in accumulation of FUS and finally nerve cell death in FUS-opathies could also shed light on the role of abnormalities in RNA processing in the wider field of neurodegeneration.
References
Aman P, Panagopoulos I, Lassen C, Fioretos T, Mencinger M, Toresson H, Hoglund M, Forster A, Rabbitts TH, Ron D, Mandahl N, Mitelman F (1996) Expression patterns of the human sarcoma-associated genes FUS and EWS and the genomic structure of FUS. Genomics 37:1–8
Andersson MK, Stahlberg A, Arvidsson Y, Olofsson A, Semb H, Stenman G, Nilsson O, Aman P (2008) The multifunctional FUS, EWS and TAF15 proto-oncoproteins show cell type-specific expression patterns and involvement in cell spreading and stress response. BMC Cell Biol 9:37
Bonifaci N, Moroianu J, Radu A, Blobel G (1997) Karyopherin beta2 mediates nuclear import of a mRNA binding protein. Proc Natl Acad Sci USA 94:5055–5060
Crozat A, Aman P, Mandahl N, Ron D (1993) Fusion of CHOP to a novel RNA-binding protein in human myxoid liposarcoma. Nature 363:640–644
Dormann D, Rodde R, Edbauer D, Bentmann E, Fischer I, Hruscha A, Than ME, Mackenzie IR, Capell A, Schmid B, Neumann M, Haass C (2010) ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J 29:2841–2857
Fujii R, Okabe S, Urushido T, Inoue K, Yoshimura A, Tachibana T, Nishikawa T, Hicks GG, Takumi T (2005) The RNA binding protein TLS is translocated to dendritic spines by mGluR5 activation and regulates spine morphology. Curr Biol 15:587–593
Fujii R, Takumi T (2005) TLS facilitates transport of mRNA encoding an actin-stabilizing protein to dendritic spines. J Cell Sci 118:5755–5765
Josephs KA, Holton JL, Rossor MN, Braendgaard H, Ozawa T, Fox NC, Petersen RC, Pearl GS, Ganguly M, Rosa P, Laursen H, Parisi JE, Waldemar G, Quinn NP, Dickson DW, Revesz T (2003) Neurofilament inclusion body disease: a new proteinopathy? Brain 126:2291–2303
Klint P, Hellman U, Wernstedt C, Aman P, Ron D, Claesson-Welsh L (2004) Translocated in liposarcoma (TLS) is a substrate for fibroblast growth factor receptor-1. Cell Signal 16:515–520
Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, Valdmanis P, Rouleau GA, Hosler BA, Cortelli P, de Jong PJ, Yoshinaga Y, Haines JL, Pericak-Vance MA, Yan J, Ticozzi N, Siddique T, Kenna-Yasek D, Sapp PC, Horvitz HR, Landers JE, Brown RH Jr (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323:1205–1208
Lagier-Tourenne C, Polymenidou M, Cleveland DW (2010) TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum Mol Genet 19:R46–R64
Lashley T, Rohrer JD, Bandopadhyay R, Fry C, Ahmed Z, Isaacs AM, Brelstaff JH, Borroni B, Warren JD, Troakes C, King A, Al-Sarraj S, Newcombe J, Quinn N, Ostergaard, SchrØder, Bojsen-MØller M, Braendgaard H, Fox N, Rossor M, Lees AJ, Holton JL, Revesz T (2011) A comparative clinical, pathological, biochemical and genetic study of fused in sarcoma proteinopathies. Brian. doi:10.1093/brain/awr160 (in press)
Lee BJ, Cansizoglu AE, Suel KE, Louis TH, Zhang Z, Chook YM (2006) Rules for nuclear localization sequence recognition by karyopherin beta 2. Cell 126:543–558
Lee HG, Ueda M, Miyamoto Y, Yoneda Y, Perry G, Smith MA, Zhu X (2006) Aberrant localization of importin alpha1 in hippocampal neurons in Alzheimer disease. Brain Res 1124:1–4
Macara IG (2001) Transport into and out of the nucleus. Microbiol Mol Biol Rev 65:570–594
Mackenzie IR, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J, Kovacs GG, Ghetti B, Halliday G, Holm IE, Ince PG, Kamphorst W, Revesz T, Rozemuller AJ, Kumar-Singh S, Akiyama H, Baborie A, Spina S, Dickson DW, Trojanowski JQ, Mann DM (2010) Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol 119:1–4
Mackenzie IR, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J, Kovacs GG, Ghetti B, Halliday G, Holm IE, Ince PG, Kamphorst W, Revesz T, Rozemuller AJ, Kumar-Singh S, Akiyama H, Baborie A, Spina S, Dickson DW, Trojanowski JQ, Mann DM (2009) Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: consensus recommendations. Acta Neuropathol 117:15–18
Mackenzie IR, Rademakers R, Neumann M (2010) TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol 9:995–1007
Munoz DG, Neumann M, Kusaka H, Yokota O, Ishihara K, Terada S, Kuroda S, Mackenzie IR (2009) FUS pathology in basophilic inclusion body disease. Acta Neuropathol 118:617–627
Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, Mackenzie IR (2009) A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain 132:2922–2931
Neumann M, Roeber S, Kretzschmar HA, Rademakers R, Baker M, Mackenzie IR (2009) Abundant FUS-immunoreactive pathology in neuronal intermediate filament inclusion disease. Acta Neuropathol 118:605–616
Prasad DD, Ouchida M, Lee L, Rao VN, Reddy ES (1994) TLS/FUS fusion domain of TLS/FUS-erg chimeric protein resulting from the t(16;21) chromosomal translocation in human myeloid leukemia functions as a transcriptional activation domain. Oncogene 9:3717–3729
Rappsilber J, Friesen WJ, Paushkin S, Dreyfuss G, Mann M (2003) Detection of arginine dimethylated peptides by parallel precursor ion scanning mass spectrometry in positive ion mode. Anal Chem 75:3107–3114
Rebane A, Aab A, Steitz JA (2004) Transportins 1 and 2 are redundant nuclear import factors for hnRNP A1 and HuR. RNA 10:590–599
Roeber S, Mackenzie IR, Kretzschmar HA, Neumann M (2008) TDP-43-negative FTLD-U is a significant new clinico-pathological subtype of FTLD. Acta Neuropathol 116:147–157
Tan AY, Manley JL (2009) The TET family of proteins: functions and roles in disease. J Mol Cell Biol 1:82–92
Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, Ganesalingam J, Williams KL, Tripathi V, Al-Saraj S, Al-Chalabi A, Leigh PN, Blair IP, Nicholson G, de Belleroche J, Gallo JM, Miller CC, Shaw CE (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323:1208–1211
Wang IF, Wu LS, Shen CK (2008) TDP-43: an emerging new player in neurodegenerative diseases. Trends Mol Med 14:479–485
Weis K (2003) Regulating access to the genome: nucleocytoplasmic transport throughout the cell cycle. Cell 112:441–451
Xu L, Massague J (2004) Nucleocytoplasmic shuttling of signal transducers. Nat Rev Mol Cell Biol 5:209–219
Yang S, Warraich ST, Nicholson GA, Blair IP (2010) Fused in sarcoma/translocated in liposarcoma: a multifunctional DNA/RNA binding protein. Int J Biochem Cell Biol 42:1408–1411
Zinszner H, Sok J, Immanuel D, Yin Y, Ron D (1997) TLS (FUS) binds RNA in vivo and engages in nucleo-cytoplasmic shuttling. J Cell Sci 110:1741–1750
Acknowledgments
The authors wish to thank the MRC London Brain Bank for Neurodegenerative Diseases, Institute of Psychiatry, King’s College, London, UK, NeuroResource, UCL Institute of Neurology, London, UK and the Department of Neuropathology, Århus Kommunehospital, Århus, Denmark for allowing them to investigate some of the cases used in this study. TR, MNR, JLH and AJL are recipients of a research grant from the Alzheimer’s Research UK, which supported this work. JLH is supported by the Reta Lila Weston Institute for Neurological Studies. The Queen Square Brain Bank receives support from the Reta Lila Weston Institute for Neurological Studies and the Progressive Supranuclear Palsy (Europe) Association and is supported by further research grants from the Multiple System Atrophy Trust and Parkinson’s UK. The Dementia Research Centre is an Alzheimer’s Research UK Coordinating Centre. This work was undertaken at UCLH/UCL who received a proportion of funding from the Department of Health’s NIHR Biomedical Research Centres funding scheme.
Author information
Authors and Affiliations
Corresponding author
Additional information
J. Brelstaff and T. Lashley contributed equally to this work.
R. Bandopadhyay and T. Revesz are shared senior authors.
Rights and permissions
About this article
Cite this article
Brelstaff, J., Lashley, T., Holton, J.L. et al. Transportin1: a marker of FTLD-FUS. Acta Neuropathol 122, 591–600 (2011). https://doi.org/10.1007/s00401-011-0863-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-011-0863-6