
Abstract
Whereas the prevalence and impact of vascular pathology in Alzheimer diease (AD) are well established, the role of vascular and Alzheimer pathologies in the progression of neurodegeneration and cognitive impairment in Parkinson disease (PD) is under discussion. A retrospective clinico-pathologic study of 100 patients with autopsy proven PD (including 44 cases with dementia/PDD) and 20 cases of dementia with Lewy bodies (DLB) confirmed essential clinical (duration of illness, Mini-Mental State Examination/MMSE, age at death) and morphologic differences between these groups; Lewy body Braak scores and Alzheimer pathologies (neuritic Braak stage, cortical Aβ plaque load, and generalized cerebral amyloid angiopathy or CAA) were significantly higher/more severe in DLB and PDD than in PD without dementia. Duration of illness showed no association to any of the examined pathologic parameters, while there was a moderate association between LB scores and neuritic Braak stages, the latter significantly increasing with age. Significant association between cerebrovascular lesions and neuritic Braak stage was seen in PDD but not in PD subjects without dementia. These data suggest an influence of Alzheimer-related lesions on the progression of the neurodegenerative process and, in particular, on cognitive decline in both PDD and DLB. On the other hand, both these factors in PD and DLB appear to be largely independent from coexistent vascular pathology, except in cases with severe cerebrovascular lesions or those related to neuritic AD pathology. Assessment of ApoE genotype in a small number of cases showed no significant differences in the severity of Aβ plaque load and CAA except for much lower intensities in non-demented ε3/3 patients. Despite increasing evidence suggesting synergistic reactions between α-synuclein (αSyn), tau and Aβ-peptides, the major protein markers of both AD and Lewy body diseases, and of both vascular pathology and AD, the molecular background and pathophysiological impact of these pathologies on the progression of neurodegeneration and development of cognitive decline in PD await further elucidation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cerebrovascular lesions (CVL) and Alzheimer-related pathology occur frequently in aging brain and coexist with Parkinson disease (PD) [42, 49, 52, 71, 72, 86]. They may be associated with the risk of PD [93] and show synergistic effects on the development of parkinsonism [40, 65], its progression and severity [49, 81], and on cognitive decline in PD patients [16, 31, 52], due to molecular and pathogenic interactions between these pathologies [20, 21, 24–26, 28, 38, 50, 63, 68, 75, 83, 89, 90, 99, 100]. More than one-third of patients with PD and dementia (PDD) display considerable superimposed, neuritic Alzheimer disease (AD) pathology corresponding to Braak and Braak [10] stages 4–6 [48]. A large retrospective study of 617 autopsy-proven cases of PD (Lewy body disease of brainstem type [73]) showed a significantly higher frequency of CVLs than in 535 age-matched controls (44.0 vs. 32.8%) that was only slightly lower than in AD [42]. In DLB, the total frequency of vascular pathology (35%) was almost equal to that in controls and lower than in both PD and AD [41, 42]. On the other hand, cerebrovascular risk factors and small CVLs were not associated with incident dementia in PD except in rare cases with additional subcortical arteriosclerotic encephalopathy (SAE) [40, 65] or mixed dementia (PD + SAE) [35, 40, 48], and the concomitant presence of white matter lesions in patients with PDD had no significant effect on cortical acetyl cholinesterase (AChE) activity [66]. The links between AD and cerebrovascular pathology [6, 18, 44, 103], the association between cognitive impairment and cerebral amyloid angiopathy (CAA) present in up to 100% of AD brains [30, 47, 95], and the influence of AD pathology on the severity and topographic distribution of CAA have been discussed extensively [4, 5]. Similarly, the clinical phenotypes and neuropathology of AD and PD may overlap [5, 22, 24, 33, 50, 67]. Previous autopsy studies reported significantly higher frequency of CAA in PDD than in those without dementia [9, 70] and in DLB with, than without, concomitant AD [101]. A recent autopsy study in PDD cases showed significantly higher neuritic Braak stages (mean 4.2 vs. 2.4; P < 0.01), higher cortical amyloid load and generalized CAA than in PD without dementia (mean grade 1.7 vs. 0.22, P < 0.001), and intermediate changes in DLB (mean Braak score 5.36; mean CAA grade 1.3) suggesting an association of CAA with cognitive decline in both PDD and DLB, particularly in cases with concomitant AD-type pathology [52].
Whereas the prevalence and impact of cerebrovascular pathology in both AD [42, 43, 44, 49, 50]) and PD are well established [41, 49], their contribution to the progression of parkinsonian neurodegeneration and the development of cognitive decline is not well understood.
The present study compares the prevalence of vascular and Alzheimer pathologies in a retrospective series of autopsy-proven PD, PDD, and DLB cases, their interrelationship and impact on disease duration, progression of the neurodegenerative disorder, and development of cognitive decline in Lewy body disease (LBD).
Materials and methods
A consecutive series of 120 patients from the files of the Vienna Neurobiology brain bank (1997–2006) with neuropathologically confirmed PD (LBD of the brainstem type) and DLB (according to current morphological criteria [42, 74]) were evaluated. The clinical data were assessed retrospectively from hospital charts with respect to major clinical symptoms (bradykinesia, rigidity, tremor, posture/gait disorder), the duration of parkinsonian symptoms (available in 100 patients), and the severity of cognitive impairment (moderate to severe dementia with Mini-Mental State Examination (MMSE) < 20). A total of 56 patients were classified as PD without dementia, 44 as PDD, and 20 as DLB. Unfortunately, the duration of dementia was not available in the majority of patients and, therefore, could not be calculated.
Neuropathological assessment was performed according to standardized methods. In 110 cases, both hemispheres fixed in 10% buffered formalin were available for histological examination, while in ten cases only the left hemisphere was formalin fixed and available for histology (the other hemisphere was deep frozen). Paraffin-embedded blocks from multiple brain regions (frontal, temporal, parietal and occipital cortex, limbic system with hippocampus, amygdala and (trans)entorhinal cortex, basal ganglia, brainstem and cerebellum) were examined using routine stains, modified Bielschowsky impregnation, and immunohistochemistry for tau protein (antibody AT-8, Innogenetics, Ghent, Belgium), β-amyloid (clone 4G8, Signet Labs, Dedham, MA, USA), and α-synuclein (αSyn) (monoclonal and polyclonal rabbit antibodies, Chemicon, Temecula, CA, USA and Hofheim, Germany). Each brain was staged for the degree of Lewy body (LB) pathology according to the scheme of Braak et al. [11, 12], for established postmortem criteria for AD, including Braak staging [10, 14], and according to the modified consortium on DLB criteria for the pathological diagnosis of DLB [74]. The Aβ plaque density in frontal and temporal cortex was assessed semiquantitatively and blinded to the clinical and pathological diagnosis by means of both Bielschowsky stain and Aβ immunohistochemistry using four separate scores (0 absence of diffuse Aβ plaques, 1+ sparse diffuse plaques, 2+ moderate numbers of diffuse and/or cored/dense plaques, 3+ frequent plaques), similar to a recent study [58]. The classification of cerebrovascular pathology is given in Table 1 (for methods and classification also see [41–43, 49]). The severity of generalized CAA was semiquantitatively assessed similar to the method described by Olichney et al. [80] (see also [3]).
ApoE genotypes were examined in 14 PD (including 6 PDD), 6 DLB cases, and 47 age-matched controls from deparaffinized cerebellar blocks using polymerase chain reaction (PCR) by enzyme digestions [7].
The statistical method applied for analyzing the associations between neuropathologic and metric (duration of illness, age, etc.) parameters was the non-parametric Kruskal–Wallis (K–W) test, while the Chi-square test was used for comparing ApoE genotypes.
Results
A summary of clinical and neuropathological data is given in Table 2.
PD without dementia
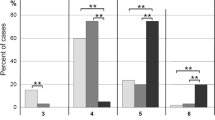
A total of 56 patients (30 female, 26 male), aged 59–91 years (mean 81.8 ± 6.4 SD) at death, met the clinical criteria for sporadic PD without dementia. The duration of illness was 4–30 years (mean 12.2 ± 6.6 SD) (n = 40). The final MMSE was 20–29 (mean 24.7 ± 3.2 SD). Brain weight was 857–1,500 g (mean 1,203 ± 123). At autopsy, PD Braak scores were 3 (n = 7), 4 (n = 34), 5 (n = 15), and none with stage 6 (mean 4.1 ± 1.2 SD; Fig. 1). Neuritic Braak stages were 0–2 (n = 30), 3 (n = 16), 4 (n = 10), and none with Braak stages 5 or 6 (mean 2.2 ± 0.4 SD; Fig. 2). Cortical Aβ plaque load was absent in 25, mild in 8, moderate in 10, and severe in 13 cases (mean 1.2; Fig. 3). Generalized CAA was absent in 48, and mild and moderate in four cases each (mean 0.21; Fig. 4). CVLs were absent in 34, mild in 15, moderate in 2, and severe in 5 brains (mean 0.68; Fig. 5). ApoE genotypes were ε3/3 in all examined eight cases, associated with cortical plaque levels ranging from 0 to 3+ (in only one single case; mean 1.5) and generalized CAA 0 to 2+ (Fig. 6).

Lewy body Braak scores in PD (total), PD without dementia, PDD, and DLB. K–W test: ** P < 0.05, * P < 0.01

Neuritic Braak stages in PD (total), PD without dementia, PDD, and DLB. K–W test: ** P < 0.05, * P < 0.01

Cortical Aβ plaque score in PD (total), PD without dementia, PDD, and DLB. K–W test: ** P < 0.05, * P < 0.01

General CAA score in PD (total), PD without dementia, PDD, and DLB. K–W test: ** P < 0.05, * P < 0.01

Cerebrovascular lesions in PD (total), PD without dementia, PDD, and DLB. K–W test: ** P < 0.05, * P < 0.01

General CAA in cases with different ApoE genotypes (n = 20). Chi-square test: ** P < 0.01
The only significant association was an increase of neuritic Braak stage with age (K–W test, P < 0.05), while it was not associated with CVLs.
PDD
A total of 44 patients (25 female, 19 male), aged between 73 and 96 years (mean 84.2 ± 6.5 SD) at death, met the clinical criteria of sporadic PD with dementia (PDD), initially presenting with motor signs and symptoms, followed by gait disorders and within 4–8 years by cognitive impairment, disorientation, hallucinations, paranoid symptoms, and finally, moderate to severe dementia with MMSE scores (around 6 months prior to death) between 0 and 20 (mean 15 ± 2.2 SD), being lower than in PD with no dementia. Unfortunately, the duration of dementia was not available in most patients. Duration of illness was 6–19 years (mean 6.6 ± 2.1 SD) (n = 40). Brain weight (n = 26) was 830–1,400 g (mean 1,123), which was significantly lower than in PD-no dementia cases (P < 0.01). At autopsy, LB Braak stages were 3 (n = 2), 4 (n = 17), 5 (n = 24), and 6 (n = 1) with a mean of 4.5 ± 0.3 SD (Fig. 1). Neuritic Braak stages ranged from 3 (n = 10) and 4 (n = 18) to 5 (n = 16) (mean 4.2 ± 0.4 SD; Fig. 2). Cortical Aβ plaque load (mean 2.1; Fig. 3) was absent in six, mild in seven, moderate and severe in 22 cases, thus being higher than in the former group. Generalized CAA was absent in 12, mild to moderate in ten and 14, respectively, and severe in eight brains (mean 1.32 ± 0.2 SD; Fig. 4). CVLs were absent in seven, mild in 22, moderate in nine and severe in six brains (mean 1.32 ± 0.3 SD; Fig. 5). There was a significant association between neuritic Braak scores and CVLs (K–W test, P < 0.01) but not between duration of illness or LB Braak scores and any other pathologic lesions.
In summary, PDD patients were significantly older at death, had a shorter duration of illness, and a lower final MMSE score. At autopsy, brain weight was lower, and there was more severe AD pathology (higher Braak stages, more severe cortical Aβ plaque load and CAA), but only slightly higher LB Braak scores that did not correlate with neuritic Braak stages.
ApoE genotypes were ε3/3 in six and ε2/3 in one PDD cases, showing cortical Aβ levels from 0 to 3+ (mean 1.8), generalized CAA negative in two, mild to moderate in three, and severe in only one brain (Fig. 6).
DLB
A total of 20 cases (ten male and female each), aged 65 to 96 years (mean 80.1 ± 4.8 SD) at death, met the morphological criteria of DLB [74]. Duration of illness was 4 to 7.5 years (mean 6.5 ± 1.3 SD) (n = 20). Most patients initially presented with parkinsonian motor signs and symptoms, followed within about 12 months or later by cognitive and psychiatric disorders, with final MMSE scores between 0 and 18 (mean 13.5 ± 3.1 SD), whereas four patients initially presented with cognitive impairment, frontal lobe syndrome, disorientation, depression, delusion, visual hallucinations, after variable periods followed by parkinsonian symptoms.
Brain weight (n = 14) was 1,000–1,500 g (mean 1,223). Neuropathology revealed PD Braak scores of 5 (n = 11) and 6 (n = 9) with a mean of 5.36 ± 0.8 SD (Fig. 1), while neuritic Braak stages were 0–2 (n = 2), 3 (n = 3), 4 (n = 4) and 5 or 6 (n = 11), with a mean of 3.8 ± 0.3 SD (Fig. 2). Thus, 15 cases corresponded to diffuse DLB without definite AD-type pathology, and five (25%) to the LB variant of AD (LBV/AD or DLB + AD [19, 33, 34, 101]). Aβ plaques were absent in frontal and temporal cortex in one single brain (with progressed neocortical LB stage 6), moderate in two, all the others showing severe Aβ plaque load (stage 4 according to [92]) (mean 3.0 ± 0.2 SD; Fig. 3), which, except in two cases with low Braak stages, correlated well with neuritic Braak stages (not shown). CAA was absent in seven brains, five of which were with severe cortical Aβ load, mild to moderate in eight and severe in five brains, either in the occipital meninges or generalized (mean 1.3 ± 0.1 SD; Fig. 4). CVLs, except for moderate to severe forms of generalized CAA (see above), were moderate lacunar state in basal ganglia in two brains (mean 1.1; Fig. 5). There was significant association between neuritic Braak stages and LB Braak scores and between both and generalized CAA but not other CVLs (K–W test, P < 0.01). Thus, DLB patients showed similar duration of illness as PDD, higher LB Braak scores than the two other groups, and similar neuritic Braak scores as PDD, but higher Aβ plaque load than both of the other two groups.
ApoE genotypes were ε3/3 in two and ε2/3 in another case of DLB without AD, with cortical plaque levels of 2+ and ε3/4 in three cases of LBV/AD or DLB + AD (neuritic Braak stages 5 and 6), with cortical Aβ plaque levels of 2+ to 3+ (mean 2.5), one negative and two with severe generalized CAA (mean 2.0; Fig. 6).
Discussion
The present study confirmed previously reported differences between PD patients with and without dementia and DLB: PDD patients were significantly older at death and had a shorter duration of illness [36, 45]. Neuropathology revealed significantly more severe AD-type pathology (both neuritic Braak stages and cortical Aβ plaque load), and generalized CAA, while the LB pathology scores [11] were only moderately increased in PDD brains (28% with score 5 and 6 vs. 17% with score 5; see Table 2). There was a significant increase of neuritic Braak stages with age, but no association between duration of illness and any of the evaluated pathologic parameters. However, there was a relationship between neuritic Braak stages and CVLs, and a moderate one between LB scores and neuritic Braak stage. These data indicate that superimposed AD-pathology in PD increases with age and is significantly more severe in PDD patients than in those without dementia, around two-thirds of them showing severe cortical Aβ plaque load (Table 2). On the other hand, recent longitudinal studies in PD, trying to correlate the pattern of levodopa response and the severity of LB distribution and other pathologies, did not show essential differences in age, duration, and manifestations of end-stage disease, suggesting that various parameters may govern the clinical and pathologic progression of PD, and that “age causes the disease process to gather pace” [56]. Moderate increase in the incidence of progressed LB scores in PDD versus PD in this cohort did not confirm findings, indicating that cognitive decline correlates with neuropathologic LB stage in PD, i.e., the risk of developing dementia increases with disease progression [13, 14], but the precise mechanisms that initiate ΑSyn aggregation and subsequently influence the propagation of the disease are poorly understood [11, 59]. By contrast, the present data indicate an association between cognitive decline and progressing AD pathology [45].
DLB patients in our cohort showed a similar duration of illness but decreased age at death than the PDD group. LB Braak scores were significantly higher than in both PD and PDD brains (5.36 vs. 4.1 and 4.2, respectively; P < 0.01); and neuritic Braak scores were similar to those in PDD but higher than in PD without dementia (mean 3.8 vs. 4.2 and 2.2, respectively; P < 0.01). Cortical Aβ plaque load in DLB brain was higher than in both PDD and PD; general CAA was similar to PDD, but significantly more severe than in PD without dementia (mean 1.4 and 1.5 vs. 0.6, respectively), while CVLs were similar to non-demented PD and less severe than in PDD (Table 2).
On the other hand, there is a positive association between LB score and neuritic Braak stage, suggesting an interaction between both pathologies, in particular between αSyn and tau (for review see [25, 27, 99]). The presence of αSyn-positive lesions in 7–71% of sporadic and familial AD even in the absence of subcortical LBs [2, 32, 68, 84, 97, 98], with involvement of other brain areas [12, 42, 46, 82, 88], the co-localization of tau and αSyn epitopes in LBs [38, 39], as well as clinical, biochemical, and morphological overlap between sporadic PD, DLB, and AD with and without LBs in the amygdala [98], suggest that the process of LB formation is triggered, at least in part, by AD pathology [38, 89]. This collision of two processes may occur in the same brain region or even within a single cell in the human brain [2, 37, 38, 68, 90] and in transgenic mice [64]. Upregulation of the PD-associated protein DJ-1 (PARK7) in tau neuronal and glial inclusions in AD and various other neurodegenerative disorders [57, 78], and the association between dopaminergic neuronal degeneration, accumulation of αSyn and tau or both proteins in LRRK2 mutations [1, 29, 102], induction of hyperphosphorylation of tau by αSyn in the MPTP model of parkinsonism [23], and the association of atypical protein kinase C (aPKC) and phospho-tau or αSyn in fibrillary tangles and LBs [91] as well as the in vitro promotion of tau aggregation by αSyn and vice versa [28], highlight the interface between the two proteins [25, 27]. The frequent relationship between the intensity of both LB and AD lesions suggests that both pathologies independently or synergistically contribute to both movement disorders and cognitive impairment or may have common origin with mutual triggering, but their pathogenic relationship and clinical impact need further clarification. Inflammatory mechanisms common in AD as well as in LB disease may also be driving processes in both disorders and explain the frequent overlap between these diseases [76, 87].
Others have suggested that amyloid rather than tau leads to increased frequency of αSyn pathology, since the latter has been shown to increase with higher density of neuritic plaques [75] and that Aβ enhanced the development of αSyn pathology in PD [83]. Recent studies demonstrated a strong association between Aβ plaque burden and cortical Lewy body density or αSyn load in LB diseases [83] or at least in a subset of PD patients [58]. Further support for this hypothesis comes from the studies in transgenic mice that developed enhanced αSyn (or tau) pathology when they were engineered to also deposit β amyloid [60, 69]. Interactions between Aβ and αSyn may be a molecular mechanism in overlapping pathology of AD and PD in DLB [63]. However, it is unclear whether there is a common underlying pathologic mechanism inducing both neurodegeneration and fibrillary protein aggregation that are typical of two or more different disease processes (double or triple amyloidosis), or if these lesions represent a common final pathology leading to neuronal degeneration.
In the present cohort, significant associations between cortical Aβ plaque load, general CAA, and neuritic Braak stages were observed, but there was no definite interrelation between these pathologies except for an increase of AD-related pathology with age.
A comparison of major clinical and AD-related morphologic changes in a series of 117 autopsy cases of LB-related disorders, AD (without other pathologies), and age-matched controls is given in Table 3. The age at death and the duration of illness did not significantly differ among the groups except for non-demented patients. MMSE scores were lowest in AD and DLB with associated severe AD (LBV/AD), non-significantly higher in PDD, much higher in DLB without severe AD, similar in non-demented PD subjects, and highest in aged controls. Neuritic Braak stages, being highest in AD and LBV/AD, similar in PDD and DLB (without AD), and lowest in non-demented PD patients and controls, correlated well with the level of cognitive impairment. These data also correlate with progressive hippocampal atrophy detected by functional neuroimaging showing a volume pattern PD > PDD > AD [17], with significant amygdalar and hippocampal atrophy in PDD [54], and with MRI studies displaying significant differences in the pattern of brain atrophy between PD, PDD, AD, and controls [15, 77]. A significantly increased plaque load in the cerebral cortex in PDD and DLB compared to PD without dementia is in agreement with previous studies [8, 53, 70], although some authors did not find any correlation between the cortical Aβ load and cognitive impairment in DLB in contrast to AD and vascular dementia [94]. Others observed cortical Aβ deposition in only less than half of the cases of PD and CERAD criteria of definite AD only in 3 out of 13 examined PD brains [72], whereas Aβ-42 plaque levels in AD were not associated with αSyn aggregation [62]. Furthermore, a lack of αSyn increased Aβ plaque accumulation in a transgenic mouse model of AD [55]. While brains of non-demented PD subjects showed either very little or only very rarely severe cortical Aβ load, almost all DLB cases had a large amount of Aβ-42 deposition that was similar in quality and quantity to that seen in AD [20]. The known protein interactions between tau and αSyn [25] are associated with loss of synaptophysin immunoreactivity in DLB, which is, however, less severe than in AD [61].
A significant increase of Aβ load in meningeal and cortical vessels (CAA) in both PDD and DLB cases compared to non-demented PD patients in the present series is also in agreement with previous studies. Mastaglia et al. [70] found deposition of Aβ in meningeal and cortical blood vessels in 38% of PD brains (without evaluation of cognitive state) compared to 25% in age-matched controls. Wu et al. [101] reported CAA in leptomeningeal vessels in 100% of AD, 58% of DLB without coexistent AD, but in 85% of DLB + AD as compared to 50% in age-matched controls. A recent study of 18 PD cases, 12 of which were with PDD, reported neuritic AD-type pathology to be restricted to limbic structures, while CAA was significantly more frequent in PDD than in non-demented PD subjects. An identical immunoreactivity of vascular and parenchymal Aβ deposits indicating similar pathomechanisms in both types of lesion, and an association between CAA and intellectual decline in PD, particularly in cases with concomitant AD-type pathology, were suggested [9].
The prevalence of CVLs ranging from 44 to 58% in non-demented PD subjects ([42]; present cohort) to 94% in PDD cases in the present cohort was significantly higher than in age-matched controls (32.8%), and only slightly lower than in AD [43, 44], while in DLB it ranged from 34.4 [41] to 40%, and thus, was less frequent than in the other disorders. Acute ischemic strokes or hemorrhages, accounting for 4.1% of PD subjects in a previous study [42], were totally absent in the present cohort. There was no association between CVLs and neuritic Braak or LB scores in the group of non-demented PD subjects, whereas it was seen in the PDD group, the latter, however, also showing correlations with increasing age. On the other hand, no relationship between the presence/severity of coexistent CVLs and the duration of illness was seen in this small cohort of PD patients. While the influence of CVLs on AD-associated pathology and related cognitive decline is well documented, the impact of CVLs on the progression of neurodegeneration in PD remains unclear, although several studies imply an association between CAA with cognitive decline in both PDD and DLB, particularly in cases with concomitant AD-type pathology [52].
In contrast to other studies showing increasing severity of CAA in carriers of ApoE ε2/4 and ε4/4 than of ε3/3 and ε2/3 alleles [51, 79, 85, 96], our data on ApoE genotype in a limited number of PD and DLB patients did not show significant differences in the severity of generalized CAA, except for a very low intensity of both lesions in non-demented ε3/3 subjects. The cortical Aβ plaque load of the whole sample had mean levels of 1.6 for ε3/3, 2.0 for ε2/3, and 2.5 for ε2/4 (two cases each), confirming previous studies reporting that mean cortical Aβ plaque density was lower in ε3/3 than in ε2 and ε4 cases [62]. Further studies of ApoE genotypes in PD, PDD, and DLB patients are in progress. In conclusion, this and other recent studies suggested synergistic reactions between αSyn and Aβ peptide as well as αSyn and tau with frequent co-occurrence of these pathologies, but both the molecular background and clinical/pathophysiological impact of these and cerebrovascular pathologies on the progression and development of cognitive impairment in LB disorders remain to be clarified.
References
Adler CH, Grover AC, Sabbagh MN, Caviness JN, Connor DJ, Beach TG (2006) Clinical and pathologic findings in PD with LRRK2 mutations: 2 cases with mild cognitive impairment and small amplitude myoclonus. Mov Disord 21(Suppl 15):S538
Arai Y, Yamazaki M, Mori O, Muramatsu H, Asano G, Katayama Y (2001) Alpha-synuclein-positive structures in cases with sporadic Alzheimer’s disease: morphology and its relationship to tau aggregation. Brain Res 888:287–296
Attems J (2005) Sporadic cerebral amyloid angiopathy: pathology, clinical implications, and possible pathomechanisms. Acta Neuropathol 110:345–359
Attems J, Jellinger KA, Lintner F (2005) Alzheimer’s disease pathology influences severity and topographical distribution of cerebral amyloid angiopathy. Acta Neuropathol 110:222–231
Attems J, Quass M, Jellinger KA, Lintner F (2007) Topographical distribution of cerebral amyloid angiopathy and its effect on cognitive decline are influenced by Alzheimer disease pathology. J Neurol Sci 257:49–55
Bailey TL, Rivara CB, Rocher AB, Hof PR (2004) The nature and effects of cortical microvascular pathology in aging and Alzheimer’s disease. Neurol Res 26:573–578
Bancher C, Egensperger R, Kosel S, Jellinger K, Graeber MB (1997) Low prevalence of apolipoprotein E epsilon 4 allele in the neurofibrillary tangle predominant form of senile dementia. Acta Neuropathol 94:403–409
Barrachina M, Dalfo E, Puig B, Vidal N, Freixes M, Castano E, Ferrer I (2005) Amyloid-beta deposition in the cerebral cortex in Dementia with Lewy bodies is accompanied by a relative increase in AbetaPP mRNA isoforms containing the Kunitz protease inhibitor. Neurochem Int 46:253–260
Bertrand E, Lewandowska E, Pasennik E, Stepien T, Szpak GM, Dymecki J, Wierzba-Bobrowicz T (2007) Cerebral amyloid angiopathy in idiopathic Parkinson’s disease. Acta Neuropathol (in press)
Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82:239–259
Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24:197–211
Braak H, Ghebremedhin E, Rub U, Bratzke H, Del Tredici K (2004) Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res 318:121–134
Braak H, Rub U, Jansen Steur EN, Del Tredici K, de Vos RA (2005) Cognitive status correlates with neuropathologic stage in Parkinson disease. Neurology 64:1404–1410
Braak H, Bohl JR, Muller CM, Rub U, de Vos RA, Del Tredici K (2006) Stanley Fahn Lecture 2005: The staging procedure for the inclusion body pathology associated with sporadic Parkinson’s disease reconsidered. Mov Disord 21:2042–2051
Burton EJ, McKeith IG, Burn DJ, Williams ED, O’Brien JT (2004) Cerebral atrophy in Parkinson’s disease with and without dementia: a comparison with Alzheimer’s disease, dementia with Lewy bodies and controls. Brain 127:791–800
Burton EJ, McKeith IG, Burn DJ, Firbank MJ, O’Brien JT (2006) Progression of white matter hyperintensities in Alzheimer disease, dementia with lewy bodies, and Parkinson disease dementia: a comparison with normal aging. Am J Geriatr Psychiatry 14:842–849
Camicioli R, Moore MM, Kinney A, Corbridge E, Glassberg K, Kaye JA (2003) Parkinson’s disease is associated with hippocampal atrophy. Mov Disord 18:784–790
de la Torre JC (2002) Alzheimer disease as a vascular disorder: nosological evidence. Stroke 33:1152–1162
Del Ser T, Hachinski V, Merskey H, Munoz DG (2001) Clinical and pathologic features of two groups of patients with dementia with Lewy bodies: effect of coexisting Alzheimer-type lesion load. Alzheimer Dis Assoc Disord 15:31–44
Deramecourt V, Bombois S, Maurage CA, Ghestem A, Drobecq H, Vanmechelen E, Lebert F, Pasquier F, Delacourte A (2006) Biochemical staging of synucleinopathy and amyloid deposition in dementia with Lewy bodies. J Neuropathol Exp Neurol 65:278–288
Desai BS, Monahan AJ, Carvey PM, Hendey B (2007) Blood–brain barrier pathology in Alzheimer’s and Parkinson’s disease: implications for drug therapy. Cell Transplant 16:285–299
Dickson DW (2000) Alzheimer–Parkinson disease overlap: neuropathology. In: Clark CM, Trojanowski JQ (eds) Neurodegenerative dementias. McGraw-Hill, New York, pp 247–259
Duka T, Rusnak M, Drolet RE, Duka V, Wersinger C, Goudreau JL, Sidhu A (2006) Alpha-synuclein induces hyperphosphorylation of Tau in the MPTP model of parkinsonism. FASEB J 20:2302–2312
Galasko D, Salmon D (2000) The Alzheimer–Parkinson’s disease connection. In: Clark CM, Trojanowski JQ (eds) Neurodegenerative dementias. McGraw-Hill, New York, pp 229–246
Galpern WR, Lang AE (2006) Interface between tauopathies and synucleinopathies: a tale of two proteins. Ann Neurol 59:449–458
Galvin JE, Pollack J, Morris JC (2006) Clinical phenotype of Parkinson disease dementia. Neurology 67:1605–1611
Geddes JW (2005) Alpha-synuclein: a potent inducer of tau pathology. Exp Neurol 192:244–250
Giasson BI, Forman MS, Higuchi M, Golbe LI, Graves CL, Kotzbauer PT, Trojanowski JQ, Lee VM (2003) Initiation and synergistic fibrillization of tau and alpha-synuclein. Science 300:636–640
Giasson BI, Covy JP, Bonini NM, Hurtig HI, Farrer MJ, Trojanowski JQ, Van Deerlin VM (2006) Biochemical and pathological characterization of Lrrk2. Ann Neurol 59:315–322
Greenberg SM, Gurol ME, Rosand J, Smith EE (2004) Amyloid angiopathy-related vascular cognitive impairment. Stroke 35:2616–2619
Guerini F, Frisoni GB, Bellwald C, Rossi R, Bellelli G, Trabucchi M (2004) Subcortical vascular lesions predict functional recovery after rehabilitation in patients with l-dopa refractory parkinsonism. J Am Geriatr Soc 52:252–256
Hamilton RL (2000) Lewy bodies in Alzheimer’s disease: a neuropathological review of 145 cases using alpha-synuclein immunohistochemistry. Brain Pathol 10:378–384
Hansen L, Salmon D, Galasko D, Masliah E, Katzman R, DeTeresa R, Thal L, Pay MM, Hofstetter R, Klauber M, Rice V, Butters N, Alford M (1990) The Lewy body variant of Alzheimer’s disease: a clinical and pathologic entity. Neurology 40:1–8
Hansen LA (1997) The Lewy body variant of Alzheimer disease. J Neural Transm Suppl 51:83–93
Haugarvoll K, Aarsland D, Wentzel-Larsen T, Larsen JP (2005) The influence of cerebrovascular risk factors on incident dementia in patients with Parkinson’s disease. Acta Neurol Scand 112:386–390
Hughes TA, Ross HF, Mindham RH, Spokes EG (2004) Mortality in Parkinson’s disease and its association with dementia and depression. Acta Neurol Scand 110:118–123
Iseki E, Marui W, Kosaka K, Ueda K (1999) Frequent coexistence of Lewy bodies and neurofibrillary tangles in the same neurons of patients with diffuse Lewy body disease. Neurosci Lett 265:9–12
Iseki E, Togo T, Suzuki K, Katsuse O, Marui W, de Silva R, Lees A, Yamamoto T, Kosaka K (2003) Dementia with Lewy bodies from the perspective of tauopathy. Acta Neuropathol 105:265–270
Ishizawa T, Mattila P, Davies P, Wang D, Dickson DW (2003) Colocalization of tau and alpha-synuclein epitopes in Lewy bodies. J Neuropathol Exp Neurol 62:389–397
Jellinger KA (1996) Parkinsonism due to Binswanger’s subcortical arteriosclerotic encephalopathy. Mov Disord 11:461–462
Jellinger KA (2003) Prevalence of vascular lesions in dementia with Lewy bodies. A postmortem study. J Neural Transm 110:771–778
Jellinger KA (2003) Prevalence of cerebrovascular lesions in Parkinson’s disease. A postmortem study. Acta Neuropathol 105:415–419
Jellinger KA, Attems J (2003) Incidence of cerebrovascular lesions in Alzheimer’s disease: a postmortem study. Acta Neuropathol 105:14–17
Jellinger KA, Mitter-Ferstl E (2003) The impact of cerebrovascular lesions in Alzheimer disease––a comparative autopsy study. J Neurol 250:1050–1055
Jellinger KA, Seppi K, Wenning GK (2003) Neuropathologic changes in Parkinson disease with late onset of dementia. Arch Neurol 60:452–453 author reply 453–454
Jellinger KA (2004) Lewy body-related alpha-synucleinopathy in the aged human brain. J Neural Transm 111:1219–1235
Jellinger KA, Attems J (2005) Prevalence and pathogenic role of cerebrovascular lesions in Alzheimer disease. J Neurol Sci 229–230:37–41
Jellinger KA (2006) The morphological basis of mental dysfunction in Parkinson’s disease. J Neurol Sci 248:167–172
Jellinger KA, Attems J (2006) Prevalence and impact of cerebrovascular pathology in Alzheimer’s disease and parkinsonism. Acta Neurol Scand 114:38–46
Jellinger KA (2007) The enigma of vascular cognitive disorder and vascular dementia. Acta Neuropathol 113:349–388
Jellinger KA, Attems J (2007) Neuropathological evaluation of mixed dementia. J Neurol Sci 257:80–87
Jellinger KA, Attems J (2008) Cerebral amyloid angiopathy in Lewy body disease. J Neural Transm. doi:10.1007/S00702-00007.00856.00708
Jendroska K, Lees AJ, Poewe W, Daniel SE (1996) Amyloid beta-peptide and the dementia of Parkinson’s disease. Mov Disord 11:647–653
Junque C, Ramirez-Ruiz B, Tolosa E, Summerfield C, Marti MJ, Pastor P, Gomez-Anson B, Mercader JM (2005) Amygdalar and hippocampal MRI volumetric reductions in Parkinson’s disease with dementia. Mov Disord 20:540–544
Kallhoff V, Peethumnongsin E, Zheng H (2007) Lack of alpha-synuclein increases amyloid plaque accumulation in a transgenic mouse model of Alzheimer’s disease. Mol Neurodegener 2:6
Kempster PA, Williams DR, Selikhova M, Holton J, Revesz T, Lees AJ (2007) Patterns of levodopa response in Parkinson’s disease: a clinico-pathological study. Brain 130:2123–2128
Kumaran R, Kingsbury A, Coulter I, Lashley T, Williams D, de Silva R, Mann D, Revesz T, Lees A, Bandopadhyay R (2007) DJ-1 (PARK7) is associated with 3R and 4R tau neuronal and glial inclusions in neurodegenerative disorders. Neurobiol Dis 28:122–132
Lashley T, Holton JL, Gray E, Kirkham K, O’Sullivan SS, Hilbig A, Wood NW, Lees AJ, Revesz T (2008) Cortical alpha-synuclein load is associated with amyloid-beta plaque burden in a subset of Parkinson’s disease patients. Acta Neuropathol. doi:10.1007/s00401-00007-00336-00400
Lee VM, Trojanowski JQ (2006) Mechanisms of Parkinson’s disease linked to pathological alpha-synuclein: new targets for drug discovery. Neuron 52:33–38
Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, Yen SH, Sahara N, Skipper L, Yager D, Eckman C, Hardy J, Hutton M, McGowan E (2001) Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 293:1487–1491
Lippa CF (2004) Synaptophysin immunoreactivity in Pick’s disease: comparison with Alzheimer’s disease and dementia with Lewy bodies. Am J Alzheimers Dis Other Demen 19:341–344
Lippa SM, Lippa CF, Mori H (2005) Alpha-Synuclein aggregation in pathological aging and Alzheimer’s disease: the impact of beta-amyloid plaque level. Am J Alzheimers Dis Other Demen 20:315–318
Mandal PK, Pettegrew JW, Masliah E, Hamilton RL, Mandal R (2006) Interaction between Abeta peptide and alpha synuclein: molecular mechanisms in overlapping pathology of Alzheimer’s and Parkinson’s in dementia with Lewy body disease. Neurochem Res 31:1153–1162
Maries E, Dass B, Collier TJ, Kordower JH, Steece-Collier K (2003) The role of alpha-synuclein in Parkinson’s disease: insights from animal models. Nat Rev Neurosci 4:727–738
Mark MH, Sage JI, Walters AS, Duvoisin RC, Miller DC (1995) Binswanger’s disease presenting as levodopa-responsive parkinsonism: clinicopathologic study of three cases. Mov Disord 10:450–454
Marshall GA, Shchelchkov E, Kaufer DI, Ivanco LS, Bohnen NI (2006) White matter hyperintensities and cortical acetylcholinesterase activity in parkinsonian dementia. Acta Neurol Scand 113:87–91
Marti MJ, Tolosa E, de la Cerda A (2007) Dementia in Parkinson’s disease. J Neurol 254(Suppl):41–48
Marui W, Iseki E, Ueda K, Kosaka K (2000) Occurrence of human alpha-synuclein immunoreactive neurons with neurofibrillary tangle formation in the limbic areas of patients with Alzheimer’s disease. J Neurol Sci 174:81–84
Masliah E, Rockenstein E, Veinbergs I, Sagara Y, Mallory M, Hashimoto M, Mucke L (2001) Beta-amyloid peptides enhance alpha-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer’s disease and Parkinson’s disease. Proc Natl Acad Sci USA 98:12245–12250
Mastaglia FL, Masters CL, Beyreuther K, Kakulas BA (1989) Deposition of Alzheimer’s disease amyloid (A4) protein in the cerebral cortex in Parkinson’s disease. Prog Clin Biol Res 317:475–484
Mastaglia FL, Johnsen RD, Kakulas BA (2002) Prevalence of stroke in Parkinson’s disease: a postmortem study. Mov Disord 17:772–774
Mastaglia FL, Johnsen RD, Byrnes ML, Kakulas BA (2003) Prevalence of amyloid-beta deposition in the cerebral cortex in Parkinson’s disease. Mov Disord 18:81–86
McKeith IG, Galasko D, Kosaka K, Perry EK, Dickson DW, Hansen LA, Salmon DP, Lowe J, Mirra SS, Byrne EJ, Lennox G, Quinn NP, Edwardson JA, Ince PG, Bergeron C, Burns A, Miller BL, Lovestone S, Collerton D, Jansen EN, Ballard C, de Vos RA, Wilcock GK, Jellinger KA, Perry RH (1996) Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology 47:1113–1124
McKeith IG, Dickson DW, Lowe J, Emre M, O’Brien JT, Feldman H, Cummings J, Duda JE, Lippa C, Perry EK, Aarsland D, Arai H, Ballard CG, Boeve B, Burn DJ, Costa D, Del Ser T, Dubois B, Galasko D, Gauthier S, Goetz CG, Gomez-Tortosa E, Halliday G, Hansen LA, Hardy J, Iwatsubo T, Kalaria RN, Kaufer D, Kenny RA, Korczyn A, Kosaka K, Lee VM, Lees A, Litvan I, Londos E, Lopez OL, Minoshima S, Mizuno Y, Molina JA, Mukaetova-Ladinska EB, Pasquier F, Perry RH, Schulz JB, Trojanowski JQ, Yamada M (2005) Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 65:1863–1872
Mikolaenko I, Pletnikova O, Kawas CH, O’Brien R, Resnick SM, Crain B, Troncoso JC (2005) Alpha-synuclein lesions in normal aging, Parkinson disease, and Alzheimer disease: evidence from the Baltimore Longitudinal Study of Aging (BLSA). J Neuropathol Exp Neurol 64:156–162
Mrak RE, Griffin WS (2007) Common inflammatory mechanisms in Lewy body disease and Alzheimer disease. J Neuropathol Exp Neurol 66:683–686
Nagano-Saito A, Washimi Y, Arahata Y, Kachi T, Lerch JP, Evans AC, Dagher A, Ito K (2005) Cerebral atrophy and its relation to cognitive impairment in Parkinson disease. Neurology 64:224–229
Neumann M, Muller V, Gorner K, Kretzschmar HA, Haass C, Kahle PJ (2004) Pathological properties of the Parkinson’s disease-associated protein DJ-1 in alpha-synucleinopathies and tauopathies: relevance for multiple system atrophy and Pick’s disease. Acta Neuropathol 107:489–496
Olichney JM, Hansen LA, Galasko D, Saitoh T, Hofstetter CR, Katzman R, Thal LJ (1996) The apolipoprotein E epsilon 4 allele is associated with increased neuritic plaques and cerebral amyloid angiopathy in Alzheimer’s disease and Lewy body variant. Neurology 47:190–196
Olichney JM, Ellis RJ, Katzman R, Sabbagh MN, Hansen L (1997) Types of cerebrovascular lesions associated with severe cerebral amyloid angiopathy in Alzheimer’s disease. Ann N Y Acad Sci 826:493–497
Papapetropoulos S, Lieberman A, Gonzalez J, Mash DC (2005) Can Alzheimer’s type pathology influence the clinical phenotype of Parkinson’s disease? Acta Neurol Scand 111:353–359
Parkkinen L, Soininen H, Alafuzoff I (2003) Regional distribution of alpha-synuclein pathology in unimpaired aging and Alzheimer disease. J Neuropathol Exp Neurol 62:363–367
Pletnikova O, West N, Lee MK, Rudow GL, Skolasky RL, Dawson TM, Marsh L, Troncoso JC (2005) Abeta deposition is associated with enhanced cortical alpha-synuclein lesions in Lewy body diseases. Neurobiol Aging 26:1183–1192
Popescu A, Lippa CF, Lee VM, Trojanowski JQ (2004) Lewy bodies in the amygdala: increase of a-synuclein aggregates in neurodegenerative diseases with tau-based inclusions. Arch Neurol 61:1915–1919
Premkumar DR, Cohen DL, Hedera P, Friedland RP, Kalaria RN (1996) Apolipoprotein E-epsilon4 alleles in cerebral amyloid angiopathy and cerebrovascular pathology associated with Alzheimer’s disease. Am J Pathol 148:2083–2095
Reitz C, Trenkwalder C, Kretzschmar K, Roesler A, Eckardstein VA, Berger K (2006) Relation of cerebral small-vessel disease and brain atrophy to mild Parkinsonism in the elderly. Mov Disord 21(11):1914–1919
Rogers J, Mastroeni D, Leonard B, Joyce J, Grover A (2007) Neuroinflammation in Alzheimer’s disease and Parkinson’s disease: are microglia pathogenic in either disorder? Int Rev Neurobiol 82:235–246
Saito Y, Kawashima A, Ruberu NN, Fujiwara H, Koyama S, Sawabe M, Arai T, Nagura H, Yamanouchi H, Hasegawa M, Iwatsubo T, Murayama S (2003) Accumulation of phosphorylated alpha-synuclein in aging human brain. J Neuropathol Exp Neurol 62:644–654
Saito Y, Ruberu NN, Sawabe M, Arai T, Kazama H, Hosoi T, Yamanouchi H, Murayama S (2004) Lewy body-related alpha-synucleinopathy in aging. J Neuropathol Exp Neurol 63:742–749
Schmidt ML, Martin JA, Lee VM, Trojanowski JQ (1996) Convergence of Lewy bodies and neurofibrillary tangles in amygdala neurons of Alzheimer’s disease and Lewy body disorders. Acta Neuropathol 91:475–481
Shao CY, Crary JF, Rao C, Sacktor TC, Mirra SS (2006) Atypical protein kinase C in neurodegenerative disease II: PKCiota/lambda in tauopathies and alpha-synucleinopathies. J Neuropathol Exp Neurol 65:327–335
Thal DR, Rub U, Schultz C, Sassin I, Ghebremedhin E, Del Tredici K, Braak E, Braak H (2000) Sequence of Abeta-protein deposition in the human medial temporal lobe. J Neuropathol Exp Neurol 59:733–748
Thanvi B, Lo N, Robinson T (2005) Vascular parkinsonism––an important cause of parkinsonism in older people. Age Ageing 34:114–119
Thomas A, Ballard C, Kenny RA, O’Brien J, Oakley A, Kalaria R (2005) Correlation of entorhinal amyloid with memory in Alzheimer’s and vascular but not Lewy body dementia. Dement Geriatr Cogn Disord 19:57–60
Tian J, Shi J, Mann DM (2004) Cerebral amyloid angiopathy and dementia. Panminerva Med 46:253–264
Trembath D, Ervin JF, Broom L, Szymanski M, Welsh-Bohmer K, Pieper C, Hulette CM (2007) The distribution of cerebrovascular amyloid in Alzheimer’s disease varies with ApoE genotype. Acta Neuropathol 113:23–31
Trembath Y, Rosenberg C, Ervin JF, Schmechel DE, Gaskell P, Pericak-Vance M, Vance J, Hulette CM (2003) Lewy body pathology is a frequent co-pathology in familial Alzheimer’s disease. Acta Neuropathol 105:484–488
Uchikado H, Lin WL, DeLucia MW, Dickson DW (2006) Alzheimer disease with amygdala Lewy bodies: a distinct form of alpha-synucleinopathy. J Neuropathol Exp Neurol 65:685–697
Wenning GK, Jellinger KA (2005) The role of alpha-synuclein and tau in neurodegenerative movement disorders. Curr Opin Neurol 18:357–362
Wirths O, Bayer TA (2003) Alpha-synuclein, Abeta and Alzheimer’s disease. Prog Neuropsychopharmacol Biol Psychiatry 27:103–108
Wu E, Lipton RB, Dickson DW (1992) Amyloid angiopathy in diffuse Lewy body disease. Neurology 42:2131–2135
Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P, Asmus F, Muller-Myhsok B, Dickson DW, Meitinger T, Strom TM, Wszolek ZK, Gasser T (2004) Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44:601–607
Zlokovic BV (2004) Clearing amyloid through the blood–brain barrier. J Neurochem 89:807–811
Acknowledgments
The authors are indebted to the colleagues of the clinical departments and Institute of Pathology of OWS Hospital (head: Prof. Dr. F. Lintner), Vienna, Austria, for the clinical and pathological data and the brain material; to Mrs. Veronika Rappelsberger for excellent laboratory work, and to Mr. Erich Mitter-Ferstl, Ph.D., for secretarial work. The study was supported in part by the Society for the Support of Research in Experimental Neurology, Vienna, Austria.
Author information
Authors and Affiliations
Corresponding author
Additional information
Dedicated to the memory of Professor Dr. Franz Seitelberger, a pioneer of modern neuropathology and neurosciences.
Rights and permissions
About this article
Cite this article
Jellinger, K.A., Attems, J. Prevalence and impact of vascular and Alzheimer pathologies in Lewy body disease. Acta Neuropathol 115, 427–436 (2008). https://doi.org/10.1007/s00401-008-0347-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-008-0347-5