
Abstract
The prevalence, morphology and pathogenesis of vascular dementia (VaD), recently termed vascular cognitive impairment, are a matter of discussion, and currently used clinical diagnostic criteria show moderate sensitivity (average 50%) and variable specificity (range 64–98%). In Western clinic-based series, VaD is suggested in 8–10% of cognitively impaired aged subjects. Its prevalence in autopsy series varies from 0.03 to 58%, with reasonable values of 8–15%, while in Japan it is seen in 22–35%. Neuropathologic changes associated with cognitive impairment include multifocal and/or diffuse disease and focal lesions: multi-infarct encephalopathy, white matter lesions or arteriosclerotic subcortical (leuko)encephalopathy, multilacunar state, mixed cortico-subcortical type, borderline/watershed lesions, rare granular cortical atrophy, post-ischemic encephalopathy and hippocampal sclerosis. They result from systemic, cardiac and local large or small vessel disease. Recent data indicate that cognitive decline is commonly associated with widespread small ischemic/vascular lesions (microinfarcts, lacunes) throughout the brain with predominant involvement of subcortical and functionally important brain areas. Their pathogenesis is multifactorial, and their pathophysiology affects neuronal networks involved in cognition, memory, behavior and executive functioning. Vascular lesions often coexist with Alzheimer disease (AD) and other pathologies. Minor cerebrovascular lesions, except for severe amyloid angiopathy, appear not essential for cognitive decline in full-blown AD, while both mild Alzheimer pathology and small vessel disease may interact synergistically. The lesion pattern of “pure” VaD, related to arteriosclerosis and microangiopathies, differs from that in mixed-type dementia (AD with vascular encephalopathy), more often showing large infarcts, which suggests different pathogenesis of both types of lesions. Due to the high variability of cerebrovascular pathology and its causative factors, no validated neuropathologic criteria for VaD are available, and a large variability across laboratories still exists in the procedures for morphologic examination and histology techniques.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Historical background and synonyms
While Alzheimer disease (AD) has become accepted as the most common cause of dementia in advanced age [268], the role of cerebrovascular disease (CVD) and ischemic brain damage for cognitive decline remains controversial and confusing. Until the 1950s and 1960s, dementia in elderly subjects was usually labeled “arteriosclerotic dementia” [319], originally conceived as an arteriosclerotic disorder arising from diffuse low cerebral perfusion [35, 274], and in 1919, Mingazzini [334] stated that this was the result of cerebral infarction, similar to the concept stressed by Fisher [163], for rev. see [423]. Tomlinson et al. [486] described the relationship between the volume of infarcted tissue and cognitive impairment, suggesting that destruction of large volumes of cortex is necessarily followed by dementia, whereas subtle cerebrovascular lesions (CVL) may or may not contribute to dementia, probably depending on their location. Hachinski et al. [196] criticized this term as both misleading and inaccurate, and coined the term “multi-infarct dementia” (MID) due to the accumulation of cerebral infarct volumes in excess of a critical threshold. Because this phenotype constitutes only a small subdivision of dementias of suggested ischemic–vascular etiology, the term “vascular dementia” (VaD) [119, 142, 291, 311, 312, 326, 332, 345, 367] and others were chosen (Table 1). Later, they were replaced by “vascular cognitive impairment” (VCI) to acknowledge that the cognitive effects of CVD may be significant, but fall short of a true dementia syndrome as defined by impairment of activities of daily living (ADL) [19, 54–58, 194, 245–247, 313, 366, 418, 460]. Others proposed the term “vascular cognitive disorder” (VCD) as a global diagnostic category, ranging from VCI to VaD [426, 430]. VCD has been recently limited to cases without dementia, i.e., cognitive impairment-no dementia (VCI-ND), but there are no universally accepted criteria for VCI [53, 56, 57, 365, 426]. Other subgroups include post-stroke dementia (PSD) [185], subcortical vascular dementia [138] and mixed AD plus CVD [326].
Although considerable progress has been made in understanding VaD, and clinical diagnostic criteria have been proposed, many questions remain open, particularly when pathologic lesions produce cognitive impairment and by what mechanisms.
Many authors consider VaD to be an ill-defined entity [118, 340]. Causal relationships between CVD and dementia are difficult to prove and morphologic substrates of cognitive impairment resulting from CVLs remain confusing, since they constitute a multifactorial disorder related to variable types, sizes and location sites of the lesions which are due to a large variety of causes [142, 143, 241, 242, 245, 246, 311, 312, 333]. A list of potential etiologies capable of producing VCD has been given [426]. Even when cerebrovascular pathology appears to be a main underlying process, the effect of damaged brain parenchyma is variable and forms a continuum of abnormalities that also can occur in the absence of dementia [154, 246, 248, 509]. Therefore, the clinical, neuroimaging and pathologic appearances may be heterogeneous; no single set of criteria proposed for the clinical diagnosis of VaD has been generally accepted (see [88]), and the phenotype remains elusive [304], making epidemiologic and treatment studies of VaD/VCI challenging. Complicating its diagnosis are other frequently co-existing pathologic entities, in particular Alzheimer-type lesions, in the aging brain [88, 97, 154, 225, 249, 255, 361, 390, 535]. This review will critically discuss current problems of the pathology and pathogenesis of acquired forms of VCD and VaD. The hereditary forms of VaD (see [257]) and the problems of mixed dementia (AD + vascular encephalopathy) have been reviewed recently (see [251]).
Clinical diagnostic criteria
Standard diagnostic criteria and assessment procedures for dementia have been published in a Practice Parameter by the American Academy of Neurology (AAN) [266] and by the Clinical Practice Committee of the American Geriatrics Society [1]. While AD can today be diagnosed with a high degree of accuracy (see [44, 175, 214, 261, 262, 266, 344]), agreement on clinical definitions of VaD is incomplete. There is no singular or specific cognitive impairment that is characteristic of VaD, and clinical criteria for VCI or VaD in general do not specify a particular neuropsychological profile.
In the preimaging era, the Hachinski Ischemic Score relied on a number of vascular signs and symptoms to gauge the likelihood of VaD versus AD [197]. Subsequent studies have shown that this score has a rather poor correlation with neuropathologic diagnosis and can be regarded as only a rough indicator of underlying pathology [161, 343]. Even the addition of CT scanning to the Score does not greatly increase its sensitivity [549]. Currently used clinical diagnostic criteria for VaD/VCI use variable data and consider variable pathogenic mechanisms and causal relationships [13, 86, 87, 197, 420, 528, 536].
All these criteria based their definition of dementia on the fact that memory deficits must be present. However, cerebral infarcts can occur in any brain territory and do not always affect memory-related circuits [42] or cause impairments in social or occupational functioning. The problem of the memory-based definition of dementia is not exclusive of VaD, but it excludes the main possible cognitive sepueloe of stroke, which in the ICD-10 classification are properly categorized as VCI [376, 528]. Some attempts to correct this problem have been made. For instance, the Cardiovascular Health Study (CHS) [303] used a definition of dementia that did not necessarily include memory loss, but a progressive or static cognitive deficit of sufficient severity to affect the subject’s ADL, and a history of normal intellectual function before the onset of cognitive abnormalities.
Modifications have been proposed to existing diagnostic criteria: VCI/VaD should be subdivided into those associated with focal neurological lesions, usually MID [196, 486], and a more diffuse disorder of “subcortical vascular dementia” (SVaD) as a clinically and pathologically homogenous syndrome, with only minimal or absent regional cerebral infarction and arising from small vessel disease (SVD) [138, 401, 424]. Integration of neuropsychological and neuroimaging data was suggested to be sufficient for the diagnosis of this more homogenous subtype [11, 401]. Regional cerebral blood flow (CBF) in subcortical and limbic areas is significantly decreased in SVaD compared to controls [449]. Further criteria have been suggested for Binswanger’s syndrome [40, 300] and for early changes of VaD termined VCI [56, 57, 194].
The newer criteria share three components: (1) evidence of dementia or cognitive impairment; (2) evidence of CVD by history or neuroimaging (CT or MRI) and (3) evidence of a relationship between cognitive impairment and CVD, e.g., temporal relationship [176, 268]. Unfortunately, these criteria are not interchangeable and may result in up to threefold differences in the number of cases classified clinically as VaD [88, 395], and may exclude a number of subjects with VaD [470]. A recent longitudinal study of VaD showed both clinical similarities and differences between pathologically defined subgroups [15]. VCI impairment harmonization standards have been published recently by the NINDS and the Canadian Stroke Network (CSN) [195].
Clinico-pathologic studies
Several class I and II studies that compared clinical diagnosis and neuropathologic findings in reference cohorts, similar to population-based studies, reported low sensitivity of clinical criteria (average 50%, range 20–89%), but high specificity (average 87%, range 64–98%), with variable interrater reliability [95, 169, 182, 183, 268, 291, 416]. A critical discrepancy among diagnostic criteria is the difference in estimating the severity of cognitive impairment necessary to make the diagnosis of dementia. Comparison of different clinical criteria for VaD showed 52% of stroke patients to meet the DSM-IV criteria for VaD, 33% of ICD-10, 27% of ADDTC and 14% of NINDS-AIREN criteria [528], while in Japan only 14% met the NINDCS-AIREN and 31% the ADDTC criteria [321]. In the CHS none of the clinical criteria for VaD identified the same group of subjects with incidental dementia—VaD classification by NINDS-AIREN, DSM-IV and ADDTC criteria was 9, 13 and 24%, respectively, and the majority of the VaD cases had AD clinical features [303]. In general, the NINDS-AIREN criteria were the most conservative ones. However, in an interobserver study, use of the operational definitions for these criteria improved agreement but only for already experienced observers [502]. Most of these figures do not take into account the more prevalent and subclinical type of cerebrovascular events, characterized by multiple or strategic lacunar infarcts and diffuse WMLs linked with small vessel dementia (SVD) [248, 383, 455]. The Mayo clinical criteria (temporal relationship between stroke and dementia or worsening of cognition, or bilateral infarction in specific regions) had 75% sensitivity and 81% specificity for autopsy-proven VaD [268]. Reclassification of 308 dementia cases from the population-based Kungsholmen Project, using vascular risk factors retrospectively, reclassified only 47% of the suggested AD cases as “pure” AD. About 26% of pure AD subjects developed a vascular disorder in the following 3 years, and among subjects with AD and a vascular component, CVD was the most common (40%) [2]. Evaluation of 3.375 participants of the CHS using MRI classified 44% of 480 incident dementia cases as possible or probable VaD by ADDTC, but there was a substantial overlap between cases classified as AD and VaD suggesting a high prevalence of mixed dementia [279]. CVD is a coincident finding in 30 to 40% of patients with autopsy-proven AD and is associated with a significantly greater likelihood of dementia for a given level of AD pathology [251, 404, 443, 444], but pathologic evidence of microvascular injury may be as frequent as Alzheimer pathology in select postmortem samples [529, 549, 550].
Accuracy of clinical diagnosis of VaD has been reported in 89 autopsy cases, in which VaD was defined pathologically by the presence of cortical infarcts in at least three areas but without subcortical lesions [183]. Using these criteria as reference standard, the clinical criteria for VaD tended to be specific but insensitive, suggesting that they underestimate the extent of ischemic brain injury. Among 110 autopsy cases, 32.8% were confirmed VaD, 42% of which had not presented with stroke, while 30% of morphologic mixed dementia cases had been clinically diagnosed as VaD by both NINDS-AIREN and ADDTC criteria [25].
In a retrospective study of consecutive autopsies of 1,050 demented patients (aged over 55 years) in Vienna, Austria, post-mortem confirmation of the clinical diagnosis of possible/probable AD was achieved in 93%, of mixed dementia in 60%, and of VaD in 52.3% [250]. Among 180 long-term followed patients in the Vienna Prospective Dementia study [30], the clinical diagnosis of AD was confirmed at autopsy in 90% (one-third each associated with vascular or Lewy pathologies) and that of VaD in only 53.3% (Jellinger, unpublished), while in other autopsy series the sensitivity for VaD ranged from 25 to 44% [513, 531].
Admittedly, the presence of CVLs found by neuroimaging techniques or autopsy does not prove that they definitely cause dementia [136, 291, 311, 374], and no VCI subtype was associated with a specific neuroimaging abnormality [418]. Therefore, the clinical diagnosis of VCI/VaD remains subjective and problematic. Controversies concerning the validity and reliability of diagnostic criteria for VaD/VCI continue, and the lack of neuropathologic criteria undermines the attempts to improve clinical and imaging criteria, and the importance of further clinico-pathologic correlations has been emphasized [136, 248, 250, 268, 291, 366].
Post-stroke dementia
Román [422] recently suggested that VaD may be the most underdiagnosed and underestimated form of dementia in the elderly. This is important because “silent” cerebral infarcts increase with advancing age and are considered major contributors to the increasing incidence of cognitive impairment [267, 506], occurring in 25–30% of patients after stroke [120, 185, 212], associated with a twofold increase in odds of dementia [293, 443]. Other recent studies confirmed that incident stroke significantly increased risk of dementia, but did not find an effect of CVD risk factors on dementia risk [170, 409]. While not all stroke patients with VCI develop dementia according to standard diagnostic criteria, such patients are at risk of a dementia syndrome in the next 3 years after stroke [212, 473], and 20–41% of them progress to cognitive impairment or dementia [101, 233, 339, 461, 527, 554]. Cognitive impairment can develop after a single ischemic lesion, after recurrent transient ischemic attacks [26] or after multiple insults, while some patients may manifest progressive cognitive decline associated with multiple “silent” ischemic lesions [143, 220, 296], and the presence of an APOE ɛ4 and ɛ2 allele is associated with greater progression of cognitive decline [28]. It is also associated with asymptomatic high-grade stenosis of the left internal carotid artery without clinically evident CVD [252]. The role of diffuse PSD has been proposed [379], although many patients with dementia identified after stroke already had cognitive impairment before [170, 212, 292]. During a 4-year follow-up the incidence of PSD increased gradually [10], but many patients with lacunar infarcts have a good functional outcome after 5 years. For older patients and those with an initial severe stroke or with additional vascular risk factors, however, the prognosis is more severe, with an increased risk for mortality, stroke recurrence and physical and cognitive decline [16, 17, 293, 307]. Dementia showed a correlation with widespread small ischemic lesions throughout the CNS [111], mainly lacunes, microinfarcts and hippocampal injury, and much less with larger infarcts; many brains showed more than one type of CVLs, although in cognitively normal aged controls similar lesions were present [102, 108, 241, 242, 509].
Because the criteria chosen to diagnose VaD will influence estimates on its incidence and prevalence, as well as its recognition and treatment, new research criteria have been proposed [136–138, 248, 250, 291, 366, 368], and the need for prospective, multi-institutional, population-based studies evaluated thoroughly and longitudinally by clinical, neuropsychological and imaging studies, with autopsy follow-up has been emphasized [193, 266, 268].
Pathologic diagnostic criteria of VaD
In contrast to recently refined morphologic criteria for AD and other neurodegenerative dementias (see [129, 221, 222, 250, 260, 318, 335, 347, 352, 363, 490]), no generally accepted neuropathologic criteria for VaD/VCI have been established up to date and no definite morphologic substrates have been included in the currently used clinical diagnostic criteria for VaD. Two important challenges exist. First, there is no accepted neuropathologic scheme for quantitating CVD in cognitive disturbances, and second, agreement on clinical definitions of VaD is limited. The ADDTC criteria did not suggest specific details for the post-mortem diagnosis of VaD but indicated that histopathologic examination of the brain with clinical evidence of dementia was necessary to confirm the presence of multiple infarcts [86]. The NINDS-AIREN criteria emphasized the heterogeneity of the VaD syndrome and its pathologic subtypes [420]. The diagnosis of “definite” VaD criteria required that the clinical probability be completed by histopathologic evidence of CVD and absence of Alzheimer-type lesions exceeding those expected for age and other conditions causing dementia. Several investigators have emphasized the inverse relation between Braak stage of neuritic Alzheimer lesions and cerebrovascular pathology [186, 239] or have used variable criteria [134, 137, 236, 268, 495].
VaD is related to a variety of pathologic lesions, the clinical significance of which and their relation to AD and other age-related changes of the brain, e.g. subcortical WMLs, remain controversial [130, 131, 136, 137, 142–144, 234, 241, 242, 246, 248, 291, 311, 312, 332, 382, 394, 454, 509]. On the other side, many elderly patients exhibit morphologic changes similar to AD, VaD or mixed dementia (MD), but do not meet the clinical criteria of dementia [179]. We are not aware of any validation study of the currently used criteria for neuropathologic diagnosis of VaD/VCI or published neuropathology data about the substrates of PSD.
The task of neuropathology is to describe the nature and severity of vascular pathology using harmonized morphologic procedures and criteria—as recently published for the assessment of AD-related lesions [8]—addressing the question, whether the CVLs present in a particular brain are of sufficient magnitude to likely contribute or are even the sole substrate of the profile which was demonstrated clinically. A proposal for the assessment of key variable to define the pathology of VaD has been made by the Newcastle group (Table 2), but the existing concepts of VaD present difficulties in generalizing clinico-pathologic correlations from patient to patient [226]. In addition, the appreciation of the presence and extent of morphologically verified vascular lesions in cognitively impaired patients may be influenced by the heterogeneity of the criteria for commonly found brain lesions and their interpretation applied in different centers, as recently shown by the large variability of answers in a semistructured questionnaire among 13 neuropathologic centers around the world [384]. The difficulty of accurately undertaking this task is formidable.
In contrast to inter-observer validation associated with pathology protocols for AD [8, 128, 201, 335–357, 389] there are no criteria, validated across different laboratories of cerebral vascular pathologies, nor for synthesizing a global “vascular pathology score” where multiple pathologies are present. The frequency of specific morphologic features of VCI depends largely on study inclusion criteria [447], and neuropathologic confirmation of a clinical diagnosis of VaD/VCI therefore remains largely subjective. It must be kept in mind that VCI/VaD has a broad spectrum of CVLs of heterogeneous pathophysiology that may result in cognitive decline, developing consensus neuropathologic criteria will be difficult.
Prevalence and epidemiology
Estimates of incidence and prevalence of VaD are affected by three major factors: choice of diagnostic criteria, availability of brain imaging and confirmation by autopsy. There is considerable lack of agreement about VaD epidemiology and prevalence. Given the difficulties in diagnosing this disorder, considerable methodologic and geographic differences, epidemiologic studies must be interpreted cautiously. Although VaD was previously considered the second most common type of dementia after AD [142, 168, 208, 253, 328, 408, 415], and is the second leading cause of death worldwide [422], in the Western world it follows AD (60–70%), dementia with Lewy bodies (DLB) (10–25%) and other non-Alzheimer dementias (8–10%) at place 3 or 4 [242, 248, 406]. A review of clinical studies showed a frequency of VaD ranging from 4.5 to 39% [258], but in most Western memory clinic-based series, it is diagnosed in about 8–10% [136, 248], with age-standardized incidence ratios (SIR), varying from 0.42 to 2.68.
Evaluation of 11 pooled European population-based studies of subjects over age 65 revealed an age-standardized prevalence of 6.4% for all dementias, 4.4% for AD and 2.6% for VaD [299]; VaD accounted for 15.8% of all dementia cases. In Canadian clinical studies, 12.1% had VaD and 12.8% mixed AD/VaD [417]. The incidence of VaD varies between 6 and 15/year/1,000 persons aged 70 and older, and increases with advancing age [209]. Among US Medicare beneficiaries VaD rose form 1991 to 1999 from 43 to 144/1000 among whites compared to 52 to 161/1000 among African Americans (3.1 to 3.3 times increase) [477]. Studies from Japan revealed that the prevalence of VaD was more than double that of AD [205, 223, 293, 494, 544], while in others, AD was two times more frequent than VaD [83], from 12 to 23% compared to 47% for AD and 22% for dementia with Lewy bodies [406].
A review of pathologic studies on the prevalence of VaD is difficult because most studies may contain referral biases because they are weighted with patients from clinical centers, where AD predominates, e.g., the CERAD group [220, 451]. The divergences in estimates of prevalence [33, 53, 299, 506] and incidence of VaD [120, 223] suggests that the concept of VaD needs further validation.
A review of autopsy studies of 2,784 patients with dementia between 1962 and 1990 revealed an overall mean risk of VaD (range 2.0–85.2%); between 1962 and 1995 the overall risk was 11.3% [311], while others classified 15–19% as “pure” VaD [337] or showed even a wider range between 9.0 and 85.2%, with a mean of 17.9% [258]. Other reports showed a prevalence of 2–9%, while in the CERAD, the Nun Study and the Florida Brain Bank series, pure VaD without other pathologies was seen in only 0.03 and 2.5%, respectively [33, 220, 455, 456]. Among 3,438 autopsy cases between 1991 and 2003, the prevalence of VaD ranged from 0.03 to 35% with a mean of 11.6%, and in a smaller community-based autopsy sample of dementia "pure" VaD was seen in 7% [414] (Table 3). For comparison, in recent autopsy series of subjects with dementia from Japanese geriatric hospitals, the incidence rates for AD, VaD and mixed or other dementias were 34, 35, 11 and 20%, respectively, in one [448] and 42, 22, 6 and 26%, respectively, in the other series [3]. Of 650 demented patients diagnosed during life as having AD, at autopsy 505 (78%) had AD; only 390 (60%) of these had AD as the only neuropathologic condition. Of the remaining 22% with no evidence of AD, 39 had Parkinson disease (PD), 25 nonspecific degenerations, 15 Pick disease, 14 multiple infarcts and 11 lacked any neuropathologic abnormality [324]. Evaluation of 363 autopsy cases of the updated Honolulu-Asia Aging Study (HAAS) showed a low correspondence between clinical and neuropathologic diagnosis, with 56% diagnosed as probable or possible AD but only 19% having neuritic plaques or NFTs as the sole or predominant dementia-related morphologic lesions. Although 16% were attributed to mixed cases during life, almost 40% were found to have significant mixtures of dementia-related lesions at autopsy [390].
Among 180 autopsy cases of the Religious Order Study (60 not cognitive impaired, 37 MCI, 83 demented), almost all patients had at least some AD pathology; cerebral infarcts were present in 35.2%, and LBD in 15.6%. Persons with MCI had intermediate levels of both AD pathology and cerebral infarctions from those without cognitive impairment and those with dementia, suggesting that MCI is related to both pathologies [41]. Neuropathologic examination of 206 demented subjects revealed AD with Lewy bodies (LBs) in 38%, AD with vascular lesions in 25%, “pure” AD and AD with LBs (with or without vascular lesions) in 13% each and 8% for pure vascular lesions [491]. In centenarians, cerebral infarcts and lacunes are frequent and are responsible, at least in part, for the high proportion of cognitive dysfunctions in these patients [207].
In a recent prospectively followed series of community-dwelling individuals involved in the Medical Research Council Cognitive Function and Ageing Study (MRC-CFAS), up to 78% of the participants presented with cerebrovascular pathology and 70% with AD-pathology upon autopsy, vascular lesions being as common in demented and nondemented individuals, although multiple CVLs were more frequent in the former group [154, 361]. An update of 456 autopsies in 41% of demented cases showed moderate or severe neurofibrillary tangles (NFTs) compared to 4% in nondemented people, 27% of whom had severe cortical neuritic plaque and 1/3 had some neocortical NFT pathologies. SVD was the most frequent lesion and mixed pathology was much more common than pure AD or vascular disease, and incidental WMLs were common in −67 to 90% [227].
A stronger correlation exists between memory impairment and NFTs [46, 322, 402] and vascular lesions [52] than with amyloid [228, 269, 479], with notable exceptions showing correlation of entorhinal Aβ42 with memory in both AD and VaD but not in DLB [481]. Circle of Willis atherosclerosis was found to be more severe in subjects with VaD and AD than in controls, while it was equivalent between elderly controls and subjects with non-AD dementia. Increasing atherosclerotic grade increased the Odds ratios (OR) for the diagnosis of both AD and VaD [39].
In a retrospective study of 1,500 consecutive autopsies of demented patients with a mean age of 83.3 ± 6.0 years in Vienna, Austria, “pure” VaD was seen in 10.8% but only in 2.2% of 850 patients with the clinical diagnosis of possible or probable AD. Alzheimer pathology was present in 40.0 and 50.7%, respectively, whereas 20–25% showed coexisting CVLs and 9% Lewy body pathology. Other disorders (Creutzfeld-Jakob disease/CJD, PD, etc.) were seen in 4.5 and 3.3%, respectively, while in 1% a morphologic basis for dementia was not found [250]. In a prospective clinico-pathologic study [30] of 180 demented patients, the prevalence of “pure” VaD was 8% compared to 46% AD, 24% AD plus minor CVD, 10% AD with Lewy pathology, 7% MD and 4% of other dementing disorders (Jellinger, unpublished data). These and other autopsy studies [143, 149, 178, 216, 338] have clearly shown the high frequency of mixed pathologies in elderly subjects with and without cognitive impairment (see [251]).
Major morphologic lesions in VCD/VaD
Pathologic changes in the brain of patients with cognitive impairment are multifold and include multifocal and/or diffuse disease and focal lesions [171, 224, 245, 246, 248, 255, 427, 509] (see Tables 4–6):
-
1.
Multifocal lesions with large territorial or borderline infarcts due to large vessels disease, distal field (watershed/borderzone) infarcts mainly related to hemodynamic events and carotid artery stenosis, microinfarcts throughout the brain, often due to embolic disease, small and medium-sized lesions mainly in functionally important brain areas, lacunes and lacunar infarcts or scars, WMLs, subcortical arteriosclerotic leukoencephalopathy (SAE) resulting from chronic ischemic hypoperfusion due to SMV and other mechanisms [132, 146, 224, 464, 517], incomplete ischemic injury, cortical pseudolaminar necrosis in cases of global ischemia and hypoperfusion, hippocampal sclerosis due to systemic and cardiovascular disease [122, 239] and multiple post-ischemic lesions.
-
2.
Focal disease with circumscribed, often strategically placed lesions, i.e., in functionally important brain areas and neuronal circuits. They are caused by many vascular and ischemic mechanisms including large arterial and small vessel diseases, cardiac embolic events, hemodynamic mechanisms and cerebral ischemia of various etiology. Complicated angiopathies such as fibromuscular dysplasia, arterial dissection, granulomatous angiitis, collagen vascular disease and other arteritides are rarer causes of VaD [142, 171, 224, 241, 242, 245, 246, 248, 255, 311, 312, 332, 351, 374, 426, 509].
Another classification distinguishes large and small vessel disease (Tables 5, 6):
-
1.
Large vessel dementia (LVD): Classical multi-infarct encephalopathy (rather rare).
-
2.
Small vessel disease (SVD): Microangiopathic (small vessel infarct) dementia (SVI), dementia with lacunes, strategic infarcts mainly in subcortical areas and WMLs [14, 101, 154, 346, 350, 520].
-
3.
Other types of VaD.
Although cause-relationships between CVLs and cognitive impairment evade strict classification [139, 144, 171, 185, 241, 242, 245, 246, 248], the major CVLs associated with cognitive impairment are summarized in Table 7. The key variables to define the pathology of VaD are summarized in Tables 2 and 7.
Classical multi-infarct encephalopathy (MIE)/large vessel disease
Single or multiple infarctions involving the areas of major cerebral arteries commonly result from atherosclerosis affecting intra-oder extracranial vessels, giving rise to local thromboembolism or hypoperfusion. These infarcts may be large (sub)territorial lesions involving much of a cerebral hemisphere. Their size is determined by assessing the two largest diameters of such a lesion or may represent multiple small lesions in cortex and/or adjacent white matter and basal ganglia, mainly in the medial cerebral artery (MCA) territories and less frequently in other supply areas or in adjacent territories, involving the dominant or both hemispheres [143, 301, 332, 408, 443] (see Table 2). Occlusion of extracranial arteries, e.g., the internal carotid artery (ICA) and the main intracranial arteries including the MCA, can lead to MIE which forms approximately 15% of VAD [69]. Medium-sized arteries in the leptomeninges and proximal perforating arteries can be involved. The damage can be worse depending upon the presence of hypertension and related CVD. Artery-to-artery embolism involves breaking of thrombi from often ulcerated lesions in extracranial arteries or heart valves. Various types of cardiogenic emboli may also find their way to the anterior and posterior cerebral circulation to cause infarcts in the respective supply areas.
The development of infarcts follows typical stages and the intensity of gliotic scars is an important consideration in judging the degree and age of infarction. Glial scarring may be seen after global ischemia, e.g., transient cardiac arrest causing lesions in vulnerable brain areas including hippocampus and neocortical (pseudo)laminar necrosis. Stroke patients with dementia had infarcts in the left hemisphere that were eight times larger than those in the right one, with a strong correlation between dementia and infarctions in the left posterior cerebral artery (PCA), anterior cerebral artery (ACA) and in parietal areas [298]. Cardiac sources, such as atrial fibrillation and myocardial infarction, provide a source for cerebral emboli, whereas most other causes, such as hematological conditions, inflammatory angiopathies/vasculitides, Sneddon’s disease [112] and familial CVD, e.g., autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) related to NOTCH3 mutations [389, 488] or cerebral amyloid angiopathy (CAA), both sporadic (see [20]) and hereditary (see [257]), usually cause multiple subcortical and/or cortical vascular lesions.
Microangiopathic small vessel lesions: lacunes and lacunar infarcts
Small vessels, including intracerebral end-arteries and arterioles, undergo progressive age-related changes [52, 283, 520], and may accelerate microvascular changes due to aging [445], which may result in lacunar or microinfarcts. The declining cerebral blood flow (CBF) and energy metabolism of the aging brain have been mainly attributed to ultrastructural alterations of the capillary walls. In aging rats, the vascular anomalies included perivascular collagen deposits, also referred to as microvascular fibrosis and basement membrane thickening [145]. Recently, age-related vessel changes in the human putamen have been demonstrated [496]. Due to the accumulation of collagen type IV, microvessels thicken and show a reduction in their lumen. Those changes likely promote occlusion or progressive stenosis with consequent acute or chronic ischemia in the dependent tissue. Alternatively, arteriosclerotic changes in small vessels in deep gray and white matter cause vessels to loose their elasticity to dilate and constrict in response to variations in systemic blood pressure or loss of autoregulation which, in turn, causes changes in cerebral perfusion. The deep cerebral structures are most vulnerable because their vessels are end arteries almost devoid of anastomoses. Small vessel pathology and acidosis acting on brain capillary endothelial cells could also lead to damage of the blood–brain barrier (BBB) with leak of fluid and macromolecules into cerebral microenvironment [392]. The presence of heavy deposits of serum proteins exclusively around the capillaries of the gray matter in cases with vascular dementia may indicate a defect of the cortical capillary system which might play a role in the clinical symptoms seen in VaD [7].
Lacunar infarcts or microinfarcts appear central to the most common cause of VCD/VaD [27, 142, 245, 246, 248, 255, 256, 509]. The term lacune appeared in the French literature in the early 1900s to describe small capsular [310] and pontine [157] infarcts with specific symptoms. Lacunar infarcts are small miliary softenings from 3 to 15 mm in diameter or small cavitations that may have more than one pathologic substrate, the most significant representing small infarcts and, less frequently, healed or reabsorbed tiny hemorrhages [162–164, 166, 171]. Seen radiologically and upon gross examination at autopsy, these lesions most frequently involve the periventricular white matter and subcortical structures, including thalamus, basal ganglia, internal capsule, pons and cerebellar white matter [166, 172]. Lacunes were classified into three types [117, 397, 398]. Type I lacunes are irregular cavities containing lipid-laden macrophages and blood vessels surrounded by a rim of gliotic, rarified brain; they are arising from small infarcts. This most significant type, resulting from narrowing or occlusion of small penetrating arteries branching directly from larger cerebral arteries [29] has been reported in 6–11% of selected brain autopsy series [162, 493]. A variant type—incomplete lacunar infarct—characterized by loss of only selectively vulnerable cellular elements without cavitation, suggests a common underlying cause, i.e., arterial obstruction, but perhaps of shorter duration or lesser severity [284], or may represent the squeal of edema-related gliosis [306]. Type II lacunes are smaller in size and distribution and contain numerous hemosiderin-laden macrophages, representing old, small hemorrhages or old hemorrhagic microinfarcts, resulting from fibrinoid vascular necrosis (Fig. 1), but are rare [282, 283]. Type III are dilatations of perivascular spaces, surrounded by a single layer of epithelial-like cells or more segments of a normal artery, and have been ascribed to small vessels permeability, interstitial fluid drainage disorders [116, 399], cerebral atrophy, mechanical stress from pulsating arterioles [219], distortion or elongation of small arteries, perivascular inflammation or other nonspecific factors. Misclassification between lacunar infarcts and enlarged perivascular spaces (EVPS) may occur, but most of these measure <2 mm, and normally surround perforating arteries entering the striatum at the anterior perforated substance [51, 61]. Their appearance in large numbers reflects focal brain atrophy around blood vessels and may lead to the so called état criblé, especially in the basal ganglia [23, 397]. EPVS are only rarely observed in the brains of young healthy adults, while they are increasingly common with aging [210]. Their presence has been associated with hypertension [90, 503], cerebral arteriosclerosis [504], PD [281], CADASIL [96] and diabetes [153].

Hypertensive microangiopathy with fibrohyalinosis and microaneurysm with perivascular hemosiderin in putamen. H&E, ×250
Lacunes were found in 36–42% of patients studied, representing the most frequent type of CVLs [419, 544]. Among 750 autopsy-proven AD brains and 562 age-matched controls, lacunar lesions were found in 32 versus 27% [237].
Hypertension has been regarded as the principal determinant of these lesions which occur in locations supplied by penetrating arterioles measuring 40–200 μm in diameter [50, 164, 166]. A classic post-mortem study of remote necroses in brains of hypertensive patients noted segmental arterial disorganization in association with 40–50 lacunes [164], caused by intracranial atherosclerosis, lipohyalinosis or segmental fibrinoid necrosis affecting small arteries with subsequent occlusion of single deep perforating vessels, whereas in one-fifth of the patients no thrombosis was found. However, most perforating branches have multiple stenoses and post-stenotic dilatation, suggesting that hemodynamic events might also play a role in the pathogenesis of lacunar state [110]. The pathogenesis of lacunar infarcts in the white matter and lenticular nucleus seems to be different, stenosis in the former and probably hemodynamic disturbances in the latter [330]. Other mechanisms are cerebral microembolism of vascular or cardiac origin, such as atrial fibrillation [142, 171].
Some recognize two distinct forms of lacunes: (1) patients with a single, symptomatic lacune having a vascular risk profile who presumably have microatheromatosis or embolism and (2) patients with multiple, usually asymptomatic, lacunes, with a higher frequency of hypertension and hypertensive SMV [50]. The pathogenesis of tissue damage in hypertension based on experimental studies has been reviewed recently [354].
Microscopic vascular pathology represents a heterogeneous group which includes two types of lesions: (1) those visualized by currently available neuroimaging methods, i.e., lacunes and WMLs [519] and (2) those that cannot be detected in vivo such as cortical microinfarcts and focal gliosis, but the increasing use of MRI to assess these lesions led to contradictory data. While some studies suggested that many lacunes identified neuropathologically or by MRI may be essentially silent from a cognitive point of view [244, 493, 506], and lacunar infarcts may be an incidental finding at neuroimaging and/or autopsy, a recent MRI-based study of 633 elderly subjects reported a significant association between lacunar scars and both the MMSE and ADAS scores, but only in cases with no or minimal disability, and therefore no or only minimal cognitive impairment were evaluated [501]. The importance of evaluation in defining the strategic importance of lacunes has been discussed recently [180].
Brain lesions associated with cognitive decline due to SMV, mainly subcortical vascular dementia [137], comprise of a tetrad of cerebral lacunes, microinfarcts, arteriosclerosis and WMLs. They are most reliable to a dementia syndrome when situated in the basal ganglia and cerebral white matter, either being single or multiple on neuroimaging or at autopsy. A cribriform pattern of multiple perivascular dilatations affecting the corpus striatum and other deep structures is referred to as “état lacunaire” or “lacunar state”.
According to the type and predominant location of SMVA, the following types of SMV-related VaD can be distinguished (Table 4):
-
1.
(Multi)lacunar state
-
2.
“Strategic” infarct dementia (SID) with borderline infarcts and other lesion
-
3.
White matter lesions and subcortical arteriosclerotic (leuko)encephalopathy (SAE)
Multilacunar state
The pathologic changes of lacunar state in basal ganglia, thalamus, hemispheral white matter and brainstem have been repeatedly reviewed [165, 282, 283, 405, 519, 520]. People with dementia are more likely to have such lesions than those without dementia, but the impact of microvascular lesions on cognitive impairment is under discussion [180].
“Strategic” infarct dementia syndrome
Focal infarctions or ischemic scars, often involving isolated functionally important brain regions, despite relatively small losses of cerebral parenchyma, may cause cognitive impairment, culminating in a dementia syndrome. Several studies have correlated cognitive impairment with the presence and emergence of lacunes, particularly in functionally important locations [476, 534]. These infarcts may be solitary but the clinico-pathologic picture is not infrequently complicated by, or dependent on, the occurrence of bilateral or multiple infarcts among which one or more may affect brain regions associated with a SID syndrome. They can be subdivided into those arising from the classical mechanisms associated with cerebral infarction (thromboembolism, atheromatous embolism, arteriosclerosis, atheromas, lipohyalinosis of small vessels) and those that manifest as focal ischemic lesions related to hypoperfusion, e.g., bilateral watershed infarcts in the cerebral hemispheres. Such strategic areas include [171, 245, 246, 248, 311, 332, 509]:
-
(a)
Watershed or borderzone infarcts: Typical infarctions in cerebral convexities may occur symmetrically or unilaterally, in circulatory borderzones between the deep and superficial branches of ACA, MCA and PCA, caused by hypotension with “misery perfusion”, i.e., diminished flow or stagnation of flow in distal vessels, or showers of microemboli. These infarcts usually occur in patients with severe atherosclerosis and extracranial carotid stenosis or occlusion and are often associated with prolonged episodes of hypotension [290]. They result in laminar necrosis of the cortex, multiple cortical and subcortical microinfarcts, often with small wedge-shaped lesions with their base to the pial surface and often extending into the white matter. They occur in the end-field territories between small superficial branches of large cerebral arteries or between deep and superficial vessel systems causing bilateral hippocampal or thalamic softening. Size and extent of these watershed infarcts depend on the amount of leptomeningeal anastomoses between the main cerebral arteries [499], and the degree of arteriosclerosis stenosis of meningeal arterial branches. The superior frontal area, between the distal supply of the ACA and MCA, and the posterior parieto-occipital junction, among ACA, MCA and PCA, are involved more often. Hypoxia also causes alterations in the hippocampal subareas CA-1 and CA-4, the outer half of the caudate nucleus and putamen and the anterior and dorsomedial nucleus of the thalamus. Bilateral ICA occlusive disease causing such lesions is frequently associated with impaired intellectual function [386].
-
(b)
Granular cortical atrophy, characterized by multiple small cortical microinfarcts and scars most often in the boundary zone between ACA and MCA in one or both hemispheres, is usually caused by hypoperfusion due to stenosis of the ICAs, cerebral microembolism to the cortex [487] or CAA, but they are a rare cause of dementia.
Other lesion patterns are:
-
(c)
Angular gyrus: Lesions involving the angular gyrus in the supply areas of ACA and MCA may occur in the dominant hemisphere or bilaterally [42].
-
(d)
Frontocingular infarcts: They involve the orbital and medial frontal cortex, in particular the territory of frontopolar and callosal marginal branches of the ACA [551].
-
(e)
Dorsal-paramedian, dorsomedial and polar areas of the thalamus are involved by unilateral and more often bilateral lesions resulting from occlusion of the thalamo-perforating artery (Fig. 2), a branch of PCA [78, 89, 103, 468] or capsular genu infarction [473], and paramedian thalamopeduncular infarcts [474].
-
(f)
Mesial temporal area of the hippocampus: Bilateral infarcts a result from circulation disorders in the supply area of the PCA [63, 140, 181, 241, 242, 245–247, 311, 551].
-
(g)
Caudate nucleus: Such lesions result from obstruction of the lateral lenticulostriatal arteries of the MCA and may include the anterior portion of putamen and anterior limb of internal capsule [75] or the head of the caudate [323] (Fig. 2).

Bilateral old infarcts in medial thalamus and old lacunar infarct in left caudate nucleus in 60-year-old hypertensive male with dementia after several TIAs
White matter lesions
Neuroimaging data and clinical relevance
Recent studies have placed emphasis on deep WML that have been increasingly detected by modern neuroimaging methods [5, 18, 34, 36, 104, 106, 150, 160, 213, 271, 302, 403, 408, 424, 436, 463, 501, 514, 518], and their relation to CVD and VAD. Synonyms include SAE or Binswanger disease [24, 47, 48, 75], diffuse white matter disease, WMLs, leukoaraiosis [198], periventricular arteriosclerotic (leuko)encephalopathy or leukomalacia, (progressive) subcortical vascular encephalopathy [424] and periventricular lucency [518]. WMLs are anatomically divided into those in a periventricular distribution and those which affect deep white matter, referred to as “deep subcortical WMLs”. Punctate and early confluent to confluent WMLs show distinguishable differences in their spatial distribution within a normal elderly population [133, 438–440].
Using CT and MRI, WMLs have been found in 22% of subjects under age 40 and in 27–60% of those over age 65 years [104], whereas in patients with AD and VaD, they are detected by MRI in almost 100% of patients [106], and enable the diagnosis of VaD with a sensitivity of 88% and a specificity of approximately 95% [213]. Their frequency is increased in patients with CVD and those at risk for vascular disease, including arterial hypertension, ischemic stroke, cardiovascular disease and diabetes mellitus [62, 295, 365, 381, 516]. The relevance of these lesions is controversial, since WMLs can be found in aged individuals with normal cognitive function with or without depression, age being the strongest predictor of their presence and severity [85, 297, 302, 404, 440, 540], as well as in patients with AD, VaD, multiple sclerosis and many other disorders [32, 518]. WMLs are more common in patients with VaD than in those with AD or healthy volunteers [385]. Longitudinal studies in white matter following ischemic stroke demonstrated improvement of the changes that continues at least into the second year following the insult [516]. WMLs and lacunes often coexist and the differential clinical significance of each of these types of lesions has been difficult to establish [180, 302, 422, 455, 540]. The relationship between APOE ɛ4 and WMLs in elderly subjects with lacunar infarcts is still controversial [31, 124, 277, 526].
Several cross-sectional studies support the view that WMLs have clinical implications even in healthy elderly people [105, 114, 173, 191, 272, 285]; their association with decline in essential cognitive abilities suggests that they should be regarded as a risk factor for cognitive impairment based on the site and volume of the lesions [91, 104, 157, 174, 308, 439, 457]. Others have only been able to confirm the association between WMLs and previous [173] or subsequent decline in cognitive function [105, 114, 278, 537]. Although longitudinal studies with serial MRI described progression of WML volumes, no association with cognitive decline was reported [59, 91, 439, 529], others observed an association with deficits of executive control [364, 401, 421, 462, 505], probably due to disruption of intra- and inter-hemispheric communications and prefrontal subcortical circuits that underly behavior, cognitive and executive cortical functions [177, 396, 401, 421, 442]. In a long-term prospective study of 68 healthy nondemented individuals the association between both periventricular and subcortical WMLs on MRI and cognitive impairment was only significant in ages 70+ [173]. While the perivascular WMLs had a relationship with cognitive deficits [80], subcortical WMLs corresponded with depressive symptoms [104, 106, 297]. Other studies showed correlations between WMLs and worse visuospatial ability and verbal memory [308]. The Rotterdam Scan Study of 1,077 elderly revealed relations between the periventricular but not the subcortical WMLs with global cortical function [60]. In patients with both WMLs and lacunes, WMLs were related only to a subscore of dementia rating scale [525], while in the LADIS study, WMLs and lacunes were both independently associated with ADAS scores, the effect of lacunes being less prominent [501]. The latter data were not confirmed by others [184, 273]. Although quantitative analysis of WMLs showed no strong association with severity of cognitive decline, it may provide a neuroimaging parameter for dementia development [81]. Posterior periventricular and corpus callosum extension of WMLs associated with MCI and AD indicate involvement of strategic white matter bundles that may contribute to the cognitive deficits seen in these syndromes [544].
Impaired cognitive domains of the speed of mental processes and memory in elderly diabetic patients were associated with WMLs and subcortical atrophy. Degenerative changes in the cerebral small vessels may constitute predictive factors for the rate of cognitive decline in these patients [5].
Neuropathologic substrates of WMLs
The morphologic lesions underlying radiologically observed WMLs include several morphologic alterations [104, 106, 150, 158, 213, 255, 482]. I n general, a morphologic triad of demyelination, axonal loss and lacunar infarcts are found mainly in the frontal, parietal and occipital white matter and in the periventricular areas. Myelin degeneration usually has a patchy confluent form sparing the subcortical U-fibers (Fig. 3). Lacunes are often scattered in the hemispheric white matter at the centers of areas of pallor (Fig. 4). There may be multiple, often asymmetrical, cavitary and noncavitary infarctions (Fig. 5) or areas of vacuolation, loss or pallor of myelin, loss of axons and oligodendroglia, areas of reactive astrocytosis, with or without macrophage reaction. Silver stains demonstrate that myelin pallor is secondary to fiber loss. Microinfarcts are generally smaller than 5 mm in diameter, and may present as a linear profile in the plane of section reflecting the course of an occluded microvessel. They are associated with astrocytic gliosis and presence of enlarged CD68-positive microglial cells. Dilated perivascular spaces may contain an insignificant and dispersed population of lipid rich or iron pigment loaden macrophages. Dilatation of the Virchow–Robin spaces (VRS) is common in disorders associated with microvascular diseases, and VRS scores were similar to scores for periventricular hyperintensity [385]. When the lesions are large and confluent there may be focal areas of incipient cavitation for which the term “incomplete white matter infarction” has been proposed [67], but a pathophysiological mechanism has not been accepted, and the term probably arose because of the lack of acceptance of a “misery perfusion” state of ischemic white matter leading to axonal and myelin loss. Vascular abnormalities include atherosclerosis of major cerebral arteries and several types of microangiopathy of the penetrating arteries, with tortuosity of thickened fibrotic or hyalinized arterioles, focal fibrinoid necrosis of vessel walls in deep gray and white matter with or without occlusions [68, 76, 150, 167, 224, 234, 235, 255, 378, 497, 509], associated with small infarcts, microaneurysms, fibrotic or thrombotic vessel occlusion [459, 539]. Collagenous thickening and occlusion of venules has been observed [66, 342]. Other lesions include small arteriovenous (AV) malformations, isolated central white matter infarcts, small foci of gliosis, vascular ectasia [518] or microvascular convolute formations [497]. Punctate hyperintensive lesions on MRI corresponding to dilated perivascular spaces are to be distinguished from extensive WMLs, the histologic correlates of which are confluent patches of white matter pallor without cavitation (Fig. 4). Under high-power histology, these areas show vacuolation and decreased numbers of oligodendroglia. Perivascular vacuolation may represent an early stage of these changes. Comparative studies of post-mortem MRI and histology showed that periventricular lesions on MRI correlated well with the severity of demyelination and astrogliosis, which were often associated with small lacunar infarcts in both the white matter and basal ganglia, suggesting a common pathophysiology, particularly arteriosclerosis of the penetrating artereries [24, 68, 150, 155, 188, 224, 355, 437, 503, 504].

Periventricular and deep white matter lesion in cerebrum semiovale and dilated lateral ventricles in 65-year-old woman with VCI. Klüver-Barrera

Focal myelin pallor in periventricular frontal white matter in patient with dysexecutive syndrome. Insert Perivascular lacunes around fibrotic arterioles in frontal white matter. Klüver-Barrera, ×200

Diffuse white matter lesions and old cystic subcortical infarction in 68-year-old hypertensive female with aphasia and subcortical dementia of Binswanger type
Attenuation of the cerebral white matter has been described into four clinical contexts: (1) SAE of Binswanger type that has been the subject of much controversy and reassessment, and some authors concluded that it should not be considered a distinct entity [76, 204, 300, 378, 471], (2) as a component of Alzheimer pathology and CAA [380], (3) as incidental finding of variable severity and debatable clinical relevance in the elderly and (4) as a feature of inherited cerebrovascular SVD syndromes [121, 257].
Selective infarction in periventricular border zones in patients with carotid stenosis and superimposed hemodynamic failure [113] and ischemic leukoencephalopathies, irrespective of the cause, e.g., carbon monoxide (CO) intoxication, acute hypoxia, AV malformations, delayed radiation encephalopathy, CAA and CADASIL [79, 159, 189, 256], share widespread demyelination of the deep white matter and periventricular necrosis, sparing the cerebral cortex and U-fibers. In AD, WMLs do not always parallel the severity of gray matter pathology and, thus, may be caused by various factors [131]. In contrast to meningeal and cortical arteries, CAA in white matter is rare, whereas fibrohyalinosis in this area is closely related with WMLs in AD, indicating their etiologic heterogeneity [485].
Cognitive impact of WMLs
The exact pathologic substrate of dementia to be associated with WMLs remains uncertain. Although WMLs and lacunes may be independently associated with cognitive dysfunction, in AD they are significantly correlated with cortical atrophy [74], and medial temporal atrophy [107, 500], and, thus, contribute to cognitive decline [441]. Together with cortical microinfarcts, WMLs may contribute to progression of dementia, but do not essentially interact with AD pathology to increase the likelihood of dementia beyond their additive effects [444]. Although the neuropathologic evaluation of focal and white matter gliosis had no clinical validity [273], regional VML volumetry may be helpful in correlating subcortical pathology and cognitive impairment [329, 498]. Periventricular WMLs predict the rate of cognitive decline [102], and they are often associated with decline in executive functioning and visual memory, even in patients without dementia [275]. Recent clinico-pathologic studies showed that even severe WMLs may be an independent risk factor for dementia [154], outlining the uncertainty about the cognitive impact of WMLs on brain aging. Synaptophysin immunoreactivity as a measure of synapse density in the cortex of SAE was almost as severely reduced as in AD, suggesting that the loss of synapses is a factor of dementia [497, 552]. Some authors have suggested that cognitive impairment associated with subcortical ischemic vascular disease is particularly the result of associated hippocampal and cortical changes [152, 349], preventing adaptive response to local changes in metabolic elements. Others showed significant correlation between WMLs scores of the right frontal regions and dementia [213].
Pathogenesis of WMLs
Several studies provided evidence that WMLs are related primarily to small vessel disease [533]. The suggestion that their pathogenesis is a chronic ischemic disorder of heterogenous pathophysiology, with hypoperfusion of the penetrating arteries supplying the white matter resulting from hypotension and reduced blood flow due to narrowing rather than occlusion of arterioles [69], was recently confirmed by experimental studies, in particular bilateral common carotid artery occlusion in the rat [84, 146, 287, 353, 381, 515] (see [148, 354]).
Structural changes of small intraparenchymal cerebral arteries and arterioles related to hypertension, altered cerebral blood flow (CBF) autoregulation and the conditions created by the unique arterial blood supply of the hemispheral white matter contribute to WML development [378]. Age-related microvascular degeneration in the periventricular white matter, mainly in the frontal and occipital areas, was recently described: Ultrastructural analysis identified the microvascular thickening as collagen deposits affecting the basement membrane. The vascular density did not correlate with the age. The basement membrane pathology significantly increased, while the number of intact microvessels gradually decreased, with advancing age in the frontal and occipital white matter [147].
MRI and PET studies showed decreased white matter CBF [271, 320, 377], which was even reduced in normal-appearing white matter [377], although their severity correlated better with the reduction in regional CBF in the cortex [271]. Others reported impaired vasodilatatory capacity in the deep white matter [64, 229]. Deep white matter hyperintensities and lacunar infarcts in basal ganglia and thalamus correlated with both a decline in mean global cortical metabolism and lowered cognitive function [465] and in patients without cortical infarctions, anterior periventricular hyperintensities reduced ipsilateral metabolic function [138]. The cortical global metabolic rates were lower in patients with subcortical strokes and cognitive impairment than in those without dementia [280], whereas others stressed the heterogeneity of CBF in subcortical VaD [541, 542].
A striking feature of WMLs is the sparing of the subcortical U-fibers, which may be related to the specific pattern of vascular supply. The periventricular regions and central white matter are feeded by long penetrating arteries coming from the pial vessels on the surface of the cortex, whereas the peripheral parts of the centrum semiovale are supplied from penetrating vessels from the cortical surface. Periventricular border/watershed zones between the long ventriculoseptal and ventriculofugal branches of the choroid and lateral striatal arteries [109] are particularly vulnerable to hypotensive episodes with lowered perfusion pressure [341]. This concept has been rejected by those who believe that the centrifugal branches are veins rather than arteries [317]. The pattern of punctate WMLs is probably the consequence of mixed etiologies. Preferred localization of the more confluent WMLs with arterial watershed areas implies a stronger ischemic component in their development [133]. Atherosclerosis in large, peripheral vessels is considered to be a predictive marker of microvascular pathology in the white matter [147, 531]. It coincides with massive microvascular fibrosis, particularly in the frontal white matter, demonstrating an age-related microvascular degeneration in the periventricular white matter with loss of medial smooth muscle cells caused by hypertension [370], which may contribute to the development of WMLs by hindering a sufficient supply of the affected sites. In SAE the frontal lobe is the site at which WM vulnerability is most pronounced, followed by parietal, occipital, and temporal lobes, and damaged nerve fibers are much more frequent than in age-matched control brains [4].
The distribution of WMLs with sparing of the subcortical U-fibers points to the pattern of cerebral edema [151]. The histological findings—spongiosis and loss of oligodendrocytes—which are also comparable to edema induced by ischemia and other mechanisms, were confirmed by experimental studies [264]. Diffuse extravasation of serum proteins throughout the white matter owing to enhanced pinocytotic activity by endothelial cells is observed without accompanying infarction in the stroke-prone spontaneously hypertensive rat; even a brief opening of the BBB may result in persistent presence of serum proteins in the white matter, including fibronectin and fibrinogen, which are capable of exerting biologic effects [432]. Breakdown of the BBB in white matter in chronic hypertension may result in focal edema with destruction of perivascular myelin [380, 452]. The edema hypothesis is consistent with the clinical observation that patients with SAE deteriorate during periods of sustained hypertension [235], while disruption of the BBB in experimental chronic cerebral hypoperfusion has not been clearly proven [148].
The molecular mechanisms of subcortical vascular WMLs concern a higher susceptibility of white matter axons to the effects of abnormal influxes of calcium that travel through various routes, including reverse Na+–Ca2+ exchange triggered by persistent Na+ channels and a parallel pathway involving Ca2+ channel causing increased endothelial permeability. Oligodendrocytes and/or myelin sheaths are more vulnerable to glutamate-triggered injury, resulting from reverse Na+-dependent glutamate transport. Some of the steps involved in these destructive events are subject to modifications by neurotransmitters, such as γ-aminobutyric acid (GABA), and by neuromodulators such as adenosine [522]. Excitotoxicity, once thought the unfortunate preserve of neurons, also contributes to white matter damage via both N-methyl-d-aspartate (NMDA) and nonNMDA glutamate receptors. However, important physiological differences are apparent in these receptors when compared to those present at the synapse [9]. Activated microglia is three times more immunoreactive for major histocompatibiity complex (MHC) class II antigen [484, 553]. The number of oligodendrocytes in the deep white matter is reduced by approximately 50% [538], and increased apoptosis of oligodendrocytes has been a major histologic correlate of WMLs [65, 287], indicating that loss of oligodendrocytes is involved in the reduction of nerve fibers. White matter reactive and fibrillary astrogliosis are common, but the number of astrocytes with light metallothionin (MT) I–II immunoreactivity in the deep white matter are reduced in contrast to normal numbers of astrocytes with strong reactivity for glial fibrillary acidic protein (GFAP) and MT I–II in subcortical white matter and cortex, suggesting topographic and biochemical differences in their dynamic plasticity. MT expression is related to regeneration, repair and/or reaction to neuronal lesions, with metal ion metabolic processing, buffering and detoxification or neuroprotection against free radicals [547]. The accumulation of APP and chromogranin A in tortuous axon indicated disturbed or blocked axonal transport [515].
In conclusion, recent evidence suggests that WMLs result from chronic ischemia due to hypoperfusion and disturbance of CBF, and, alternatively, recurrent edema resulting from severe disturbance of the BBB [68, 156, 245, 246, 381], but its exact etiology is unclear.
Post-ischemic encephalopathy
The sequelae of local or diffuse hypoxia and ischemia resulting from different causes can be separated into three major groups according to their predominant distribution pattern:
-
1.
Cortical laminar necrosis: Damage to cerebral cortex with laminar necroses and their sequelae may arise from cardiac or respiratory arrest (hypotension, anesthesia accidents, cardiac an/or respiratory failure, shock). They appear frequently at the arterial border zones [69, 256] and are often associated with diffuse white matter damage and cerebellar atrophy.
-
2.
Multiple post-ischemic lesions occur in case of dramatic systemic blood flow failure, combined with focal narrowing of large and small brain-feeding vessels, leading to disseminated or systemic post-ischemic lesions in cerebral cortex, subcortical white matter and basal ganglia. Combined cortico-cortical encephalopathies with multiple cortical and subcortical (micro)infarcts, resulting from various causes, may be associated with VaD.
-
3.
Hippocampal sclerosis (HS): This specific type of lesion defined as severe gliosis and neuronal loss in the CA1 region of the hippocampus and in the subiculum is a rare cause of cognitive impairment mimicking AD, and cognitive decline is often featured by marked memory impairment, but often without frank dementia [6]. Its prevalence in autopsy series of very old subjects with dementia ranged from 0.4 to 26% [22, 122, 123, 238, 289]. It predominantly co-occurs with AD [94], but is associated with a variety of disorders [38], in particular frontotemporal lobar degeneration (FTLD) and tauopathies [206, 314] and is extremely rare in oldest–old nondemented patients [314]. Hippocampal damage/necrosis, ranging from selective neuronal loss and gliosis to frank infarction, is often accompanied by multiple small infarcts in other brain regions, leukoencephalopathy or both. Current data showed that patients with HS are significantly older than those without HS and had more coronary artery disease, suggesting that related occult hypoxic–ischemic episodes may represent pathogenic factors [22, 314]. It has been occasionally observed in elderly individuals after receiving general anesthesia. Age-associated and other disease-related processes, such as atherosclerosis and cardiac failure, explain why the elderly cannot tolerate hypoperfusion of these vulnerable diseases like younger, healthy adults [425]. The morphologic differences between HS of hypoxic–ischemic etiology and HS associated with FTLD have recently been discussed, the former showing more marked neuronal and synaptic loss and greater reactive gliosis than HS associated with neurodegeneration [12].
Factors involved in VaD
Volume of brain destruction
Although it is understandable that subjects with large brain infarcts might experience intellectual decline, the idea that the diagnosis of VaD requires a brain tissue loss exceeding 100 ml is a persistent component of neuromythology. Tomlinson et al. [486] showed that although all patients with brain tissue losses of more than 100 ml suffered from dementia and that infarct volumes between 50 and 100 ml produced dementia less consistently, they observed several cases of dementia with infarcts of lesser volumes. Those totaling over 20 ml were significantly more frequent in subjects with dementia than in controls, and a marked difference between the two groups was present at 50 ml tissue loss cut-off. According to their study, which has never been replicated, a relatively small aggregate volume of brain infarct may or may not contribute to dementia, probably depending on its location, whereas destruction of a larger volume of cortex is usually followed by dementia. Therefore, these authors proposed the concept of strategic sites of infarcts. A quantitative MRI study demonstrated that total cerebral infarct area and cortical involvement were significantly larger in stroke patients than those without dementia [298]. PET studies also showed a correlation between metabolic impairment of frontal and temporomedial cortex and the total volume of hypometabolic regions to dementia severity in both VaD and AD, whereas VaD cases also showed metabolic impairment of subcortical regions not present in AD, suggesting some distinction with FDG PET between these two types of dementia [325, 331]. Studies measuring macroscopic infarcts in VaD brains revealed mean volumes of infarcted brain of 39 ml (range 1–229 ml) in one study [550] and 40.7 ml with a range of 6.9–220 ml in another [135], whereas patients with AD and AD plus VaD had infarct volumes of less than 10 ml. In patients who showed only vascular lesions on histologic examination with senile plaques below the level necessary for diagnosis of AD, the total volume of infarcted brain was more than twofold greater in subjects with than without dementia; only 3 patients had brain lesions larger than 100 g, and 17 had smaller volumes within the range of patients without dementia, suggesting that dementia is not directly and consistently related to the volume of infarction [115]. These data were confirmed by recent studies that found only a nonsignificant trend for lobar infarcts to occupy more cerebral hemispheral volume in VaD than in patients without overt dementia [143, 241, 242, 509].
Location of cerebrovascular lesions
The location of CVLs is probably more important than the volume of tissue destruction. Multiple brain regions have been implicated in VaD. Infarction in the left hemisphere and bilateral necroses with more involvement of the dominant hemisphere increase the risk of dementia after stroke [115, 134, 298]. Vascular lesions in the angular gyrus of the dominant hemisphere showed clinical similarities with AD [37, 99]. Cognitive impairment after stroke was more frequently associated with lesions in the left ACA and PCA territories [298, 475], and after left or bilateral PCA occlusion [99]. Bilateral (paramedian) thalamic infarction (Fig. 3) is often associated with memory deficit and “subcortical dementia” [78, 103, 468], as are lacunar infarcts in basal ganglia, especially in the head of the caudate [45, 79] and in the inferior genu of the anterior capsule, interrupting corticothalamic and thalamocortical pathways [474, 476]. However, whether selective lesions of the thalamus constitute a distinct dementia entity remains uncertain [336]. In patients with VaD, infarcts in hippocampus have been observed in 48%, in temporal lobe in 91% and in basal ganglia in 83% [374], and both hippocampal infarcts and sclerosis, either alone or in combination with other vascular lesions, have been related to dementia [122, 311]. Although the entorhinal cortex and hippocampus are less affected by subcortical CVLs than by AD [125], VaD resulting from microvascular pathology showed significant hippocampal neuronal loss [276] related to impaired microcirculation due to decreased microvessel diameters [52], and hippocampal atrophy may increase the development of PSD [93].
Number of CVLs
Only few studies addressed the important problem of the number of lesions in VaD and whether severe large infarcts are more likely to cause dementia than multiple small lesions. Erkinjuntti et al. [134] found that the mean number of infarcts in VaD was 5.8, compared with 0.2 in mixed AD plus VaD, whereas others reported a mean number of 6.7 CVLs in patients with dementia compared with 3.2 in those without cognitive impairment [115]. This significance reached statistical significance in the ACA and MCA territory. Although infarct location, size and numbers are important, other factors, such as age, systemic disease, other brain lesions, the degree of aging changes, the extent of WMLs, medial temporal lobe atrophy and level of education, etc. are involved in determining intellectual decline [394].
Importance of small vascular lesions
Comparing the neuropathologic findings in elderly subjects without considerable Alzheimer pathology and nondemented controls, Esiri et al. [143] saw correlations of microvascular brain damage with dementia. Severe lacunar state, microinfarcts and CAA had a greater prevalence in the dementia group which had less frequent macroscopic infarcts than the nondemented. There was a nonsignificant trend for the ratio of infarcted versus noninfarcted tissue in one cerebral hemisphere to be higher in the dementia group. Without differences in severity of extracerebral atheroma, the dementia group more often revealed arteriosclerosis and hypertensive microangiopathy. Examination of 19 different regions from 52 human brains showed that the expansion of CAA and arteriosclerosis/lipohyalinosis of small cerebral vessels was correlated with an increase of dementia, with amyloid β phases and neurofibrillary stages, suggesting that widespread small vessel changes are an important component of AD [480]. There was a correlation of dementia with widespread small ischemic lesions throughout the CNS, lacunes, microinfarcts and hippocampal injury being much more frequent than cystic infarcts greater than 1 cm in diameter [509]. Many brains showed more than one type of CVLs, most associated with severe atherosclerosis and arteriosclerosis. However, in two cognitively normal controls, similar multiple CVLs were seen. These data indicating that lacunes and microinfarcts are the most common neuropathologic features of VaD were confirmed in a study of 130 elderly subjects (age 80–92 years) [248]. Among demented subjects, 29% had large old infarcts, 57% multiple subcortical lacunes, 9.5% cortical and subcortical microinfarcts and 4.5% hippocampal sclerosis. Of the patients with MCI, 65% had cystic infarcts, 30% lacunar and 5% microinfarcts, while of the cognitively normal age-matched controls, 2/3 had cystic infarcts, 30% multiple subcortical lacunes with preserved thalamus and 1 multiple old microinfarcts (Table 8).
The impact of small CVLs has been examined in several autopsy series with vascular pathology confined to microinfarcts [273] and lacunes [184]. Among 45 patients (age 62–100 years, 13 with no dementia), there was a strong association between the extent of cerebral microinfarcts and cognitive findings. In the second study including 72 patients (mean age 83.4 years) with lacunes, deep white matter and periventricular demyelination contributed equally to cognitive dysfunction, and, contrary to earlier neuroimaging studies, the relationship between CDR scores and deep and periventricular WMLs was no longer significant after controlling for lacunes in a multivariant model [184]. In both univariant and multivariant models, thalamic and basal ganglia, but not deep white matter lacunes, significantly predicted cognitive impairment [180].
A similar preponderance of cortical and subcortical microinfarcts was observed in a cohort of longitudinally followed autopsy cases of AD, 36 with concomitant small cerebral infarcts with volumes less than 10 ml, which had no impact on cognitive decline [286]. These data, which were at variance to others suggesting a contribution to cognitive decline of CBLs with volumes of even less than 1 ml [92], were confirmed by studies in a larger sample showing that in AD with minor CVLs, the majority of lesions were lacunes in basal ganglia and/or white matter, and multiple microinfarcts [240, 248].
Evaluation of the type and topographic pattern of CVLs in a large autopsy series of demented subjects, in cases with “pure” VaD, i.e., those without essential concomitant Alzheimer-type or other pathologies, showed a significantly higher frequency of small subcortical lesions (lacunes and microinfarcts representing 71%) than of large infarcts involving one or both cerebral hemispheres (29%) (Table 9). This pattern differed considerably from that in cases of mixed dementia (combination of definite AD with vascular encephalopathy), where 64% revealed large, often lobar infarcts or multiple cortical and subcortical infarcts larger than 10 ml in diameter involving one or both hemispheres, whereas lacunes and small subcortical microinfarcts were seen in only 36% (Table 10), which suggests different pathogenic mechanisms between both types of disorders [248].
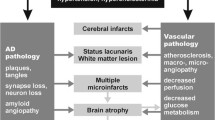
These and other data indicate that subcortical lacunes and multiple disseminated infarcts are the most common morphologic features of VAD, while large cystic infarcts are less common [224, 241, 242, 245, 246, 248, 255]. Recent studies comparing CBF, cerebral metabolic rate of oxygen (CMRO2) and vascular reactivity (VR) in VaD showed significant reduction as compared to AD, mainly in frontal lobes, suggesting that patchy reduction of CBF and CMRO2 seem to be distinct features of VaD [356]. The vicious circle of factors causing VaD is summarized in Figs. 6 and 7.

Scheme of cerebral microvascular pathology and its deleterious consequences in the aging brain with a vicious circle of factors causing VaD. Modified from [145]

Schematic interplay of pathogenic factors causing vascular cognitive impairment/dementia
Hemorrhagic dementia
Primary intracerebral hemorrhages are uncommon causes of dementia. Most occur in basal ganglia and thalamus, although a few are located in cerebral cortex and white matter. These hemorrhages may vary in size. Massive basal ganglia bleeds with rupture into the ventricles or brainstem compression are often fatal. Multiple small or slit-like hemorrhages in cortex and white mater may lead to cognitive impairment [311, 332]. In addition to hypertension and atherosclerosis, the majority of dementing disorders related to cerebral hemorrhages and/or hemorrhagic infarcts occur in sporadic and familial conditions associated with CAA [411–413], and other hereditary forms of VaD [257].
Sporadic cerebral amyloid angiopathy
CAA is defined as deposition of amyloid β peptide (Aβ) in the walls of meningeal and intracortical (parenchymal) arteries, arterioles, capillaries and rarely veins, with thickening of their walls and degeneration of smooth muscle cells (SMC), sometimes with additional spread into the surrounding neuropil or depositions in the glia limitans or capillary wall, referred to as capillary CAA (capCAA) [20]. The origin of Aβ in blood vessel walls is poorly understood, and several mechanisms have been proposed: (1) systemic hypothesis suggesting that Aβ, at least partially, originates from circulation [410–412]; (2) vascular hypothesis suggesting production of Aβ by SMCs within the vessel walls and/or pericytes [410, 532]; (3) drainage hypothesis, suggesting that Aβ produced by neurons is transported in the course of perivascular drainage, is strongly supported by transgenic animal models [20, 215, 523, 524]; (4) other pathogenetic mechanisms (see [413]). Although hereditary forms of CAA caused by mutations of different genesis are known, CAA is most commonly sporadic. Its incidence increases with age to almost 100% after 80 years, and ranges from 70 to 97.6% in AD [20]. Infiltration of vessel layers by Aβ and loss of SMCs, fibrinoid necrosis and formation of microaneurysms are the mechanisms by which CAA progresses and becomes symptomatic [411–413, 510]; two types of sporadic CAA have been described [477].
Many different approaches have been used to evaluate the severity of CAA (see [20]). In routine neuropathology two grading systems are commonly used. Olichney et al. [371] proposed the scale: 0—no Aβ-positive vessels; 1—scattered Aβ positivity in either leptomeningeal or intracortical vessels; 2—strong, circumferential Aβ positivity in either some meningeal or intracortical vessels; 3—widespread, strong, circumferential Aβ deposition in both; 4—same as 3 with additional dyshoric changes. This system has a rather quantitative approach, whereas Vonsattel et al. [512] graded CAA with respect to the severity of pathologic changes in a given blood vessel: mild—Aβ restricted to the tunica media without significant destruction of SMCs; moderate—tunica media replaced by amyloid and is thicker than normal; severe—extensive amyloid deposition with focal wall fragmentation or double barreling of the vessel wall, microaneurysm formation, fibrinoid necrosis and leakage of blood through the vessel wall. Despite the practical value of these systems, they have some limitations. The first links meningeal and intracortical involvement, and does not allow scoring cases with strong positivity in parenchymal vessels but without involvement of meningeal vessels, the other does not distinguish between meningeal and intracortical involvement. Another system distinguishes between meningeal and parenchymal involvement and also allows screening CAA in different regions. Meningeal and intracortical vessels are scored separately: 0—no Aβ positive vessels; 1—mild (scattered positivity in a few vessels); 2—moderate (scattered positivity in many vessels or strong positivity in a few ones); 3—severe (strong positivity in many vessels), 4—severe with dysoric changes in intracortical vessels. For each region, both the meningeal and intracortical score is assessed separately, and the overall severity of CAA is calculated as mean values of all scores [20, 21]. A similar protocol has been suggested recently by an international working group (Table 11). To date, however, no validated and generally accepted neuropathologic criteria for rating CAA are available, and “this lack makes it difficult to compare results across different populations and studies, and clearly outlines an obstacle to be addressed by the investigators in this field” [190].
CAA, now recognized as a major cause of nonhypertensive, nonatherosclerotic CVLs including lobar or multiple cortical hemorrhages and multiple infarcts in the elderly [20, 230, 232, 508], has a pathogenic role in dementia and can be associated with vasculitic process, usually a granulomatous, giant cell arteritis [411–413, 510]. CAA-related intracerebral hemorrhages are seen in 5–20% of all spontaneous (nontraumatic) bleeds in elderly subjects [243, 512]. Both acute and recurrent CAA-related cerebral hemorrhages are mainly located in the frontoparietal (35%), parietal (11–26%), temporal and occipital regions (5–18%), in basal ganglia (5–10%), with multiple bleeds in about 10%, and multiple simultaneous hemorrhages may occur [217, 413]. Microbleeds in AD, detectable in vivo by MRI [71] may be associated with CAA, but not with hypertension or CVD [358]. The co-localization of hem, a marker of cerebral bleeding, and amyloid as a marker of plaques, has suggested that senile plaques are sites of microhemorrhages, both lesions being more frequent in demented than in non-demented subjects [98]. CAA is also associated with ischemic infarctions, the frequency of which increases with its severity, and the combination of CAA and hypertension is highly associated with cerebral infarcts, suggesting an additive injury to blood vessels [72, 372, 373].
CAA may also be a cause of WMLs [469]. The severity of WMLs is significantly correlated with fibrohyalinosis of deep vessels, but neither with the age at onset nor the scores of CAA, suggesting that although CAA is an independent risk factor for WMLs in AD, its role is limited in comparison to that of fibrohyalinosis of white matter microvessels, which may or may not be associated with CAA, thus indicating the pathogenetic heterogeneity of WMLs [485]. There is progressive increase in WMLs in subjects with CAA, and the association of WMLs with incident lobar hemorrhages suggests that the white matter damage may reflect a progressive microangiopathy due to CAA [82]. In any case, coexistent pathology is a much more common cause of dementia in sporadic CAA, which is explained by the closely related pathobiologies of both conditions [141]. Several studies suggested severe CAA to be an independent risk for cognitive decline [154, 190, 361, 543, 551], whereas others did not confirm a higher prevalence of CAA in demented subjects [391]. Advanced CAA causes clinically important vascular dysfunction [190]. Since it frequently occurs in subjects with cortical infarcts but only mild Alzheimer pathology, it has been suggested that CAA represents an underestimated variant of VaD [199]. Microinfarcts in cerebral cortex associated with severe CAA could be a primary pathologic substrate of VaD [200]. Studies in a 95+ cohort, showing a strong association between CAA and APOE ɛ4 as well as dementia, suggest that the contribution of CAA to dementia is largely dependent of APOE ɛ4 [472], and long term studies showed that plasma Aβ-40 concentration is independently associated with WMLs in subjects with AD, mild cognitive impairment (MCI) or CAA [192], but the relations between these factors and the potential contribution of vascular dysfunction and CAA in cognitive decline remain debatable [288].
Although the role of CAA as an independent, primary cause of dementia is unclear, it is widely assumed that it has an aggravating effect on other pathologies and may lower the threshold for overt dementia, especially in AD, where subjects with only medium neuritic pathology, e.g., Braak stages III and IV, but considerable CAA were clinically demented [249, 391].
Aβ-related hereditary CAAs (hereditary cerebral hemorrhage/HCH)
Most of these disorders that are associated with dementia combine CAA with multiple cerebral hemorrhages or infarcts, and are caused by mutations of various genes.
Familial CAA can be caused by deposition of Aβ due to mutations of the APP gene located on chromosome 20. They include the Flemish (A692G), Dutch (E693Q causing HCH-WA-d), Arctic (E693G), Italian (E693K) and Iowa (D694N) mutations. The Dutch form (HCHA-D), the Islandic type (HCHWA-I), the Flemish, Italian and Arctic mutations, all inherited as autosomal traits, show dementia and leukodystrophy with multiple recurrent hemorrhages or hemorrhagic infarcts mainly in the subcortical white matter, completed by diffuse plaques [187, 362, 521]. Neurofibrillary pathology is minimal in HCHWAD, dementia being largely independent from tangles and plaques but attributable to multiple CAA-related CVLs [360]. These hereditary forms are rare and include about a dozen pedigrees with 150 post-mortem cases [393]. APP locus duplication explained <2% of familial, nonautosomal dominant early onset AD with severe CAA [453].
Other mutated proteins such as variant transthyretin (ATTS), variant gelsolin (Agel), variant cystatin (Acys), amyloid-Bri (Abri), amyloid-Dan (Adan) or disease-associated prion protein (PrPsc) are also implicated in CNS amyloidosis with CAA [411–413]. In some familial forms, such as in familial British dementia (FBD) and familial Danish dementia (FDD), CAA is much more widespread and affects most CNS areas [218, 265, 270].
CAA due to deposition of ATTR (amyloid transthyrenin), resulting from more than 60 mutations of the TTR gene, located on chromosome 18, shows characteristic morphologic phenotypes with prominent amyloid deposition and CAA in leptomeninges, cerebral parenchyma and vitreous vessels [43, 49].
Gelsolin-related amyloidosis of the Finish type, due to mutations (D187N amino acid mutation and others) of the gelsolin gene on chromosome 9, in addition to MCI and other lesions show widespread CAA, with deposition of gelsolin-related amyloid in almost all organs and diffuse myelin loss in centrum semiovale [202, 410–412].
Familial British dementia (FBD) associated with point mutation in the BRI gene located on chromosome 13, dementia, ataxia and spastic tetraparesis, in addition to NFTs and neuroils threads in hippocampus shows widespread CAA and Binswanger type WMLs but only rare cerebral hemorrhages [411, 412, 507].
Familial Danish dementia (FDD), associated with duplication between codons 265 and 266 on the BR12 gene, with cataracts, deafness, ataxia and dementia shows CAA in almost all CNS regions and hippocampal and perivascular amyloid plaques and tangles, mimicking pathologic aspects of AD [411, 412].
Other types of hereditary VaD
A number of monogenic diseases is associated with hereditary forms of VAD. They include hereditary MID, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), cerebral autosomal recessive arteriopathy with subcortital infarcts and leukoencephalopathy (CARASIL, Maeda syndrome), cerebroretinal vasculopathy, hereditary endotheliopathy with retinopathy, nephropathy and stroke and hereditary vascular retinopathy (CRV and HERNS) and MELAS syndrome.
Hereditary MID in a Swedish family with onset between 29 and 38 years of age at autopsy revealed multiple cystic infarcts in gray and white mater and pons with cerebral cortical atrophy resulting from occlusion of small arteries and arterioles [458], but its genetics are unknown [256].
CADASIL with clinical onset in midadult life and a history of small strokes, migraine, progressive dementia and pseudobulbar palsy is caused by a defective NOTCH3 gene on chromosome 19q12. Neuropathology shows multiple small, deep infarcts, diffuse leukoencephalopathy [546] and widespread vasculopathy causing fibrotic thickening of the arteriolar walls with or without obliteration of small arteries (see [256, 257, 330]). The vascular lesions consisting of granular osmiophilic material (GOM) in SMCs are also seen (with electron microscopy) in other organs (vessels in spleen, kidney, muscle, skin) allowing diagnosis by skin biopsies [316] or by a histochemical method to demonstrate the extracellular domain of NOTCH3 [254]. In CADASIL with white matter infarcts, cholinergic denervation was seen in parietal, frontal and occipital regions, and the entorhinal and temporal neocortex usually affected in AD were preserved [327], whereas a recent study reported prominent cholinergic deficits in the frontal and temporal cortex [259]. Cortical neuronal apoptosis involved in cortical atrophy appears related to the burden of subcortical ischemic lesions [511]. Transgenic animal models of CADASIL are available (see [257]).
CARASIL, another nonhypertensive, nonamyloid small vessel arteriopathy with similar lacunar infarcts as in CADASIL, but with different vascular pathology, recessive inheritance, pseudobulbar palsy and orthopedic problems, pathologically shows severe changes of small penetrating arteries of white matter and basal ganglia, less severe in brainstem and meninges. So far no genetic defect has been reported.
Two other very rare hereditary small vessel diseases are CRV and HERNS, showing progressive visual impairment, headache and strokes with neurologic deficits and cognitive decline to dementia, have been mapped to locus 3p21 [375]. Brain biopsies showed fibrosis of small cerebral arteries with fibrous thrombi but no vasculitis and infarction and gliosis of the surrounding brain (see [257].
A totally different disorder related to mitochondrial encephalopathies is MELAS syndrome (mitochondrial myopathy encephalopathy, lactic acidosis and stroke-like episodes), caused by a A-to-G mutation at nucleotide 3243 in the mitochondrial tRNA LEU(UUR) gene in approximately 80% of patients [400], which is clinically characterized by stroke-like episodes, seizures and dementia with onset before 40 years of age. Pathology shows generalized mitochondrial microangiopathy without paracrystalline aggregates [348]. The diffuse atrophic brain shows multifocal and asymmetric, occasionally “punched-out” or cystic infarct-like lesions in cortex and subcortical white matter. Cerebral lesions are distributed in pseudolaminar, pancortical and cortico-subcortical patterns; deep cortex with increased vascularity, spongy changes in white matter and rare small infarcts in basal ganglia. The meningeal and cortical arteries and arterioles are patent, whereas myocytes and small artery endothelium show degenerative changes with increased numbers of mitochondria. These data indicate that mitochondrial vasculopathy is responsible for the CVLs in MELAS [369], although others questioned the hypothesis that defective mitochondria are responsible for the infarct-like lesions [305].
Pathophysiology of VaD
VAD/VCI is thought to be caused by focal and multifocal vascular and/or ischemic lesions involving various, often strategically/functionally important brain areas with deafferentation of frontal and limbic cortical structures and interruption of thalamo-cortical, striato-cortical, cortico-cortical as well as ascending pathways caused by lesions in basal ganglia, thalamus, white matter and subfrontal areas. VaD may present with memory loss resulting from interruption by ischemic injury of segments of the Papez circuit comprising the hippocampus, fornix, mamillary body, mamillothalamic tract (fasciculus of Vicq d’Azyr), anterior thalamus and cingulate gyrus [127]. Memory loss also occurs with ischemic damage to the medio-basal forebrain, in particular cholinergic neurons in septal and basal nuclei and their projections, in the vicinity of septal gray and nucleus basalis Meynert and destruction of cholinergic loops in the deep white matter [428]. These lesions may sometimes be confused clinically with AD. The pattern of cognitive impairment is consistent with models of disturbed cortical and subcortical neuronal circuits [275]. Complex interactions between subcortical lesions and radiologically detected changes in cortex and hippocampus in producing cognitive decline have been shown [152], while others suggested significant correlations to cortical lesions and frontal atrophy [70, 213]. WMLs impair frontal functions regardless of their location [492], are associated with neocortical more than entorhinal and hippocampal atrophy [126], that is more severe in AD than in VaD [548], and increase the risk of dementia, particularly in patients with lacunar infarcts [409, 525]. Strategically situated small lesions that may represent a mixture of large and small vessel disease rather than an isolated entity may destruct thalamo-cortical, striatocortical and prefrontal-basal ganglia pathways, involving cognition, memory and behavior [42, 76, 103, 248, 290, 301, 332, 425, 468, 473, 550]. The suggestion that cognitive impairment associated with lacunes in basal gray and white matter may result from disruption of subcortical frontal circuits [100, 407] has been confirmed by recent studies on the impact of subcortical lacunes and microinfarcts on cognitive dysfunction [180, 184, 273] and by experimental models of ischemia [359].
The above findings indicate that damage in the subcortical gray matter may directly influence cognitive performance, whereas the frequent development of lacunes within the deep white matter in brain aging appears not sufficient to induce dementia. Frontal impairment in VaD is responsible for greater executive dysfunction in comparison with AD patients who show more severe impairment of attentional shifting and working memory [73].
Neurochemical studies showed that VaD is associated with abnormalities in key neurotransmitter systems, in particular the basal forebrain cholinergic system, while earlier studies showed a significant reduction of substance P-like immunoreactivity, choline acetyltransferase (ChAT) activity and muscarinic receptors in the hippocampus of MID and AD/MID brains [431], but preserved nicotinic receptors in VaD [315]. There is emerging evidence that this system is affected in SVaD related to diffuse WMLs involving the central axonal radiation fields for this projection pathway [446, 467], such lesions may cause widespread disconnection of cholinergic projections to the center [467]. Since cholinergic mechanisms play a role in the regulation of regional blood flow in the brain [203, 433, 434], dysfunction of the cholinergic system caused by extensive WMLs and other vascular lesions may cause decreased CBF and hypoperfusion that are critical in the pathogenesis of VaD [425], and may cause cognitive impairment. Preclinical [263, 483] and clinical evidence [315, 466] suggested that cholinergic deficit may be associated with VaD [311, 428], and evidence of cholinergic changes from animal models of CVD/VaD have been reviewed recently (see [429]). AD and mixed dementia (AD + VaD) usually have greater deficits of ChAT activity in temporal cortex than age-matched controls and patients with “pure” VaD [388], but a recent study showed severe cholinergic deficits in frontal and temporal cortices in CADASIL cases [259]. There is paucity of data related to other specific neurotransmitter deficits in VaD. The involvement of the supraoptic and tuberomamillary nuclei [231] and the corresponding reduction in vasopressin and histamine may have clinical correlates (e.g., incontinence in VaD). An important neurotransmitter involved is the striatonigral dopaminergic system giving rise to vascular pseudoparkinsonism [449].
Conclusions and future aspects
Recent autopsy data confirm that (1) VaD is a nonfrequent entity, in the course of which multiple ischemia and/or vascular brain lesions of variable etiology, pathogenesis, extent and location result in progressive cognitive and memory impairment (Fig. 7). While most of the currently used clinical guidelines for diagnosis of VCI/VaD show variable sensitivity and specificity, and recent Mayo Clinic criteria [268] await further validation; at present, no generally accepted and validated neuropathologic criteria are available. Further criteria should be more evidence-based, but how the VCD/VaD debate will evolve, remains unclear [266]. Cognitive impairment appears to correlate with widespread or even focally limited vascular lesions throughout the brain. “Pure” VaD that should only be diagnosed in the absence of Alzheimer-type lesions beyond age-related levels and other concomitant pathologies is rather rare, but ischemic/vascular brain lesions even of smaller volume, particularly in functionally important (“strategic”) areas and neuronal loops, may aggravate the neuropsychologic deficit in aging brain and AD. (2) The concept that VaD is determined primarily by the volume of infarcted brain is oversimplistic and was not confirmed by a majority of neuroimaging and autopsy studies. (3) Although disputed as an isolated nosologic entity, VaD/VCI is correlated with multiple vascular/ischemic brain lesions, due to a multitude of causes including large and small vessel disease, cardiovascular and systemic disorders, that need to be determined more precisely. (4) Sporadic and genetic/familial CAA and other hereditary vascular disorders related to a multitude of gene defects frequently cause cerebral hemorrhages and other CVLs which may result in dementia. (5) MD resulting from combined AD and VaD (or other pathologies) is a diagnostic challenge for which neither validated clinical criteria, generally accepted and validated neuropathologic guidelines nor exact epidemiologic data are available. (6) The histologic pattern of vascular brain lesions in “pure” VaD, mainly related to multiple small CVLs in subcortical brain regions or multiple cortico-subcortical microinfarcts, differs considerably from that in MD that more often shows larger hemispheral infarcts, suggesting different pathogenic mechanisms. (7) Hippocampal sclerosis, often accompanied with AD lesions or multiple other CVLs, is a not infrequent form of VaD in very old subjects. (8) Neuropathologic examination of brains in which vascular pathologies are identified necessitates careful documentation of the nature, severity, anatomical extent and location of the vascular lesions.
Additional quantitative studies in larger series including not only the assessment of Alzheimer-type lesions but also estimates of numbers of both synapses and microvascular lesions as well as characterization of microvessel abnormalities are needed to give further insights into the structural substrates of cognitive impairment in the elderly and to develop standardized neuropathologic criteria for the diagnosis of both vascular and mixed dementia. Future development of homogenous and harmonized neuropathological definitions and procedures in classifying vascular lesions and of methods to more accurately characterize the independent severity of vascular and neurodegenerative brain lesions remain an important priority for the future. Accurate diagnosis of both VCI/VaD and MD and further exploration of the structural substrates of cognitive impairment in order to get better insights into the impact of CVLs and their interrelationship to Alzheimer and other pathologies on cognitive function are among the most important challenges of modern neuroscience.
References
AGS Clinical Practice Committee (2003) Guidelines abstracted from the American Academy of Neurology’s Dementia Guidelines for early detection, diagnosis, and management of dementia. J Am Geriatr Soc 51:869–873
Aguero-Torres H, Kivipelto M, von Strauss E (2006) Rethinking the dementia diagnoses in a population-based study: what is Alzheimer’s disease and what is vascular dementia? A study from the Kungsholmen Project. Dement Geriatr Cogn Disord 22:244–249
Akatsu H, Takahashi M, Matsukawa N, Ishikawa Y, Kondo N, Sato T, Nakazawa H, Yamada T, Okada H, Yamamoto T, Kosaka K (2002) Subtype analysis of neuropathologically diagnosed patients in a Japanese geriatric hospital. J Neurol Sci 196:63–69
Akiguchi I, Tomimoto H, Wakita H, Kawamoto Y, Matsuo A, Ohnishi K, Watanabe T, Budka H (2004) Topographical and cytopathological lesions analysis of the white matter in Binswanger's disease brains. Acta Neuropathol (Berl) 107:563–570
Akisaki T, Sakurai T, Takata T, Umegaki H, Araki A, Mizuno S, Tanaka S, Ohashi Y, Iguchi A, Yokono K, Ito H (2006) Cognitive dysfunction associates with white matter hyperintensities and subcortical atrophy on magnetic resonance imaging of the elderly diabetes mellitus Japanese elderly diabetes intervention trial (J-EDIT). Diab Metab Res Rev 22:376–384
Ala TA, Beh GO, Frey WH 2nd (2000) Pure hippocampal sclerosis: a rare cause of dementia mimicking Alzheimer’s disease. Neurology 54:843–848
Alafuzoff I, Adolfsson R, Grundke-Iqbal I, Winblad B (1985) Perivascular deposits of serum proteins in cerebral cortex in vascular dementia. Acta Neuropathol Berl 66:292–298
Alafuzoff I, Pikkarainen M, Al-Sarraj S, Arzberger T, Bell J, Bodi I, Bogdanovic N, Budka H, Bugiani O, Ferrer I, Gelpi E, Giaccone G, Graeber MB, Hauw JJ, Kamphorst W, King A, Kopp N, Korkolopoulou P, Kovacs GG, Meyronet D, Parchi P, Patsouris E, Preusser M, Ravid R, Roggendorf W, Seilhean D, Streichenberger N, Thal DR, Kretzschmar H (2006) Interlaboratory comparison of assessments of Alzheimer disease-related lesions: a study of the BrainNet Europe Consortium. J Neuropathol Exp Neurol 65:740–757
Alix JJ (2006) Recent biochemical advances in white matter ischaemia. Eur Neurol 56:74–77
Altieri M, Di Piero V, Pasquini M, Gasparini M, Vanacore N, Vicenzini E, Lenzi GL (2004) Delayed poststroke dementia: a 4-year follow-up study. Neurology 62:2193–2197
Alvarez-Sauco M, Molto-Jorda JM, Morera-Guitart J, Frutos-Alegria MT, Matias-Guiu Guia J (2005) [An update on the diagnosis of vascular dementia]. Rev Neurol 41:484–492
Amador-Ortiz C, Ahmed Z, Zehr C, Dickson DW (2006) Hippocampal sclerosis in the elderly differs from hippocampal sclerosis in frontal lobe degeneration. Acta Neuropathol (in press)
American Psychiatric Association (1994) Diagnostic and statistical manual of mental disorders, 4th edn. American Psychiatric Association, Washington
Andin U, Gustafson L, Passant U, Brun A (2005) A clinico-pathological study of heart and brain lesions in vascular dementia. Dement Geriatr Cogn Disord 19:222–228
Andin U, Gustafson L, Brun A, Passant U (2006) Clinical manifestations in neuropathologically defined subgroups of vascular dementia. Int J Geriatr Psychiatry 21:688–697
Appelros P, Samuelsson M, Lindell D (2005) Lacunar infarcts: functional and cognitive outcomes at five years in relation to MRI findings. Cerebrovasc Dis 20:34–40
Appelros P, Viitanen M (2005) What causes increased stroke mortality in patients with prestroke dementia? Cerebrovasc Dis 19:323–327
Artero S, Tiemeier H, Prins ND, Sabatier R, Breteler MM, Ritchie K (2004) Neuroanatomical localisation and clinical correlates of white matter lesions in the elderly. J Neurol Neurosurg Psychiatry 75:1304–1308
Arvanitakis Z, Hachinski V (1999) Vascular cognitive impairment: what else do we need to learn? In: Sisodia S (ed) Alzheimer disease, 2nd edn. Lippincott Williams & Wilkins, Philadelphia, pp 147–160
Attems J (2005) Sporadic cerebral amyloid angiopathy: pathology, clinical implications, and possible pathomechanisms. Acta Neuropathol (Berl) 110:345–359
Attems J, Konig C, Huber M, Lintner F, Jellinger KA (2005) Cause of death in demented and non-demented elderly inpatients; an autopsy study of 308 cases. J Alzheimers Dis 8:57–62
Attems J, Jellinger KA (2006) Hippocampal sclerosis in Alzheimer disease and other dementias. Neurology 66:775
Awad IA, Johnson PC, Spetzler RF, Hodak JA (1986) Incidental subcortical lesions identified on magnetic resonance imaging in the elderly. II. Postmortem pathological correlations. Stroke 17:1090–1097
Babikian V, Ropper AH (1987) Binswanger’s disease: a review. Stroke 18:2–12
Bacchetta JP, Kovari E, Merlo M, Canuto A, Herrmann FR, Bouras C, Gold G, Hof PR, Giannakopoulos P (2006) Validation of clinical criteria for possible vascular dementia in the oldest-old. Neurobiol Aging. DOI 10.1016/j.neurobiolaging.2006.1002.1006 (in press)
Bakker FC, Klijn CJM, van der Grond J, Kappelle LJ, Jennekens-Schinkel A (2004) Cognition and quality of life in patients with carotid artery occlusion. A follow-up study. Neurology 62:2230–2235
Ballard C, McKeith I, O’Brien J, Kalaria R, Jaros E, Ince P, Perry R (2000) Neuropathological substrates of dementia and depression in vascular dementia, with a particular focus on cases with small infarct volumes. Dement Geriatr Cogn Disord 11:59–65
Ballard CG, Morris CM, Rao H, O’Brien JT, Barber R, Stephens S, Rowan E, Gibson A, Kalaria RN, Kenny RA (2004) APOE epsilon4 and cognitive decline in older stroke patients with early cognitive impairment. Neurology 63:1399–1402
Bamford JM, Warlow CP (1988) Evolution and testing of the lacunar hypothesis. Stroke 19:1074–1082
Bancher C, Jellinger K, Lassmann H, Fischer P, Leblhuber F (1996) Correlations between mental state and quantitative neuropathology in the Vienna Longitudinal Study on Dementia. Eur Arch Psychiatry Clin Neurosci 246:137–146
Barber R, Gholkar A, Scheltens P, Ballard C, McKeith IG, Morris CM, O’Brien JT (1999) Apolipoprotein E epsilon4 allele, temporal lobe atrophy, and white matter lesions in late-life dementias. Arch Neurol 56:961–965
Barber R, Scheltens P, Gholkar A, Ballard C, McKeith I, Ince P, Perry R, O’Brien J (1999) White matter lesions on magnetic resonance imaging in dementia with Lewy bodies, Alzheimer’s disease, vascular dementia, and normal aging. J Neurol Neurosurg Psychiatry 67:66–72
Barker WW, Luis CA, Kashuba A, Luis M, Harwood DG, Loewenstein D, Waters C, Jimison P, Shepherd E, Sevush S, Graff-Radford N, Newland D, Todd M, Miller B, Gold M, Heilman K, Doty L, Goodman I, Robinson B, Pearl G, Dickson D, Duara R (2002) Relative frequencies of Alzheimer disease, Lewy body, vascular and frontotemporal dementia, and hippocampal sclerosis in the State of Florida Brain Bank. Alzheimer Dis Assoc Disord 16:203–212
Barkhof F, Scheltens P (2002) Imaging of white matter lesions. Cerebrovasc Dis 13(Suppl 2):21–30
Barret AM (1913) Presenile, arteriosclerotic and senile disorders of the brain and cord. In: White WA, Jellife SA (eds) The modern treatment of nervous and mental diseases. Kimpton, London, pp 675–709
Bartzokis G, Cummings JL, Sultzer D, Henderson VW, Nuechterlein KH, Mintz J (2003) White matter structural integrity in healthy aging adults and patients with Alzheimer disease: a magnetic resonance imaging study. Arch Neurol 60:393–398
Basso A, Capitani E, Luzzatti C, Spinnler H (1981) Intelligence and left hemisphere disease. The role of aphasia, apraxia and size of lesion. Brain 104:721–734
Beach TG, Sue L, Scott S, Layne K, Newell A, Walker D, Baker M, Sahara N, Yen SH, Hutton M, Caselli R, Adler C, Connor D, Sabbagh M (2003) Hippocampal sclerosis dementia with tauopathy. Brain Pathol 13:263–278
Beach TG, Wilson JR, Sue LI, Newell A, Poston M, Cisneros R, Pandya Y, Esh C, Connor DJ, Sabbagh M, Walker DG, Roher AB (2007): Circle of Willis atherosclerosis: association with Alzheimer's disease, neuritic plaques and neurofibrillary tangles. Acta Neuropathol (Brl) 113:13–21
Bennett DA, Wilson RS, Gilley DW, Fox JH (1990) Clinical diagnosis of Binswanger’s disease. J Neurol Neurosurg Psychiatry 53:961–965
Bennett DA, Schneider JA, Bienias JL, Evans DA, Wilson RS (2005) Mild cognitive impairment is related to Alzheimer disease pathology and cerebral infarctions. Neurology 64:834–841
Benson DF, Cummings JL (1982) Angular gyrus syndrome simulating Alzheimer’s disease. Arch Neurol 39:616–620
Benson MD (1996) Leptomeningeal amyloid and variant transthyretins. Am J Pathol 148:351–354
Berg L, McKeel DW Jr, Miller JP, Storandt M, Rubin EH, Morris JC, Baty J, Coats M, Norton J, Goate AM, Price JL, Gearing M, Mirra SS, Saunders AM (1998) Clinicopathologic studies in cognitively healthy aging and Alzheimer’s disease: relation of histologic markers to dementia severity, age, sex, and apolipoprotein E genotype. Arch Neurol 55:326–335
Bhatia KP, Marsden CD (1994) The behavioural and motor consequences of focal lesions of the basal ganglia in man. Brain 117:859–876
Bierer LM, Hof PR, Purohit DP, Carlin L, Schmeidler J, Davis KL, Perl DP (1995) Neocortical neurofibrillary tangles correlate with dementia severity in Alzheimer’s disease. Arch Neurol 52:81–88
Binswanger O (1894) Die Abgrenzung der allgemeinen progressiven Paralyse. Berliner Klin Wochenschr 31:1103–1105, 1137–1139, 1180–1186
Blass JP, Hoyer S, Nitsch R (1991) A translation of Otto Binswanger’s article, ‘The delineation of the generalized progressive paralyses’. 1894. Arch Neurol 48:961–972
Blevins G, Macaulay R, Harder S, Fladeland D, Yamashita T, Yazaki M, Hamidi Asl K, Benson MD, Donat JR (2003) Oculoleptomeningeal amyloidosis in a large kindred with a new transthyretin variant Tyr69His. Neurology 60:1625–1630
Boiten J, Lodder J (2002) Risk factors for lacunar stroke. In: Donnan GA, Norrving B, Bogousslavski J (eds) Subcortical stroke. Oxford University Press, Oxford, pp 87–97
Bokura H, Kobayashi S, Yamaguchi S (1998) Distinguishing silent lacunar infarction from enlarged Virchow–Robin spaces: a magnetic resonance imaging and pathological study. J Neurol 245:116–122
Bouras C, Kovari E, Herrmann FR, Rivara CB, Bailey TL, von Gunten A, Hof PR, Giannakopoulos P (2006) Stereologic analysis of microvascular morphology in the elderly: Alzheimer disease pathology and cognitive status. J Neuropathol Exp Neurol 65:235–244
Bowler J (2003) Epidemiology: identifying vascular cognitive impairment. Int Psychogeriatr 15(Suppl 1):115–122
Bowler JV, Hachinski V (1995) Vascular cognitive impairment: a new approach to vascular dementia. In: Hachinski V (ed) Baillière’s clinical neurology. Cerebrovascular disease. Baillière Tindall, London, pp 357–376
Bowler JV, Hachinski V (1995) Vascular cognitive impairment: a new approach to vascular dementia. Baillieres Clin Neurol 4:357–376
Bowler JV, Hachinski V (eds) (2003) Vascular cognitive impairment. Preventable dementia. Oxford University Press, Oxford
Bowler JV, Hachinski V (2003) Current criteria for vascular dementia—a critical appraisal. In: Bowler JV, Hachinski V (eds) Vascular cognitive impairment. Preventable dementia. Oxford University Press, Oxford, pp 1–11
Bowler JV (2005) Vascular cognitive impairment. J Neurol Neurosurg Psychiatry 76(Suppl 5):35–44
Bracco L, Campani D, Baratti E, Lippi A, Inzitari D, Pracucci G, Amaducci L (1993) Relation between MRI features and dementia in cerebrovascular disease patients with leukoaraiosis: a longitudinal study. J Neurol Sci 120:131–136
Bracco L, Piccini C, Moretti M, Mascalchi M, Sforza A, Nacmias B, Cellini E, Bagnoli S, Sorbi S (2005) Alzheimer’s disease: role of size and location of white matter changes in determining cognitive deficits. Dement Geriatr Cogn Disord 20:358–366
Braffman BH, Zimmerman RA, Trojanowski JQ, Gonatas NK, Hickey WF, Schlaepfer WW (1988) Brain MR: pathologic correlation with gross and histopathology. 1. Lacunar infarction and Virchow–Robin spaces. AJR Am J Roentgenol 151:551–558
Breteler MM, van Swieten JC, Bots ML, Grobbee DE, Claus JJ, van den Hout JH, van Harskamp F, Tanghe HL, de Jong PT, van Gijn J et al (1994) Cerebral white matter lesions, vascular risk factors, and cognitive function in a population-based study: the Rotterdam Study. Neurology 44:1246–1252
Brion S, Pragier G, Guerin R, Teitgen M (1969) Syndrome de Korsakoff par ramollisement bilatéral du fornix. Rev Neurol 120:255–262
Brown MM, Pelz DM, Hachinski VC (1990) White matter vasodilatory reserve is impaired in patients with cerebrovascular disease and diffuse periventricular lacunes. J Neurol 237:157
Brown WR, Moody DM, Thore CR, Challa VR (2000) Cerebrovascular pathology in Alzheimer’s disease and leukoaraiosis. Ann N Y Acad Sci 903:39–45
Brown WR, Moody DM, Challa VR, Thore CR, Anstrom JA (2002) Venous collagenosis and arteriolar tortuosity in leukoaraiosis. J Neurol Sci 203–204:159–163
Brun A, Englund E (1986) A white matter disorder in dementia of the Alzheimer type: a pathoanatomical study. Ann Neurol 19:253–262
Brun A, Fredriksson K, Gustafson L (1992) Pure subcortical arteriosclerotic encephalopathy (Binswanger’s disease): a clinicopathological study. Part I: pathological features. Cerebrovasc Dis 2:87–92
Brun A (1994) Pathology and pathophysiology of cerebrovascular dementia: pure subgroups of obstructive and hypoperfusive etiology. Dementia 5:145–147
Burton E, Ballard C, Stephens S, Kenny RA, Kalaria R, Barber R, O’Brien J (2003) Hyperintensities and fronto-subcortical atrophy on MRI are substrates of mild cognitive deficits after stroke. Dement Geriatr Cogn Disord 16:113–118
Cabrejo L, Chassagne P, Doucet J, Laquerriere A, Puech N, Hannequin D (2006) Sporadic cerebral amyloidotic angiopathy. Rev Neurol (Paris) 162:1059–1067
Cadavid D, Mena H, Koeller K, Frommelt RA (2000) Cerebral beta amyloid angiopathy is a risk factor for cerebral ischemic infarction. A case control study in human brain biopsies. J Neuropathol Exp Neurol 59:768–773
Cannata AP, Alberoni M, Franceschi M, Mariani C (2002) Frontal impairment in subcortical ischemic vascular dementia in comparison to Alzheimer’s disease. Dement Geriatr Cogn Disord 13:101–111
Capizzano AA, Acion L, Bekinschtein T, Furman M, Gomila H, Martinez A, Mizrahi R, Starkstein SE (2004) White matter hyperintensities are significantly associated with cortical atrophy in Alzheimer’s disease. J Neurol Neurosurg Psychiatry 75:822–827
Caplan LR (1995) Binswanger’s disease—revisited. Neurology 45:626–633
Caplan LR, Helgason CM (1995) Caudate infarcts. In: Bogousslavsky J (ed) Lacunar and other subcortical infarctions. Oxford University Press, Oxford, pp 117–130
Caplan LR (2002) Caudate infarcts. In: Bogousslavsky J (ed) Subcortical stroke, 2nd edn. Oxford University Press, Oxford, pp 208–223
Castaigne P, Lhermitte F, Buge A, Escourolle R, Hauw JJ, Lyon-Caen O (1981) Paramedian thalamic and midbrain infarct: clinical and neuropathological study. Ann Neurol 10:127–148
Chabriat H, Bousser MG (2002) Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. In: Bogousslavsky J (ed) Subcortical stroke, 2nd edn. Oxford University Press, Oxford, pp 116–126
Challa VR, Thore CR, Moody DM, Anstrom JA, Brown WR (2004) Increase of white matter string vessels in Alzheimer’s disease. J Alzheimers Dis 6:379–383
Chen YF, Wang H, Chu Y, Huang YC, Su MY (2006) Regional quantification of white matter hyperintensity in normal aging, mild cognitive impairment, and Alzheimer’s disease. Dement Geriatr Cogn Disord 22:177–184
Chen YW, Gurol ME, Rosand J, Viswanathan A, Rakich SM, Groover TR, Greenberg SM, Smith EE (2006) Progression of white matter lesions and hemorrhages in cerebral amyloid angiopathy. Neurology 67:83–87
Chiu HF, Lam LC, Chi I, Leung T, Li SW, Law WT, Chung DW, Fung HH, Kan PS, Lum CM, Ng J, Lau J (1998) Prevalence of dementia in Chinese elderly in Hong Kong. Neurology 50:1002–1009
Cho KO, La HO, Cho YJ, Sung KW, Kim SY (2006) Minocycline attenuates white matter damage in a rat model of chronic cerebral hypoperfusion. J Neurosci Res 83:285–291
Christiansen P, Larsson HB, Thomsen C, Wieslander SB, Henriksen O (1994) Age dependent white matter lesions and brain volume changes in healthy volunteers. Acta Radiol 35:117–122
Chui HC, Victoroff JI, Margolin D, Jagust W, Shankle R, Katzman R (1992) Criteria for the diagnosis of ischemic vascular dementia proposed by the State of California Alzheimer’s Disease Diagnostic and Treatment Centers. Neurology 42:473–480
Chui HC, Mack W, Jackson JE, Mungas D, Reed BR, Tinklenberg J, Chang FL, Skinner K, Tasaki C, Jagust WJ (2000) Clinical criteria for the diagnosis of vascular dementia: a multicenter study of comparability and interrater reliability. Arch Neurol 57:191–196
Chui HC (2006) Vascular cognitive impairment: today and tomorrow. Alzheimer’s Dement 2:185–194
Chukwudelunzu FE, Meschia JF, Graff-Radford NR, Lucas JA (2001) Extensive metabolic and neuropsychological abnormalities associated with discrete infarction of the genu of the internal capsule. J Neurol Neurosurg Psychiatry 71:658–662
Cole FM, Yates PO (1968) Comparative incidence of cerebrovascular lesions in normotensive and hypertensive patients. Neurology 18:255–259
Cook IA, Leuchter AF, Morgan ML, Dunkin JJ, Witte E, David S, Mickes L, O’Hara R, Simon S, Lufkin R, Abrams M, Rosenberg S (2004) Longitudinal progression of subclinical structural brain disease in normal aging. Am J Geriatr Psychiatry 12:190–200
Corbett A, Bennett H, Kos S (1994) Cognitive dysfunction following subcortical infarction. Arch Neurol 51:999–1007
Cordoliani-Mackowiak MA, Henon H, Pruvo JP, Pasquier F, Leys D (2003) Poststroke dementia: influence of hippocampal atrophy. Arch Neurol 60:585–590
Corey-Bloom J, Sabbagh MN, Bondi MW, Hansen L, Alford MF, Masliah E, Thal LJ (1997) Hippocampal sclerosis contributes to dementia in the elderly. Neurology 48:154–160
Cosentino SA, Jefferson AL, Carey M, Price CC, Davis-Garrett K, Swenson R, Libon DJ (2004) The clinical diagnosis of vascular dementia: A comparison among four classification systems and a proposal for a new paradigm. Clin Neuropsychol 18:6–21
Coulthard A, Blank SC, Bushby K, Kalaria RN, Burn DJ (2000) Distribution of cranial MRI abnormalities in patients with symptomatic and subclinical CADASIL. Br J Radiol 73:256–265
Crystal H, Dickson D (2002) Cerebral infarcts in patients with autopsy proven Alzheimer’s disease (Abstract). Neurobiol Aging 23:207
Cullen KM, Kócsi Z, Stone J (2006) Microvascular pathology in the aging human brain: Evidence that senile plaques are sites of microhaemorrhages. Neurobiol Aging 27:1786–1796
Cummings JL, Benson DF (1988) Psychological dysfunction accompanying subcortical dementias. Annu Rev Med 39:53–61
Cummings JL (1993) Frontal-subcortical circuits and human behavior. Arch Neurol 50:873–880
D’Abreu A, Ott BR (2005) Poststroke dementia. In: Paul RH, Cohen R, Ott BR, Salloway S (eds) Vascular dementia: cerebrovascular mechanisms and clinical management. Human Press Inc. Totowa, pp 231–241
Davis DG, Schmitt FA, Wekstein DR, Markesbery WR (1999) Alzheimer neuropathologic alterations in aged cognitively normal subjects. J Neuropathol Exp Neurol 58:376–388
de Freitas GR, Bogousslavsky J (2002) Thalamic infarcts. In: Bogousslavsky J (ed) Subcortical stroke, 2nd edn. Oxford University Press, Oxford, pp 255–285
de Groot JC, de Leeuw FE, Oudkerk M, van Gijn J, Hofman A, Jolles J, Breteler MM (2000) Cerebral white matter lesions and cognitive function: the Rotterdam Scan Study. Ann Neurol 47:145–151
de Groot JC, de Leeuw FE, Oudkerk M, van Gijn J, Hofman A, Jolles J, Breteler MM (2002) Periventricular cerebral white matter lesions predict rate of cognitive decline. Ann Neurol 52:335–341
de Leeuw FE, de Groot JC, Oudkerk M, Witteman JC, Hofman A, van Gijn J, Breteler MM (2002) Hypertension and cerebral white matter lesions in a prospective cohort study. Brain 125:765–772
de Leeuw FE, Korf E, Barkhof F, Scheltens P (2006) White matter lesions are associated with progression of medial temporal lobe atrophy in Alzheimer disease. Stroke 37:2248–2252
de Mendonca A, Ribeiro F, Guerreiro M, Palma T, Garcia C (2005) Clinical significance of subcortical vascular disease in patients with mild cognitive impairment. Eur J Neurol 12:125–130
de Reuck J, Schaumburg HH (1972) Periventricular atherosclerotic leukoencephalopathy. Neurology 22:1094–1097
de Reuck J, Van der Eecken H (1976) The topography of infarcts in the lacunar state. In: Reivich M (ed) Cerebral vascular disease, vol 7. Thieme Stuttgart, pp 162–165
de Reuck J, Sieben G, de Coster W, vander Eecken H (1982) Dementia and confusional state in patients with cerebral infarcts. A clinicopathological study. Eur Neurol 21:94–97
de Reuck J, de Reus R, de Koninck J (1987) Sneddon’s syndrome A not unusual cause of stroke in young women. In: Ott E (ed) Cerebral vascular disease. Excerpta medica, vol 6, Amsterdam, pp 171–174
de Reuck J (1994) Neuropathology of vascular dementia. In: Scheltens P (ed) Current issues in neurodegenerative diseases, vol 6: vascular dementia. ICG Amsterdam, pp 9–15
DeCarli C, Miller BL, Swan GE, Reed T, Wolf PA, Carmelli D (2001) Cerebrovascular and brain morphologic correlates of mild cognitive impairment in the National Heart, Lung, and Blood Institute Twin Study. Arch Neurol 58:643–647
del Ser T, Bermejo F, Portera A, Arredondo JM, Bouras C, Constantinidis J (1990) Vascular dementia. A clinicopathological study. J Neurol Sci 96:1–17
Derouesne C, Gray F, Escourelle R, Castaigne P (1987) ‘Expanding cerebral lacunae’ in a hypertensive patient with normal pressure hydrocephalus. Neuropathol Appl Neurobiol 13:309–320
Derouesne C, Poirier J (1999) [Cerebral lacunae: still under debate]. Rev Neurol Paris 155:823–831
Derouesne C (2005) Vascular dementia: the dubious disease (French). Psychol Neuropsychiatr Vieil 3:89–96
Desmond DW (1996) Vascular dementia: a construct in evolution. Cerebrovasc Brain Metab Rev 8:296–325
Desmond DW, Moroney JT, Sano M, Stern Y (2002) Incidence of dementia after ischemic stroke: results of a longitudinal study. Stroke 33:2254–2260
Dichgans M, Mayer M, Uttner I, Bruning R, Muller-Hocker J, Rungger G, Ebke M, Klockgether T, Gasser T (1998) The phenotypic spectrum of CADASIL: clinical findings in 102 cases. Ann Neurol 44:731–739
Dickson DW, Davies P, Bevona C, Van Hoeven KH, Factor SM, Grober E, Aronson MK, Crystal HA (1994) Hippocampal sclerosis: a common pathological feature of dementia in very old (> or = 80 years of age) humans. Acta Neuropathol (Berl) 88:212–221
Dickson DW (2001) Neuropathology of Alzheimer’s disease and other dementias. Clin Geriatr Med 17:209–228
Doody RS, Azher SN, Haykal HA, Dunn JK, Liao T, Schneider L (2000) Does APO epsilon4 correlate with MRI changes in Alzheimer’s disease? J Neurol Neurosurg Psychiatry 69:668–671
Du AT, Schuff N, Laakso MP, Zhu XP, Jagust WJ, Yaffe K, Kramer JH, Miller BL, Reed BR, Norman D, Chui HC, Weiner MW (2002) Effects of subcortical ischemic vascular dementia and AD on entorhinal cortex and hippocampus. Neurology 58:1635–1641
Du A-T, Schuff N, Chao LL, Kornak J, Ezekiel F, Jagust WJ, Kramer JH, Reed BR, Miller BL, Norman D, Chui HC, Weiner MW (2005) White matter lesions are associated with cortical atrophy more than entorhinal and hippocampal atrophy. Neurobiol Aging 26:553–559
Dupont S (2003) [The anatomy of episodic memory: evolution of concepts]. Morphologie 87:5–9
Duyckaerts C, Delaere P, Hauw JJ, Abbamondi-Pinto AL, Sorbi S, Allen I, Brion JP, Flament-Durand J, Duchen L, Kauss J et al (1990) Rating of the lesions in senile dementia of the Alzheimer type: concordance between laboratories. A European multicenter study under the auspices of EURAGE. J Neurol Sci 97:295–323
Duyckaerts C, Dickson DW (2003) Neuropathology of Alzheimer´s disease. In: Dickson DW (ed) Neurodegeneration. The molecular pathology of dementia and movement disorders. ISN Neuropath Press, Basel, pp 47–65
Englund E (2000) White matter pathology in vascular dementia. In: Folstein M (ed) Cerebrovascular disease and dementia. pathology, neuropsychiatry and management. Martin Dunitz Publ, London, pp 77–84
Englund E (2002) Neuropathology of white matter lesions in vascular cognitive impairment. Cerebrovasc Dis 13(Suppl 2):11–15
Englund E (2004) White matter pathology of vascular dementia. In: O’Brien J, Ames D, Gustafson L, Foctin M, Chui E (eds) Vascular dementia. M. Dunitz, London, pp 117–130
Enzinger C, Smith S, Fazekas F, Drevin G, Ropele S, Nichols T, Behrens T, Schmidt R, Matthews PM (2006) Lesion probability maps of white matter hyperintensities in elderly individuals: results of the Austrian stroke prevention study. J Neurol 253:1064–1070
Erkinjuntti T, Haltia M, Palo J, Sulkava R, Paetau A (1988) Accuracy of the clinical diagnosis of vascular dementia: a prospective clinical and post-mortem neuropathological study. J Neurol Neurosurg Psychiatry 51:1037–1044
Erkinjuntti T, Benavente O, Eliasziw M, Munoz DG, Sulkava R, Haltia M, Hachinski V (1996) Diffuse vacuolization (spongiosis) and arteriolosclerosis in the frontal white matter occurs in vascular dementia. Arch Neurol 53:325–332
Erkinjuntti T (1999) Cerebrovascular dementia: pathophysiology, diagnosis and treatment. CNS Drugs 12:35–48
Erkinjuntti T (2000) Classification and criteria. In: Folstein M (ed) Cerebrovascular disease and dementia. Pathology, neuropsychiatry and management. Martin Dunitz Publ, London, pp 99–113
Erkinjuntti T, Inzitari D, Pantoni L, Wallin A, Scheltens P, Rockwood K, Roman GC, Chui H, Desmond DW (2000) Research criteria for subcortical vascular dementia in clinical trials. J Neural Transm Suppl 59:23–30
Erkinjuntti T (2002) Subcortical vascular dementia. Cerebrovasc Dis 13(Suppl 2):58–60
Escourolle R, Gray F (1975) Les accidents vasculaires du systeme limbique. In: 7th Congr Neuropathol Budapest. Excerpta Medica Amsterdam
Esiri MM, Wilcock GK (1986) Cerebral amyloid angiopathy in dementia and old age. J Neurol Neurosurg Psychiatry 49:1221–1226
Esiri MM, Hyman BT, Beyreuther K, Masters CL (1997) Vascular dementia. In: Lantos P (ed) Greenfield’s neuropathology, 6th edn. Arnold Publishing, London, pp 204–210
Esiri MM, Wilcock GK, Morris JH (1997) Neuropathological assessment of the lesions of significance in vascular dementia. J Neurol Neurosurg Psychiatry 63:749–753
Esiri MM (2000) Which vascular lesions are of importance in vascular dementia? Ann N Y Acad Sci 903:239–243
Farkas E, Luiten PG (2001) Cerebral microvascular pathology in aging and Alzheimer’s disease. Prog Neurobiol 64:575–611
Farkas E, Donka G, De Vos RA, Mihaly A, Bari F, Luiten PG (2004) Experimental cerebral hypoperfusion induces white matter injury and microglial activation in the rat brain. Acta Neuropathol (Berl) 108:57–64
Farkas E, de Vos RA, Donka G, Jansen Steur EN, Mihaly A, Luiten PG (2006) Age-related microvascular degeneration in the human cerebral periventricular white matter. Acta Neuropathol (Berl) 111:150–157
Farkas E, Luiten PG (2007) Permanent, bilateral common carotid artery occlusion in the rat: a model for chronic cerebral hypoperfusion-related neurodegenerative disease. Brain Res (in press)
Fayet G, Hauw JJ, Delaere P, He Y, Duyckaerts C, Beck H, Forette F, Gallinari C, Laurent M, Moulias R et al (1994) Neuropathology of 20 centenarians. I: Clinical data. Rev Neurol 150:16–21
Fazekas F, Schmidt R, Kleinert R, Kapeller P, Roob G, Flooh E (1998) The spectrum of age-associated brain abnormalities: their measurement and histopathological correlates. J Neural Transm Suppl 53:31–39
Feigin L, Poport N (1963) Neuropathological changes late in cerebral edema. The relationship to trauma, hypertensive disease and Binswanger’s encephalopathy. J Neuropathol Exp Neurol 22:500–511
Fein G, Di Sclafani V, Tanabe J, Cardenas V, Weiner MW, Jagust WJ, Reed BR, Norman D, Schuff N, Kusdra L, Greenfield T, Chui H (2000) Hippocampal and cortical atrophy predict dementia in subcortical ischemic vascular disease. Neurology 55:1626–1635
Ferguson SC, Blane A, Perros P, McCrimmon RJ, Best JJ, Wardlaw J, Deary IJ, Frier BM (2003) Cognitive ability and brain structure in type 1 diabetes: relation to microangiopathy and preceding severe hypoglycemia. Diabetes 52:149–156
Fernando MS, Ince PG (2004) Vascular pathologies and cognition in a population-based cohort of elderly people. J Neurol Sci 226:13–17
Fernando MS, O’Brien JT, Perry RH, English P, Forster G, McMeekin W, Slade JY, Golkhar A, Matthews FE, Barber R, Kalaria RN, Ince PG (2004) Comparison of the pathology of cerebral white matter with post-mortem magnetic resonance imaging (MRI) in the elderly brain. Neuropathol Appl Neurobiol 30:385–395
Fernando MS, Simpson JE, Matthews F, Brayne C, Lewis CE, Barber R, Kalaria RN, Forster G, Esteves F, Wharton SB, Shaw PJ, O’Brien JT, Ince PG (2006) White matter lesions in an unselected cohort of the elderly: molecular pathology suggests origin from chronic hypoperfusion injury. Stroke 37:1391–1398
Ferrand J (1902) Essai sur l’hémiplégie des viellards. Les lacunes de désintégration cérébrale. Thesis, Rousset, Paris
Ferrer I, Bella R, Serrano MT, Marti E, Guionnet N (1990) Arteriolosclerotic leucoencephalopathy in the elderly and its relation to white matter lesions in Binswanger’s disease, multi-infarct encephalopathy and Alzheimer’s disease. J Neurol Sci 98:37–50
Filley CM, Thompson LL, Sze CI, Simon JA, Paskavitz JF, Kleinschmidt-DeMasters BK (1999) White matter dementia in CADASIL. J Neurol Sci 163:163–167
Firbank MJ, Minett T, O’Brien JT (2003) Changes in DWI and MRS associated with white matter hyperintensities in elderly subjects. Neurology 61:950–954
Fischer P, Lassmann H, Jellinger K, Simanyi M, Bancher C, Travniczek-Marterer A, Gatterer G, Danielczyk W (1991) [Alzheimer dementia. A clinical long-term study with quantitative neuropathology]. Wien Med Wochenschr 141:455–462
Fisher CM (1965) The vascular lesion in lacunae. Trans Am Neurol Assoc 90:243–245
Fisher CM (1968) Dementia in cerebral vascular disease. In: Whisnant J (ed) Cerebral vascular disease. Sixth Princeton Conference. Greene and Stratton, New York, pp 232–236
Fisher CM (1968) The arterial lesions underlying lacunes. Acta Neuropathol Berl 12:1–15
Fisher CM (1979) Capsular infarcts: the underlying vascular lesions. Arch Neurol 36:65–73
Fisher CM (1982) Lacunar strokes and infarcts: a review. Neurology 32:871–876
Fisher CM (1989) Binswanger’s encephalopathy: a review. J Neurol 236:65–79
Fratiglioni L, Grut M, Forsell Y, Viitanen M, Grafstrom M, Holmen K, Ericsson K, Backman L, Ahlbom A, Winblad B (1991) Prevalence of Alzheimer’s disease and other dementias in an elderly urban population: relationship with age, sex, and education. Neurology 41:1886–1892
Galasko D, Hansen LA, Katzman R, Wiederholt W, Masliah E, Terry R, Hill LR, Lessin P, Thal LJ (1994) Clinical–neuropathological correlations in Alzheimer’s disease and related dementias. Arch Neurol 51:888–895
Gamaldo A, Moghekar A, Kilada S, Resnick SM, Zonderman AB, O’Brien R (2006) Effect of a clinical stroke on the risk of dementia in a prospective cohort. Neurology 67:1363–1369
Garcia JH, Brown GG (1992) Vascular dementia: neuropathologic alterations and metabolic brain changes. J Neurol Sci 109:121–131
Garcia JH, Lassen NA, Weiller C, Sperling B, Nakagawa J (1995) Ischemic stroke and incomplete infarction. Stroke 27:761–765
Garde E, Mortensen EL, Krabbe K, Rostrup E, Larsson HB (2000) Relation between age-related decline in intelligence and cerebral white-matter hyperintensities in healthy octogenarians: a longitudinal study. Lancet 356:628–634
Garde E, Lykke Mortensen E, Rostrup E, Paulson OB (2005) Decline in intelligence is associated with progression in white matter hyperintensity volume. J Neurol Neurosurg Psychiatry 76:1289–1291
Gauthier S (ed) (2007) Clinical diagnosis and management of Alzheimer’s disease, 3rd edn. Informa Healthcare, London
Gertz HJ, Wolf H, Arendt T (2002) [Vascular dementia]. Nervenarzt 73:393–404
Geschwind N (1965) Disconnexion syndromes in animals and man. I. Brain 88:237–294
Giannakopoulos P, Hof PR, Surini M, Michel JP, Bouras C (1993) Quantitative immunohistochemical analysis of the distribution of neurofibrillary tangles and senile plaques in the cerebral cortex of nonagenarians and centenarians. Acta Neuropathol 85:602–610
Giannakopoulos P, Hof PR, Michel JP, Guimon J, Bouras C (1997) Cerebral cortex pathology in aging and Alzheimer’s disease: a quantitative survey of large hospital-based geriatric and psychiatric cohorts. Brain Res Brain Res Rev 25:217–245
Giannakopoulos P, Gold G, Kövari E, von Gunten A, Imhof A, Bouras C, Hof PR (2007) Assessing the cognitive impact of Alzheimer disease pathology and vascular burden in the aging brain: the Geneva experience. Acta Neuropathol 113:1–12
Glees P, Griffith GH (1953) Bilateral destruction of the hippocampus in case of dementia. Monatsschr Psychiatr Neurol 123:193–204
Gold G, Giannakopoulos P, Montes-Paixao Junior C, Herrmann FR, Mulligan R, Michel JP, Bouras C (1997) Sensitivity and specificity of newly proposed clinical criteria for possible vascular dementia. Neurology 49:690–694
Gold G, Bouras C, Canuto A, Bergallo MF, Herrmann FR, Hof PR, Mayor PA, Michel JP, Giannakopoulos P (2002) Clinicopathological validation study of four sets of clinical criteria for vascular dementia. Am J Psychiatry 159:82–87
Gold G, Kovari E, Herrmann FR, Canuto A, Hof PR, Michel JP, Bouras C, Giannakopoulos P (2005) Cognitive consequences of thalamic, basal ganglia, and deep white matter lacunes in brain aging and dementia. Stroke 36:1184–1188
Gorelick PB (1997) Status of risk factors for dementia associated with stroke. Stroke 28:459–463
Goulding J, Signorini D, Chatterjee S, Nicoll J, Stewart J, Morris R, Lammie G (1999) Inverse relation between Braak stage and cerebrovascular pathology in Alzheimer predominant dementia. J Neurol Neurosurg Psychiatry 67:654–657
Grabowski TJ, Cho HS, Vonsattel JP, Rebeck GW, Greenberg SM (2001) Novel amyloid precursor protein mutation in an Iowa family with dementia and severe cerebral amyloid angiopathy. Ann Neurol 49:697–705
Grafton ST, Sumi SM, Stimac GK, Alvord EC Jr, Shaw CM, Nochlin D (1991) Comparison of postmortem magnetic resonance imaging and neuropathologic findings in the cerebral white matter. Arch Neurol 48:293–298
Gray F, Dubas F, Roullet E, Escourolle R (1985) Leukoencephalopathy in diffuse hemorrhagic cerebral amyloid angiopathy. Ann Neurol 18:54–59
Greenberg SM, Gurol ME, Rosand J, Smith EE (2004) Amyloid angiopathy-related vascular cognitive impairment. Stroke 35:2616–2619
Gunning-Dixon FM, Raz N (2003) Neuroanatomical correlates of selected executive functions in middle-aged and older adults: a prospective MRI study. Neuropsychologia 41:1929–1941
Gurol ME, Irizarry MC, Smith EE, Raju S, Diaz-Arrastia R, Bottiglieri T, Rosand J, Growdon JH, Greenberg SM (2006) Plasma beta-amyloid and white matter lesions in AD, MCI, and cerebral amyloid angiopathy. Neurology 66:23–29
Gustafson I, Passant U (2000) Clinical pathological correlates. In: Folstein M (ed) Cerebrovascular disease and dementia. Pathology, neuropsychiatry and management. Martin Dunitz Publ, London, pp 85–97
Hachinski V (2006) Commentary on “Vascular cognitive impairment: today and tomorrow.” Vascular cognitive impairment: yesterday, today, and tomorrow. Alzheimers Dement 2:198–199
Hachinski V, Iadecola C, Petersen RC, Breteler MM, Nyenhuis DL, Black SE, Powers WJ, DeCarli C, Merino JG, Kalaria RN, Vinters HV, Holtzman DM, Rosenberg GA, Dichgans M, Marler JR, Leblanc GG (2006) National Institute of Neurological Disorders and Stroke-Canadian Stroke Network vascular cognitive impairment harmonization standards. Stroke 37:2220–2241
Hachinski VC, Lassen NA, Marshall J (1974) Multi-infarct dementia. A cause of mental deterioration in the elderly. Lancet 2:207–210
Hachinski VC, Iliff L, Zihlka E, Du Boulay G, McAllister V, Marshall J, Russel R, Symon L (1975) Cerebral blood flow in dementia. Arch Neurol 32:632–637
Hachinski VC, Potter P, Merskey H (1987) Leuko-araiosis. Arch Neurol 44:21–23
Haglund M, Sjobeck M, Englund E (2004) Severe cerebral amyloid angiopathy characterizes an underestimated variant of vascular dementia. Dement Geriatr Cogn Disord 18:132–137
Haglund M, Passant U, Sjobeck M, Ghebremedhin E, Englund E (2006) Cerebral amyloid angiopathy and cortical microinfarcts as putative substrates of vascular dementia. Int J Geriatr Psychiatry 21:681–687
Halliday G, Ng T, Rodriguez M, Harding A, Blumbergs P, Evans W, Fabian V, Fryer J, Gonzales M, Harper C, Kalnins R, Masters CL, McLean C, Milder DG, Pamphlett R, Scott G, Tannenberg A, Kril J (2002) Consensus neuropathological diagnosis of common dementia syndromes: testing and standardising the use of multiple diagnostic criteria. Acta Neuropathol (Berl) 104:72–78
Haltia M, Ghiso J, Prelli F, Gallo G, Kiuru S, Somer H, Palo J, Frangione B (1990) Amyloid in familial amyloidosis, Finnish type, is antigenically and structurally related to gelsolin. Am J Pathol 136:1223–1228
Hamel E (2004) Cholinergic modulation of the cortical microvascular bed. Prog Brain Res 145:171–178
Hanyu H, Tanaka Y, Shimizu S, Takasaki M, Fujita H, Kaneko N, Yamamoto Y, Harada M (2003) Cerebral microbleeds in Binswanger’s disease: a gradient-echo T2*-weighted magnetic resonance imaging study. Neurosci Lett 340:213–216
Hasegawa K, Homma A, Imai Y (1986) An epidemiological study of age-related dementia in the community. Int J Geriatr Psychiatry 1:94–105
Hatanpaa KJ, Blass DM, Pletnikova O, Crain BJ, Bigio EH, Hedreen JC, White CL 3rd, Troncoso JC (2004) Most cases of dementia with hippocampal sclerosis may represent frontotemporal dementia. Neurology 63:538–542
Hauw JJ, Zekry D, Seilhean D, Forette B, Gallinari C, Laurent M, Moulias R, Piette F, Sachet A, Duyckaerts C (2002) Neuropathology of the cerebral vessels in centenarians. J Mal Vasc 27:S13–S18.
Hebert R, Brayne C (1995) Epidemiology of vascular dementia. Neuroepidemiology 14:240–257
Hebert R, Lindsay J, Verreault R, Rockwood K, Hill G, Dubois MF (2000) Vascular dementia: incidence and risk factors in the Canadian study of health and aging. Stroke 31:1487–1493
Heier LA, Bauer CJ, Schwartz L, Zimmerman RD, Morgello S, Deck MD (1989) Large Virchow–Robin spaces: MR-clinical correlation. AJNR Am J Neuroradiol 10:929–936
Henon H, Pasquier F, Durieu I, Godefroy O, Lucas C, Lebert F, Leys D (1997) Preexisting dementia in stroke patients. Baseline frequency, associated factors, and outcome. Stroke 28:2429–2436
Henon H, Durieu I, Guerouaou D, Lebert F, Pasquier F, Leys D (2001) Poststroke dementia: incidence and relationship to prestroke cognitive decline. Neurology 57:1216–1222
Hentschel F, Kreis M, Damian M, Krumm B (2003) [Microangiopathic lesions of white matter. Quantitation of cerebral MRI findings and correlation with psychological tests]. Nervenarzt 74:355–361
Herholz K, Perani D, Morris JC (2006) The dementias: early diagnosis and evaluation. Taylor & Francis, New York
Herzig MC, Winkler DT, Burgermeister P, Pfeifer M, Kohler E, Schmidt SD, Danner S, Abramowski D, Sturchler-Pierrat C, Burki K, van Duinen SG, Maat-Schieman ML, Staufenbiel M, Mathews PM, Jucker M (2004) Abeta is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat Neurosci 7:954–960
Heyman A, Fillenbaum G, Welsh-Bohmer KA et al (1998) Cerebral infarcts in patients with autopsy-proven Alzheimer’s disease, CERAD, Part XVIII (Abstr.). Neurology 51:159–162
Hill MD, Silver FL, Austin PC, Tu JV (2000) Rate of stroke recurrence in patients with primary intracerebral hemorrhage. Stroke 31:123–127
Holton JL, Lashley T, Ghiso J, Braendgaard H, Vidal R, Guerin CJ, Gibb G, Hanger DP, Rostagno A, Anderton BH, Strand C, Ayling H, Plant G, Frangione B, Bojsen-Moller M, Revesz T (2002) Familial Danish dementia: a novel form of cerebral amyloidosis associated with deposition of both amyloid-Dan and amyloid-beta. J Neuropathol Exp Neurol 61:254–267
Hughes W (1965) Hypothesis—origin of lacunes. Lancet 2:19–21
Hulette C, Nochlin D, McKeel D, Morris JC, Mirra SS, Sumi SM, Heyman A (1997) Clinical-neuropathologic findings in multi-infarct dementia: a report of six autopsied cases. Neurology 48:668–672
Hyman BT, Trojanowski JQ (1997) Consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J Neuropathol Exp Neurol 56:1095–1097
Hyman BT (1998) New neuropathological criteria for Alzheimer disease. Arch Neurol 55:1174–1176
Ikeda M, Hokoishi K, Maki N, Nebu A, Tachibana N, Komori K, Shigenobu K, Fukuhara R, Tanabe H (2001) Increased prevalence of vascular dementia in Japan: a community-based epidemiological study. Neurology 57:839–844
Ince P (2005) Acquired forms of vascular dementia. In: Kalimo H (ed) Cerebrovascular diseases. ISN Neuropath Press, Basel, pp 316–323
Ince PG, McArthur FK, Bjertness E, Torvik A, Candy JM, Edwardson JA (1995) Neuropathological diagnoses in elderly patients in Oslo: Alzheimer’s disease, Lewy body disease, vascular lesions. Dementia 6:162–168
Ince PG, Fernando MS (2003) Neuropathology of vascular cognitive impairment and vascular dementia. Int Psychogeriatr 15(Suppl 1):71–75
Ince PG, Fernando MS, Matthews F, Brayne C, Lowe JS, Esiri MM, O’Brien JT (2006) White matter lesions in an unselected cohort of the elderly: prevalence, relation to other pathologies and risk factors (Abstract). Brain Pathol 16(Suppl 1):286
Ingelsson M, Fikumoto H, Newell K, Hyman BT, Irizarry MC (2003) Lack of correlation between biochemical and neuropathological amyloid measures in the Alzheimer brain. In: Iqbal K, Winblad B (eds) Alzheimer’s disease and related disorders. Ana Aslan Intl Acad of Aging Bucharest, Romania, pp 193–201
Isaka Y, Okamoto M, Ashida K, Imaizumi M (1994) Decreased cerebrovascular dilatory capacity in subjects with asymptomatic periventricular hyperintensities. Stroke 25:375–381
Ishii N, Nishihara Y, Horie A (1984) Amyloid angiopathy and lobar cerebral haemorrhage. J Neurol Neurosurg Psychiatry 47:1203–1210
Ishunina TA, Kamphorst W, Swaab DF (2004) Metabolic alterations in the hypothalamus and basal forebrain in vascular dementia. J Neuropathol Exp Neurol 63:1243–1254
Itoh Y, Yamada M, Hayakawa M, Otomo E, Miyatake T (1993) Cerebral amyloid angiopathy: a significant cause of cerebellar as well as lobar cerebral hemorrhage in the elderly. J Neurol Sci 116:135–141
Ivan CS, Seshadri S, Beiser A, Au R, Kase CS, Kelly-Hayes M, Wolf PA (2004) Dementia after stroke: the Framingham Study. Stroke 35:1264–1268
Janota I, Mirsen TR, Hachinski VC, Lee DH, Merskey H (1989) Neuropathologic correlates of leuko-araiosis. Arch Neurol 46:1124–1128
Jellinger K, Neumayer E (1964) Progressive subcorticale Encephalopathie Binswanger. Eine klinisch-neuropathologische Studie. Arch Psych Z Ges Neurol 205:523–554
Jellinger K, Danielczyk W, Fischer P, Gabriel E (1990) Clinicopathological analysis of dementia disorders in the elderly. J Neurol Sci 95:239–258
Jellinger K, Mitter-Ferstl E (2003) The impact of cerebrovascular lesions in Alzheimer disease. A comparative autopsy study. J Neurol 150:1050–1055
Jellinger KA (1994) Hippocampal sclerosis: a common pathological feature of dementia in very old humans. Acta Neuropathol Berl 88:599
Jellinger KA (2000) Inverse correlation between cerebrovascular lesions and Braak stage letter. J Neurol Neurosurg Psychiatry 68:799–800
Jellinger KA (2001) Small concomitant cerebrovascular lesions are not important for cognitive decline in severe Alzheimer disease. Arch Neurol 58:520–521
Jellinger KA (2002) Vascular–ischemic dementia: an update. J Neural Transm Suppl 1–23
Jellinger KA (2002) The pathology of ischemic–vascular dementia: an update. J Neurol Sci 203–204:153–157
Jellinger KA (2002) Alzheimer disease and cerebrovascular pathology: an update. J Neural Transm 109:813–836
Jellinger KA, Attems J (2003) Incidence of cerebrovascular lesions in Alzheimer’s disease: a postmortem study. Acta Neuropathol (Berl) 105:14–17
Jellinger KA (2004) Pathology and pathophysiology of vascular cognitive impairment. A critical update. Panminerva Med 46:217–226
Jellinger KA (2004) The neuropathologic substrates of vascular–ischemic dementia. In: Paul RH, Cohen R, Ott BR, Salloway S (eds) Current clinical neurology. Vascular dementia: cerebrovascular mechanisms and clinical management. Humana Press, Totowa, pp 23–57
Jellinger KA (2004) Pathophysiology and pathogenesis of vascular cognitive impairment: a critical update. In: Clark LV (ed) Trends in atherosclerosis research. Nova Sciences Publishers, Hauppauge, pp 189–234
Jellinger KA (2005) Understanding the pathology of vascular cognitive impairment. J Neurol Sci 229–230:57–63
Jellinger KA, Attems J (2005) Prevalence and pathogenic role of cerebrovascular lesions in Alzheimer’s disease. J Neurol Sci 229–230:37–41
Jellinger KA (2006) A view on early diagnosis of dementias from neuropathology. In: Herholz K, Perani D, Morris CM (eds) The dementias: early diagnosis and evaluation. Taylor & Francis, New York, pp 311–428
Jellinger KA (2007) The enigma of mixed dementia. Alzheimers Dement (in press)
Johnston SC, O’Meara ES, Manolio TA, Lefkowitz D, O’Leary DH, Goldstein S, Carlson MC, Fried LP, Longstreth WT Jr (2004) Cognitive impairment and decline are associated with carotid artery disease in patients without clinically evident cerebrovascular disease. Ann Intern Med 140:237–247
Jorm AF (2000) Epidemiology: meta-analysis. In: Folstein M (eds) Cerebrovascular disease and dementia. Pathology, neuropsychiatry and management. Martin Dunitz Publ, London, pp 85–97
Joutel A, Favrole P, Labauge P, Chabriat H, Lescoat C, Andreux F, Domenga V, Cecillon M, Vahedi K, Ducros A, Cave-Riant F, Bousser MG, Tournier-Lasserve E (2001) Skin biopsy immunostaining with a NOTCH3 monoclonal antibody for CADASIL diagnosis. Lancet 358:2049–2051
Kalaria RN, Kenny RA, Ballard CG, Perry R, Ince P, Polvikoski T (2004) Towards defining the neuropathological substrates of vascular dementia. J Neurol Sci 226:75–80
Kalimo H, Ruchoux MM, Viitanen M, Kalaria RN (2002) CADASIL: a common form of hereditary arteriopathy causing brain infarcts and dementia. Brain Pathol 12:371–384
Kalimo H, Kalaria RN (2005) Hereditary forms of vascular dementia. In: Kalimo H (ed) Pathology & genetics. Cerebrovascular diseases. ISN Neuropath Press, Basel, pp 324–334
Kase CS (1991) Epidemiology of multi-infarct dementia. Alzheimer Dis Assoc Disord 5:71–76
Keverne JS, Low WC, Ziabreva I, Court JA, Oakley AE, Kalaria RN (2006) Cholinergic neuronal deficits in CADASIL. Stroke. DOI 10.1161/1101.STR.0000251787.0000290695.0000251705
Khachaturian ZS (1985) Diagnosis of Alzheimer’s disease. Arch Neurol 42:1097–1105
Khachaturian ZS (2005) Diagnosis of Alzheimer’s disease: two decades of progress. Alzheimers Dement 1:93–98
Khachaturian ZS (2006) Diagnosis of Alzheimer’s disease: two decades of progress. J Alzheimers Dis 9:409–415
Kimura S, Saito H, Minami M, Togashi H, Nakamura N, Nemoto M, Parvez HS (2000) Pathogenesis of vascular dementia in stroke-prone spontaneously hypertensive rats. Toxicology 153:167–178
Kirkpatrick JB, Hayman LA (1987) White-matter lesions in MR imaging of clinically healthy brains of elderly subjects: possible pathologic basis. Radiology 162:509–511
Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergstrom M, Savitcheva I, Huang GF, Estrada S, Ausen B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA, Langstrom B (2004) Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol 55:306–319
Knopman DS, DeKosky ST, Cummings JL, Chui H, Corey-Bloom J, Relkin N, Small GW, Miller B, Stevens JC (2001) Practice parameter: diagnosis of dementia (an evidence-based review). Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 56:1143–1153
Knopman DS, Rocca WA, Cha RH, Edland SD, Kokmen E (2002) Incidence of vascular dementia in Rochester, Minn, 1985–1989. Arch Neurol 59:1605–1610
Knopman DS, Parisi JE, Boeve BF, Cha RH, Apaydin H, Salviati A, Edland SD, Rocca WA (2003) Vascular dementia in a population-based autopsy study. Arch Neurol 60:569–575
Knowles RB, Gomez-Isla T, Hyman BT (1998) Abeta associated neuropil changes: correlation with neuronal loss and dementia. J Neuropathol Exp Neurol 57:1122–1130
Knudsen KA, Rosand J, Karluk D, Greenberg SM (2001) Clinical diagnosis of cerebral amyloid angiopathy: validation of the Boston criteria. Neurology 56:537–539
Kobari M, Meyer JS, Ichijo M, Oravez WT (1990) Leukoaraiosis: correlation of MR and CT findings with blood flow, atrophy, and cognition. AJNR Am J Neuroradiol 11:273–281
Koga H, Yuzuriha T, Yao H, Endo K, Hiejima S, Takashima Y, Sadanaga F, Matsumoto T, Uchino A, Ogomori K, Ichimiya A, Uchimura H, Tashiro N (2002) Quantitative MRI findings and cognitive impairment among community dwelling elderly subjects. J Neurol Neurosurg Psychiatry 72:737–741
Kövari E, Gold G, Herrmann FR, Canuto A, Hof PR, Michel JP, Bouras C, Giannakopoulos P (2004) Cortical microinfarcts and demyelination significantly affect cognition in brain aging. Stroke 35:410–414
Kraepelin E (1910) Das senile und präsenile Irresein. In: Psychiatrie: Ein Lehrbuch für Studierende und Ärzte. Johann Ambrosius Barth Leipzig, pp 533–632
Kramer JH, Reed BR, Mungas D, Weiner MW, Chui HC (2002) Executive dysfunction in subcortical ischaemic vascular disease. J Neurol Neurosurg Psychiatry 72:217–220
Kril JJ, Patel S, Harding AJ, Halliday GM (2002) Patients with vascular dementia due to microvascular pathology have significant hippocampal neuronal loss. J Neurol Neurosurg Psychiatry 72:747–751
Kuller LH, Shemanski L, Manolio T, Haan M, Fried L, Bryan N, Burke GL, Tracy R, Bhadelia R (1998) Relationship between APOE, MRI findings, and cognitive function in the Cardiovascular Health Study. Stroke 29:388–398
Kuller LH, Lopez OL, Newman A, Beauchamp NJ, Burke G, Dulberg C, Fitzpatrick A, Fried L, Haan MN (2003) Risk factors for dementia in the cardiovascular health cognition study. Neuroepidemiology 22:13–22
Kuller LH, Lopez OL, Jagust WJ, Becker JT, DeKosky ST, Lyketsos C, Kawas C, Breitner JC, Fitzpatrick A, Dulberg C (2005) Determinants of vascular dementia in the Cardiovascular Health Cognition Study. Neurology 64:1548–1552
Kwan LT, Reed BR, Eberling JL, Schuff N, Tanabe J, Norman D, Weiner MW, Jagust WJ (1999) Effects of subcortical cerebral infarction on cortical glucose metabolism and cognitive function. Arch Neurol 56:809–814
Laitinen LV, Chudy D, Tengvar M, Hariz MI, Bergenheim AT (2000) Dilated perivascular spaces in the putamen and pallidum in patients with Parkinson’s disease scheduled for pallidotomy: a comparison between MRI findings and clinical symptoms and signs. Mov Disord 15:1139–1144
Lammie GA (2002) Pathology of lacunar infarction. In: Bogousslavsky J (ed) Subcortical stroke. Oxford University Press, Oxford, pp 38–46
Lammie GA (2002) Hypertensive cerebral small vessel disease and stroke. Brain Pathol 12:358–370
Lassen NA (1982) Incomplete cerebral infarction—focal incomplete ischemic tissue necrosis not leading to emollision. Stroke 13:522–523
Leaper SA, Murray AD, Lemmon HA, Staff RT, Deary IJ, Crawford JR, Whalley LJ (2001) Neuropsychologic correlates of brain white matter lesions depicted on MR images:1921 Aberdeen Birth Cohort. Radiology 221:51–55
Lee JH, Olichney JM, Hansen LA, Hofstetter CR, Thal LJ (2000) Small concomitant vascular lesions do not influence rates of cognitive decline in patients with Alzheimer disease. Arch Neurol 57:1474–1479
Lee JH, Park SY, Shin YW, Hong KW, Kim CD, Sung SM, Kim KY, Lee WS (2006) Neuroprotection by cilostazol, a phosphodiesterase type 3 inhibitor, against apoptotic white matter changes in rat after chronic cerebral hypoperfusion. Brain Res 1082:182–191
Lee JM, Markus HS (2006) Does the white matter in Alzheimer disease and cerebral amyloid angiopathy? Neurology 66:6–7
Leverenz JB, Agustin CM, Tsuang D, Peskind ER, Edland SD, Nochlin D, DiGiacomo L, Bowen JD, McCormick WC, Teri L, Raskind MA, Kukull WA, Larson EB (2002) Clinical and neuropathological characteristics of hippocampal sclerosis: a community-based study. Arch Neurol 59:1099–1106
Leys D, Bogousslavsky J (1994) Mechanisms of vascular dementia. In: Scheltens P (ed) Current issues in neurodegenerative diseases, vol 6: vascular dementia. ICG Amsterdam, pp 121–132
Leys D, Englund E, Erkinjuntti T (2002) Vascular dementia. In: Erkinjuntti T (ed) Evidence based dementia practice. Blackwell Science Publishing, Oxford, pp 260–287
Leys D, Henon H (2004) Many patients with dementia identified after stroke already had dementia present before. J Neurol 251:609–610
Leys D, Henon H, Mackowiak-Cordoliani MA, Pasquier F (2005) Poststroke dementia. Lancet Neurol 4:752–759
Li G, Shen YC, Chen CH, Zhau YW, Li SR, Lu M (1991) A three-year follow-up study of age-related dementia in an urban area of Beijing. Acta Psychiatr Scand 83:99–104
Liao D, Cooper L, Cai J, Toole J, Bryan N, Burke G, Shahar E, Nieto J, Mosley T, Heiss G (1997) The prevalence and severity of white matter lesions, their relationship with age, ethnicity, gender, and cardiovascular disease risk factors: the ARIC Study. Neuroepidemiology 16:149–162
Liebetrau M, Steen B, Hamann GF, Skoog I (2004) Silent and symptomatic infarcts on cranial computerized tomography in relation to dementia and mortality: a population-based study in 85-year-old subjects. Stroke 35:1816–1820
Lind K, Jonsson M, Karlsson I, Sjogren M, Wallin A, Edman A (2006) Depressive symptoms and white matter changes in patients with dementia. Int J Geriatr Psychiatry 21:119–125
Liu CK, Miller BL, Cummings JL, Mehringer CM, Goldberg MA, Howng SL, Benson DF (1992) A quantitative MRI study of vascular dementia. Neurology 42:138–143
Lobo A, Launer LJ, Fratiglioni L, Andersen K, Di Carlo A, Breteler MM, Copeland JR, Dartigues JF, Jagger C, Martinez-Lage J, Soininen H, Hofman A (2000) Prevalence of dementia and major subtypes in Europe: a collaborative study of population-based cohorts. Neurologic Diseases in the Elderly Research Group. Neurology 54:S4–S9
Loeb C (2000) Binswanger’s disease is not a single entity. Neurol Sci 21:343–348
Loeb C (2001) Neuropathological correlates of vascular dementia. In: Toole J (ed) Vascular dementia. Futura Publishing Armonk, NY, pp 59–75
Longstreth WT Jr, Manolio TA, Arnold A, Burke GL, Bryan N, Jungreis CA, Enright PL, O’Leary D, Fried L (1996) Clinical correlates of white matter findings on cranial magnetic resonance imaging of 3301 elderly people. The Cardiovascular Health Study. Stroke 27:1274–1282
Lopez OL, Kuller LH, Becker JT, Jagust WJ, DeKosky ST, Fitzpatrick A, Breitner J, Lyketsos C, Kawas C, Carlson M (2005) Classification of vascular dementia in the Cardiovascular Health Study Cognition Study. Neurology 64:1539–1547
Lopez OL (2006) Commentary on “Vascular cognitive impairment: today and tomorrow”. Alzheimers Dement 2:200–201
Love S, Hilton DA (1996) Assessment of the distribution of mitochondrial ribosomal RNA in melas and in thrombotic cerebral infarcts by in situ hybridization. J Pathol 178:182–189
Ma CK, Olsson Y (1997) The role of chronic brain edema in the formation of lacunes in Binswanger’s encephalopathy. Histopathology and immunohistochemical observations. Cerebrovasc Dis 7:324–331
Mackowiak-Cordoliani MA, Bombois S, Memin A, Henon H, Pasquier F (2005) Poststroke dementia in the elderly. Drugs Aging 22:483–493
Maclullich AM, Wardlaw JM, Ferguson KJ, Starr JM, Seckl JR, Deary IJ (2004) Enlarged perivascular spaces are associated with cognitive function in healthy elderly men. J Neurol Neurosurg Psychiatry 75:1519–1523
Mann DM, Yates PO, Marcyniuk B (1986) The nucleus basalis of Meynert in multi-infarct (vascular) dementia. Acta Neuropathol (Berl) 71:332–337
Marie P (1901) Des foyers lacunaires de désintégration et de différents autres états cavitaires du cerveau. Rev Med 21:281–298
Markesbery WR (1998) Vascular dementia. In: Markesbery W (ed) Neuropathology of dementing disorders. Arnold Publishers, London, pp 293–311
Markesbery WR (2001) Overview of vascular dementia. In: Winblad B (ed) Alzheimer disease: advances in etiology, pathogenetics and therapy. Wiley, Paris, pp 205–220
Markesbery WR (2006) Commentary on “Vascular cognitive impairment: today and tomorrow”. Alzheimers Dement 2:205–206
Marshall GA, Mendez MF, Fairbanks L, Cummings JL, Vinters HV (2005) Presence of hippocampal sclerosis in the elderly and co-occurrence with different dementias. Ann Neurol 58(Suppl 9):S16
Martin-Ruiz C, Court J, Lee M, Piggott M, Johnson M, Ballard C, Kalaria R, Perry R, Perry E (2000) Nicotinic receptors in dementia of Alzheimer, Lewy body and vascular types. Acta Neurol Scand Suppl 176:34–41
Mayer M, Straube A, Bruening R, Uttner I, Pongratz D, Gasser T, Dichgans M, Muller-Hocker J (1999) Muscle and skin biopsies are a sensitive diagnostic tool in the diagnosis of CADASIL. J Neurol 246:526–532
Mayer PL, Kier EL (1991) The controversy of the periventricular white matter circulation: a review of the anatomic literature. AJNR Am J Neuroradiol 12:223–228
McKeith IG, Dickson DW, Lowe J, Emre M, O’Brien JT, Feldman H, Cummings J, Duda JE, Lippa C, Perry EK, Aarsland D, Arai H, Ballard CG, Boeve B, Burn DJ, Costa D, Del Ser T, Dubois B, Galasko D, Gauthier S, Goetz CG, Gomez-Tortosa E, Halliday G, Hansen LA, Hardy J, Iwatsubo T, Kalaria RN, Kaufer D, Kenny RA, Korczyn A, Kosaka K, Lee VM, Lees A, Litvan I, Londos E, Lopez OL, Minoshima S, Mizuno Y, Molina JA, Mukaetova-Ladinska EB, Pasquier F, Perry RH, Schulz JB, Trojanowski JQ, Yamada M (2005) Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 65:1863–1872
McMenemey WH (1961) The dementias and progressive diseases of the basal ganglia. In: Greenfield JG (ed) Neuropathology, 3rd edn. E. Arnold, London, pp 475–521
Meguro K, Hatazawa J, Yamaguchi T, Itoh M, Matsuzawa T, Ono S, Miyazawa H, Hishinuma T, Yanai K, Sekita Y et al (1990) Cerebral circulation and oxygen metabolism associated with subclinical periventricular hyperintensity as shown by magnetic resonance imaging. Ann Neurol 28:378–383
Meguro K, Ishii H, Yamaguchi S, Ishizaki J, Shimada M, Sato M, Hashimoto R, Shimada Y, Meguro M, Yamadori A, Sekita Y (2002) Prevalence of dementia and dementing diseases in Japan: the Tajiri project. Arch Neurol 59:1109–1114
Mena R, Wischik CM, Novak M, Milstein C, Cuello AC (1991) A progressive deposition of paired helical filaments (PHF) in the brain characterizes the evolution of dementia in Alzheimer’s disease. An immunocytochemical study with a monoclonal antibody against the PHF core. J Neuropathol Exp Neurol 50:474–490
Mendez MF, Adams NL, Lewandowski KS (1989) Neurobehavioral changes associated with caudate lesions. Neurology 39:349–354
Mendez MF, Mastri AR, Sung JH, Frey WH (1992) Clinically diagnosed Alzheimer disease: neuropathologic findings in 650 cases. Alzheimer Dis Assoc Disord 6:35–43
Mendez MF, Ottowitz W, Brown CV, Cummings JL, Perryman KM, Mandelkern MA (1999) Dementia with leukoaraiosis: clinical differentiation by temporoparietal hypometabolism on (18)FDG-PET imaging. Dement Geriatr Cogn Disord 10:518–525
Merino JG, Hachinski V (2005) Diagnosis of vascular dementia. In: Paul RH, Cohen R, Ott BR, Salloway S (eds) Vascular dementia: cerebrovascular mechanisms and clinical management. Human Press Inc, Totowa, pp 57–71
Mesulam M, Siddique T, Cohen B (2003) Cholinergic denervation in a pure multi-infarct state: observations on CADASIL. Neurology 60:1183–1185
Meyer JS, Rauch GM, Lechner H, Loeb C (eds) (2001) Vascular Dementia. Futura Publishing, Armonk
Meyer JS, Huang J, Chowdhury M (2005) MRI abnormalities associated with mild cognitive impairments of vascular (VMCI) versus neurodegenerative (NMCI) types prodromal for vascular and Alzheimer’s dementias. Curr Alzheimer Res 2:579–585
Miao Q, Paloneva T, Tuisku S, Roine S, Poyhonen M, Viitanen M, Kalimo H (2006) Arterioles of the lenticular nucleus in CADASIL. Stroke 37:2242–2247
Mielke R, Herholz K, Grond M, Kessler J, Heiss WD (1992) Severity of vascular dementia is related to volume of metabolically impaired tissue. Arch Neurol 49:909–913
Mikol J (2001) Vascular dementia. In: Duckett S, De La Torre JC (eds) Pathology of the aging human nervous system, 2nd edn. Oxford University Press, Oxford, pp 101–121
Mikol J, Henin D, Baudrimont M, Gaulier A, Bacri D, Tillier JN, Davous P (2001) Atypical CADASIL phenotypes and pathological findings in two new French families (French). Rev Neurol Paris 157:655–667
Mingazzini G (1913) Anatomia clinica die centri nervosi. UTET, Torino, pp 548–573
Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L (1991) The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 41:479–486
Mirra SS, Hyman BT (2002) Vascular dementia. In: Lantos P (ed) Greenfield’s neuropathology, 7th edn. Arnold Publishing, London, pp 241–245
Mirsen T, Hachinski V (1988) The epidemiology and classification of vascular and multi-infarct dementia. In: Toole J (ed) Vascular and multi-infarct dementia. Futura Publishing, Mount Kisco, pp 61–76
Mizutani T, Shimada H (1992) Neuropathological background of twenty-seven centenarian brains. J Neurol Sci 108:168–177
Mok VC, Wong A, Lam WW, Fan YH, Tang WK, Kwok T, Hui AC, Wong KS (2004) Cognitive impairment and functional outcome after stroke associated with small vessel disease. J Neurol Neurosurg Psychiatry 75:560–566
Moncayo J, Bogousslavski J (1996) Vascular dementia: persisting controversies and questions. Eur J Neurol 3:299–308
Moody DM, Bell MA, Challa VR (1990) Features of the cerebral vascular pattern that predict vulnerability to perfusion or oxygenation deficiency: an anatomic study. AJNR Am J Neuroradiol 11:431–439
Moody DM, Brown WR, Challa VR, Anderson RL (1995) Periventricular venous collagenosis: association with leukoaraiosis. Radiology 194:469–476
Moroney JT, Bagiella E, Desmond DW, Hachinski VC, Molsa PK, Gustafson L, Brun A, Fischer P, Erkinjuntti T, Rosen W, Paik MC, Tatemichi TK (1997) Meta-analysis of the Hachinski Ischemic Score in pathologically verified dementias. Neurology 49:1096–1105
Morris JC, Heyman A, Mohs RC, Hughes JP, van Belle G, Fillenbaum G, Mellits ED, Clark C (1989) The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part I. Clinical and neuropsychological assessment of Alzheimer’s disease. Neurology 39:1159–1165
Morris JH (1997) Vascular dementia. In: Morris J (ed) The neuropathology of dementia. Cambridge University Press, Cambridge, pp 137–171
Moser DJ, Kanz JE, Garrett KD (2005) White matter hyperintensities and cognition. In: Paul RH, Cohen R, Ott BR, Salloway S (eds) Vascular dementia: cerebrovascular mechanisms and clinical management. Human Press Inc, Totowa, pp 223–229
Mott RT, Dickson DW, Trojanowski JQ, Zhukareva V, Lee VM, Forman M, Van Deerlin V, Ervin JF, Wang DS, Schmechel DE, Hulette CM (2005) Neuropathologic, biochemical, and molecular characterization of the frontotemporal dementias. J Neuropathol Exp Neurol 64:420–428
Muller-Hocker J, Hubner G, Bise K, Forster C, Hauck S, Paetzke I, Pongratz D, Kadenbach B (1993) Generalized mitochondrial microangiopathy and vascular cytochrome c oxidase deficiency. Occurrence in a case of MELAS syndrome with mitochondrial cardiomyopathy-myopathy and combined complex I/IV deficiency. Arch Pathol Lab Med 117:202–210
Mungas D, Jagust WJ, Reed BR, Kramer JH, Weiner MW, Schuff N, Norman D, Mack WJ, Willis L, Chui HC (2001) MRI predictors of cognition in subcortical ischemic vascular disease and Alzheimer’s disease. Neurology 57:2229–2235
Mungas D (2005) Contributions of subcortical lacunar infarcts to cognitive impairment in older persons. In: Paul RH, Cohen R, Ott BR, Salloway S (eds) Vascular dementia: cerebrovascular mechanisms and clinical management. Human Press Inc, Totowa, pp 211–222
Munoz DG (1991) The pathological basis of multi-infarct dementia. Alzheimer Dis Assoc Disord 5:77–90
Munoz DG, Dickson DW, Bergeron C, Mackenzie IR, Delacourte A, Zhukareva V (2003) The neuropathology and biochemistry of frontotemporal dementia. Ann Neurol 54(Suppl 5):S24–28
Nag S (1993) Cerebral endothelial mechanisms in increased permeability in chronic hypertension. Adv Exp Med Biol 331:263–266
Nag S, Robertson DM (2005) The brain in hypertension. In: Kalimo H (ed) Cerebrovascular diseases. ISN Neuropath Press, Basel, pp 286–292
Nagara H, Inoue T, Koga T, Kitaguchi T, Tateishi J, Goto I (1987) Formalin fixed brains are useful for magnetic resonance imaging (MRI) study. J Neurol Sci 81:67–77
Nagata K, Maeda T, Kato H, Satoh Y, Nakase T (2006) Hemodynamic pathophysiology in vascular dementia (Abstract). Ann Neurol 60(Suppl 3):S9
Nagy Z, Esiri MM, Joachim C, Jobst KA, Morris JH, King EM, Hindley NJ, McDonald B, Litchfield S, Barnetson L, Smith AD (1998) Comparison of pathological diagnostic criteria for Alzheimer disease. Alzheimer Dis Assoc Disord 12:182–189
Nakata-Kudo Y, Mizuno T, Yamada K, Shiga K, Yoshikawa K, Mori S, Nishimura T, Nakajima K, Nakagawa M (2006) Microbleeds in Alzheimer disease are more related to cerebral amyloid angiopathy than cerebrovascular disease. Dement Geriatr Cogn Disord 22:8–14
Naritomi H (1991) Experimental basis of multi-infarct dementia: memory impairments in rodent models of ischemia. Alzheimer Dis Assoc Disord 5:103–111
Natté R, Maat-Schieman ML, Haan J, Bornebroek M, Roos RA, van Duinen SG (2001) Dementia in hereditary cerebral hemorrhage with amyloidosis-Dutch type is associated with cerebral amyloid angiopathy but is independent of plaques and neurofibrillary tangles. Ann Neurol 50:765–772
Neuropathology G (2001) Neuropathology Group of the Medical Research Council Cognitive Function and Ageing Study (MRC CFAS). Pathological correlates of late-onset dementia in a multicentre, community-based population in England and Wales. Lancet 357:169–175
Nilsberth C, Westlind-Danielsson A, Eckman CB, Condron MM, Axelman K, Forsell C, Stenh C, Luthman J, Teplow DB, Younkin SG, Naslund J, Lannfelt L (2001) The ‘Arctic’ APP mutation (E693G) causes Alzheimer’s disease by enhanced Abeta protofibril formation. Nat Neurosci 4:887–893
O’Brien J, McKeith I, Ames D, Chiu E (2006) Dementia with lewy bodies and Parkinson’s disease dementia. Taylor & Francis, London
O’Brien JT, Wiseman R, Burton EJ, Barber B, Wesnes K, Saxby B, Ford GA (2002) Cognitive associations of subcortical white matter lesions in older people. Ann N Y Acad Sci 977:436–444
O’Brien JT, Erkinjuntti T, Reisberg B, Roman G, Sawada T, Pantoni L, Bowler JV, Ballard C, DeCarli C, Gorelick PB, Rockwood K, Burns A, Gauthier S, DeKosky ST (2003) Vascular cognitive impairment. Lancet Neurol 2:89–98
O’Brien JT (2006) Vascular cognitive impairment. Am J Geriatr Psychiatry 14:724–733
O’Brien MD (1988) Vascular dementia is underdiagnosed. Arch Neurol 45:797–798
O’Brien MD (1994) How does cerebrovascular disease cause dementia. Dementia 5:133–136
Ohama E, Ohara S, Ikuta F, Tanaka K, Nishizawa M, Miyatake T (1987) Mitochondrial angiopathy in cerebral blood vessels of mitochondrial encephalomyopathy. Acta Neuropathol (Berl) 74:226–233
Okeda R, Murayama S, Sawabe M, Kuroiwa T (2004) Pathology of the cerebral artery in Binswanger’s disease in the aged: observation by serial sections and morphometry of the cerebral arteries. Neuropathology 24:21–29
Olichney JM, Hansen LA, Hofstetter CR, Grundman M, Katzman R, Thal LJ (1995) Cerebral infarction in Alzheimer’s disease is associated with severe amyloid angiopathy and hypertension. Arch Neurol 52:702–708
Olichney JM, Ellis RJ, Katzman R, Sabbagh MN, Hansen L (1997) Types of cerebrovascular lesions associated with severe cerebral amyloid angiopathy in Alzheimer’s disease. Ann N Y Acad Sci 826:493–497
Olichney JM, Hansen LA, Hofstetter CR, Lee JH, Katzman R, Thal LJ (2000) Association between severe cerebral amyloid angiopathy and cerebrovascular lesions in Alzheimer disease is not a spurious one attributable to apolipoprotein E epsilon4. Arch Neurol 57:869–874
Olsson Y, Brun A, Englund E (1996) Fundamental pathological lesions in vascular dementia. Acta Neurol Scand Suppl 168:31–38
Ophoff RA, DeYoung J, Service SK, Joosse M, Caffo NA, Sandkuijl LA, Terwindt GM, Haan J, van den Maagdenberg AM, Jen J, Baloh RW, Barilla-LaBarca ML, Saccone NL, Atkinson JP, Ferrari MD, Freimer NB, Frants RR (2001) Hereditary vascular retinopathy, cerebroretinal vasculopathy, and hereditary endotheliopathy with retinopathy, nephropathy, and stroke map to a single locus on chromosome 3p21.1-p21.3. Am J Hum Genet 69:447–453
Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby LC, Jouanny P, Dubois B, Eisner L, Flitman S, Michel BF, Boada M, Frank A, Hock C (2003) Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology 61:46–54
O’Sullivan M, Lythgoe DJ, Pereira AC, Summers PE, Jarosz JM, Williams SC, Markus HS (2002) Patterns of cerebral blood flow reduction in patients with ischemic leukoaraiosis. Neurology 59:321–326
Pantoni L, Garcia JH (1995) The significance of cerebral white matter abnormalities 100 years after Binswanger’s report. A review. Stroke 26:1293–1301
Pantoni L, Garcia JH, Brown GG (1996) Vascular pathology in three cases of progressive cognitive deterioration. J Neurol Sci 135:131–139
Pantoni L, Garcia JH (1997) Pathogenesis of leukoaraiosis: a review. Stroke 28:652–659
Pantoni L (2002) Pathophysiology of age-related cerebral white matter changes. Cerebrovasc Dis 13(Suppl 2):7–10
Pantoni L, Palumbo V, Sarti C (2002) Pathological lesions in vascular dementia. Ann N Y Acad Sci 977:279–291
Pantoni L (2003) Subtypes of vascular dementia and their pathogenesis: a critical overview. In: Bowler JV, Hachinski V (eds) Vascular cognitive impairment—preventable dementia. Oxford University Press, New York, pp 217–229
Pantoni L, Sarti C, Alafuzoff I, Jellinger K, Munoz DG, Ogata J, Palumbo V (2006) Postmortem examination of vascular lesions in cognitive impairment: a survey among neuropathological services. Stroke 37:1005–1009
Patankar TF, Mitra D, Varma A, Snowden J, Neary D, Jackson A (2005) Dilatation of the Virchow–Robin space is a sensitive indicator of cerebral microvascular disease: study in elderly patients with dementia. Am J Neuroradiol 26:1512–1520
Paulson GW, Kapp J, Cook W (1966) Dementia associated with bilateral carotid artery disease. Geriatrics 21:159–166
Paulus W, Bancher C, Jellinger K (1992) Interrater reliability in the neuropathologic diagnosis of Alzheimer’s disease. Neurology 42:329–332
Perry E, Ziabreva I, Perry R, Aarsland D, Ballard C (2005) Absence of cholinergic deficits in “pure” vascular dementia. Neurology 64:132–133
Peters N, Opherk C, Danek A, Ballard C, Herzog J, Dichgans M (2005) The pattern of cognitive performance in CADASIL: a monogenic condition leading to subcortical ischemic vascular dementia. Am J Psychiatry 162:2078–2085
Petrovitch H, Ross GW, Steinhorn SC, Abbott RD, Markesberry W, Davis DG, Nelson J, Hardman J, Masaki KH, Vogt MR, Launer LJ, White LR (2005) AD lesions and infarcts in demented and no-demented Japanese–American men. Ann Neurol 57:98–103
Pfeifer LA, White LR, Ross GW, Petrovitch H, Launer LJ (2002) Cerebral amyloid angiopathy and cognitive function: the HAAS autopsy study. Neurology 58:1629–1634
Pirchl M, Marksteiner J, Humpel C (2006) Effects of acidosis on brain capillary endothelial cells and cholinergic neurons: relevance to vascular dementia and Alzheimer’s disease. Neurol Res 28:657–664
Plant G, Ghiso J, Holton JL, Frangione B, Revesz T (2004) Familial and sporadic cerebral amyloid angiopathies associated with dementia and the BRI dementias. In: Esiri MM, Lee VMY, Trojanowski JQ (eds) The neuropathology of dementia. Cambridge University Press, Cambridge, pp 330–352
Pohjasvaara T, Mantyla R, Salonen O, Aronen HJ, Ylikoski R, Hietanen M, Kaste M, Erkinjuntti T (2000) How complex interactions of ischemic brain infarcts, white matter lesions, and atrophy relate to poststroke dementia. Arch Neurol 57:1295–1300
Pohjasvaara T, Mantyla R, Ylikoski R, Kaste M, Erkinjuntti T (2000) Comparison of different clinical criteria (DSM-III, ADDTC, ICD-10, NINDS-AIREN, DSM-IV) for the diagnosis of vascular dementia. National Institute of Neurological Disorders and Stroke-Association Internationale pour la Recherche et l’Enseignement en Neurosciences. Stroke 31:2952–2957
Pohjasvaara T, Leskela M, Vataja R, Kalska H, Ylikoski R, Hietanen M, Leppavuori A, Kaste M, Erkinjuntti T (2002) Post-stroke depression, executive dysfunction and functional outcome. Eur J Neurol 9:269–275
Poirier J, Derouesne C (1984) Cerebral lacunae. A proposed new classification. Clin Neuropathol 3:266
Poirier J, Derouesne C (1985) [The concept of cerebral lacunae from 1838 to the present]. Rev Neurol Paris 141:3–17
Pollock H, Hutchings M, Weller RO, Zhang ET (1997) Perivascular spaces in the basal ganglia of the human brain: their relationship to lacunes. J Anat 191:337–346
Powers JM, DeVivo DC (2002) Peroxismal and mitochindrial disorders. In: Lantos P (ed) Greenfield’s neuropathology, 7th edn. Arnold Publishing, London, pp 737–797
Price CC, Jefferson AL, Merino JG, Heilman KM, Libon DJ (2005) Subcortical vascular dementia: integrating neuropsychological and neuroradiologic data. Neurology 65:376–382
Price JL, Morris JC (1999) Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Ann Neurol 45:358–368
Prins ND, Den Heijer T, Hofman A, Koudstaal PJ, Jolles J, Clarke R, Breteler MM (2002) Homocysteine and cognitive function in the elderly: the Rotterdam Scan Study. Neurology 59:1375–1380
Prins ND, van Dijk EJ, den Heijer T, Vermeer SE, Koudstaal PJ, Oudkerk M, Hofman A, Breteler MM (2004) Cerebral white matter lesions and the risk of dementia. Arch Neurol 61:1531–1534
Pullicino PM (1993) Pathogenesis of lacunar infarcts and small deep infarcts. Adv Neurol 62:125–140
Rahkonen T, Eloniemi-Sulkava U, Rissanen S, Vatanen A, Viramo P, Sulkava R (2003) Dementia with Lewy bodies according to the consensus criteria in a general population aged 75 years or older. J Neurol Neurosurg Psychiatry 74:720–724
Reed BR, Eberling JL, Mungas D, Weiner M, Jagust WJ (2001) Frontal lobe hypometabolism predicts cognitive decline in patients with lacunar infarcts. Arch Neurol 58:493–497
Reed BR, Eberling JL, Mungas D, Weiner M, Kramer JH, Jagust WJ (2004) Effects of white matter lesions and lacunes on cortical function. Arch Neurol 61:1545–1550
Regan C, Katona C, Walker Z (2006) Relationship of vascular risk to the progression of Alzheimer disease. Neurology 67:1357–1362
Revesz T, Holton JL, Lashley T, Plant G, Rostagno A, Ghiso J, Frangione B (2002) Sporadic and familial cerebral amyloid angiopathies. Brain Pathol 12:343–357
Revesz T, Ghiso J, Lashley T, Plant G, Rostagno A, Frangione B, Holton JL (2003) Cerebral amyloid angiopathies: a pathologic, biochemical, and genetic view. J Neuropathol Exp Neurol 62:885–898
Revesz T, Ghiso J, Plant G, Holton JL, Frangione B (2003) Inherited amyloidosis and neurodegenerations in Familial British and Danish Dementia. In: Dickson DW (ed) Neurodegeneration. The molecular pathology of dementia and movement disorders. ISN Neuropath Press, Basel, pp 380–385
Revesz T, Ghiso J, Plant G, Lashley T, Rostagno A, Frangione B, Holton JL (2005) Cerebral amyloid angiopathy. In: Kalimo H (ed) Pathology & genetics. Cerebrovascular diseases. ISN Neuropath Press, Basel, pp 94–102
Riekse RG (2004) Autopsy-confirmed “pure” vascular dementia in a comunity-based sample of dementia (Abstr.) Neurobiol Aging 52:S125
Rocca WA, Hofman A, Brayne C, Breteler MM, Clarke M, Copeland JR, Dartigues JF, Engedal K, Hagnell O, Heeren TJ et al (1991) The prevalence of vascular dementia in Europe: facts and fragments from 1980–1990 studies. EURODEM-Prevalence Research Group. Ann Neurol 30:817–824
Rocca WA, Knopman DS (2003) Prevalence and incidence patterns of vascular dementia. In: Bowler JV, Hachinski V (eds) Vascular cognitive impairment. Preventable dementia. Oxford University Press, Oxford, pp 21–32
Rockwood K, Wentzel C, Hachinski V, Hogan DB, MacKnight C, McDowell I (2000) Prevalence and outcomes of vascular cognitive impairment. Vascular Cognitive Impairment Investigators of the Canadian Study of Health and Aging. Neurology 54:447–451
Rockwood K, Black SE, Song X, Hogan DB, Gauthier S, MacKnight C, Vandorpe R, Guzman A, Montgomery P, Kertesz A, Bouchard RW, Feldman H (2006) Clinical and radiographic subtypes of vascular cognitive impairment in a clinic-based cohort study. J Neurol Sci 240:7–14
Román GC (1981) Lagunas cerebrales. Estudio clínico y neurópatológico de 100 cases fatales. Rev Facultad Ciencias Medicas/Univ Nac Córdoba, vol 39, pp 115–129
Román GC, Tatemichi TK, Erkinjuntti T, Cummings JL, Masdeu JC, Garcia JH, Amaducci L, Orgogozo JM, Brun A, Hofman A et al (1993) Vascular dementia: diagnostic criteria for research studies. Report of the NINDS-AIREN International Workshop. Neurology 43:250–260
Román GC, Royall DR (1999) Executive control function: a rational basis for the diagnosis of vascular dementia. Alzheimer Dis Assoc Disord 13(Suppl 3):S69–80
Román GC (2002) Vascular dementia may be the most common form of dementia in the elderly. J Neurol Sci 203–204:7–10
Román GC (2002) On the history of lacunes, état criblé, and the white matter lesions of vascular dementia. Cerebrovasc Dis 13(Suppl 2):1–6
Román GC, Erkinjuntti T, Wallin A, Pantoni L, Chui HC (2002) Subcortical ischaemic vascular dementia. Lancet Neurol 1:426–436
Román GC (2004) Brain hypoperfusion: a critical factor in vascular dementia. Neurol Res 26:454–458
Román GC, Sachdev P, Royall DR, Bullock RA, Orgogozo JM, Lopez-Pousa S, Arizaga R, Wallin A (2004) Vascular cognitive disorder: a new diagnostic category updating vascular cognitive impairment and vascular dementia. J Neurol Sci 226:81–87
Román GC (2005) Clinical forms of vascular dementia. In: Paul RH, Cohen R, Ott BR, Salloway S (eds) Vascular dementia: cerebrovascular mechanisms and clinical management. Human Press Inc, Totowa, pp 7–21
Román GC, Kalaria RN (2006) Vascular determinants of cholinergic deficits in Alzheimer disease and vascular dementia. Neurobiol Aging 27:1769–1785
Rossi R, Joachim C, Geroldi C, Esiri MM, Smith AD, Frisoni GB (2005) Pathological validation of a CT-based scale for subcortical vascular disease. The OPTIMA study. Dementia Geriatr Cogn Disord 19:61–66
Sachdev P (1999) Vascular cognitive disorder. Int J Geriatr Psychiatry 14:402–403
Sakurada T, Alufuzoff I, Winblad B, Nordberg A (1990) Substance P-like immunoreactivity, choline acetyltransferase activity and cholinergic muscarinic receptors in Alzheimer’s disease and multi-infarct dementia. Brain Res 521:329–332
Salahuddin TS, Johansson BB, Kalimo H, Olsson Y (1988) Structural changes in the rat brain after carotid infusions of hyperosmolar solutions: a light microscopic and immunohistochemical study. Neuropathol Appl Neurobiol 14:467–482
Sato A, Sato Y, Uchida S (2001) Regulation of regional cerebral blood flow by cholinergic fibers originating in the basal forebrain. Int J Dev Neurosci 19:327–337
Sato A, Sato Y, Uchida S (2004) Activation of the intracerebral cholinergic nerve fibers originating in the basal forebrain increases regional cerebral blood flow in the rat’s cortex and hippocampus. Neurosci Lett 361:90–93
Scheinberg P (1988) Dementia due to vascular disease—a multifactorial disorder. Stroke 19:1291–1299
Scheltens P, Barkhof F, Leys D, Pruvo JP, Nauta JJ, Vermersch P, Steinling M, Valk J (1993) A semiquantative rating scale for the assessment of signal hyperintensities on magnetic resonance imaging. J Neurol Sci 114:7–12
Scheltens P, Barkhof F, Leys D, Wolters EC, Ravid R, Kamphorst W (1995) Histopathologic correlates of white matter changes on MRI in Alzheimer’s disease and normal aging. Neurology 45:883–888
Schmidt R, Schmidt H, Kapeller P, Lechner A, Fazekas F (2002) Evolution of white matter lesions. Cerebrovasc Dis 13(Suppl 2):16–20
Schmidt R, Enzinger C, Ropele S, Schmidt H, Fazekas F (2003) Progression of cerebral white matter lesions:6-year results of the Austrian Stroke Prevention Study. Lancet 361:2046–2048
Schmidt R, Scheltens P, Erkinjuntti T, Pantoni L, Markus HS, Wallin A, Barkhof F, Fazekas F (2004) White matter lesion progression: a surrogate endpoint for trials in cerebral small-vessel disease. Neurology 63:139–144
Schmidt R, Ropele S, Enzinger C, Petrovic K, Smith S, Schmidt H, Matthews PM, Fazekas F (2005) White matter lesion progression, brain atrophy, and cognitive decline: The Austrian stroke prevention study. Ann Neurol 58:610–616
Schmidtke K, Hull M (2005) Cerebral small vessel disease: how does it progress? J Neurol Sci 229–230:13–20
Schneider JA, Wilson RS, Cochran EJ, Bienias JL, Arnold SE, Evans DA, Bennett DA (2003) Relation of cerebral infarctions to dementia and cognitive function in older persons. Neurology 60:1082–1088
Schneider JA, Wilson RS, Bienias JL, Evans DA, Bennett DA (2004) Cerebral infarctions and the likelihood of dementia from Alzheimer disease pathology. Neurology 62:1148–1155
Schroeter ML, Bucheler MM, Preul C, Scheid R, Schmiedel O, Guthke T, von Cramon DY (2005) Spontaneous slow hemodynamic oscillations are impaired in cerebral microangiopathy. J Cereb Blood Flow Metab 25:1675–1684
Selden NR, Gitelman DR, Salamon-Murayama N, Parrish TB, Mesulam MM (1998) Trajectories of cholinergic pathways within the cerebral hemispheres of the human brain. Brain 121(Pt 12):2249–2257
Selnes OA, Vinters HV (2006) Vascular cognitive impairment. Nat Clin Pract Neurol 2:538–547
Seno H, Ishino H, Inagaki T, Iijima M, Kaku K, Inata T (1999) A neuropathological study of dementia in nursing homes over a 17-year period, in Shimane Prefecture, Japan. Gerontology 45:44–48
Shim YS, Yang D-W, Kim B-S, Shon YM, Chung Y-A (2006) Comparison of regional cerebral blood flow in two subsets of subcortical ischemic vascular dementia: Statistical parametric mapping analysis of SPECT. J Neurol Sci 250:85–91
Sibon I, Fenelon G, Quinn NP, Tison F (2004) Vascular parkinsonism. J Neurol 251:513–524
Skoog I, Nilsson L, Palmertz B, Andreasson LA, Svanborg A (1993) A population-based study of dementia in 85-year-olds. N Engl J Med 328:153–158
Skoog I (1998) A review on blood pressure and ischaemic white matter lesions. Dement Geriatr Cogn Disord 9(Suppl 1):13–19
Sleegers K, Brouwers N, Gijselinck I, Theuns J, Goossens D, Wauters J, Del-Favero J, Cruts M, van Duijn CM, Van Broeckhoven C (2006) APP duplication is sufficient to cause early onset Alzheimer’s dementia with cerebral amyloid angiopathy. Brain 129:2977–2983
Smith EE, Gurol ME, Eng JA, Engel CR, Nguyen TN, Rosand J, Greenberg SM (2004) White matter lesions, cognition, and recurrent hemorrhage in lobar intracerebral hemorrhage. Neurology 63:1606–1612
Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR (1997) Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA 277:813–817
Snowdon DA, Markesbery WR (1999) The prevalence of neuropathologically confirmed vascular dementia: findings from the Nun study. In: 1st International congress on vascular dementia, Geneva, Switzerland, October 1999. Monduzzi Editore S.p.A Bologna, Italy, pp 19–24
Soderlund H, Nyberg L, Adolfsson R, Nilsson LG, Launer LJ (2003) High prevalence of white matter hyperintensities in normal aging: relation to blood pressure and cognition. Cortex 39:1093–1105
Sourander P, Walinder J (1977) Hereditary multi-infarct dementia. Morphological and clinical studies of a new disease. Acta Neuropathol (Berl) 39:247–254
Spangler KM, Challa VR, Moody DM, Bell MA (1994) Arteriolar tortuosity of the white matter in aging and hypertension. A microradiographic study. J Neuropathol Exp Neurol 53:22–26
Srikanth V, Saling MM, Thrift AG (2007) Vascular cognitive impairment. In: Gilman S (ed) Neurobiology of disease. Elsevier, Amsterdam, pp 223–233
Srikanth VK, Anderson JF, Donnan GA, Saling MM, Didus E, Alpitsis R, Dewey HM, Macdonell RA, Thrift AG (2004) Progressive dementia after first-ever stroke: a community-based follow-up study. Neurology 63:785–792
Starkstein SE, Sabe L, Vazquez S, Teson A, Petracca G, Chemerinski E, Di Lorenzo G, Leiguarda R (1996) Neuropsychological, psychiatric, and cerebral blood flow findings in vascular dementia and Alzheimer’s disease. Stroke 27:408–414
Stenset V, Johnsen L, Kocot D, Negaard A, Skinningsrud A, Gulbrandsen P, Wallin A, Fladby T (2006) Associations between white matter lesions, cerebrovascular risk factors, and low CSF Abeta42. Neurology 67:830–833
Stys PK (2004) White matter injury mechanisms. Curr Mol Med 4:113–130
Sultzer DL, Mahler ME, Cummings JL, Van Gorp WG, Hinkin CH, Brown C (1995) Cortical abnormalities associated with subcortical lesions in vascular dementia. Clinical and position emission tomographic findings. Arch Neurol 52:773–780
Swartz RH, Black SE (2002) How common is vascular compromise of cholinergic white matter pathways in a memory clinic sample? J Neurol Sci 203–204:281
Swartz RH, Sahlas DJ, Black SE (2003) Strategic involvement of cholinergic pathways and executive dysfunction: does location of white matter signal hyperintensities matter? J Stroke Cerebrovasc Dis 12:29–36
Szirmai I, Vastagh I, Szombathelyi E, Kamondi A (2002) Strategic infarcts of the thalamus in vascular dementia. J Neurol Sci 203–204:91–97
Tabaton M, Caponnetto C, Mancardi G, Loeb C (1991) Amyloid beta protein deposition in brains from elderly subjects with leukoaraiosis. J Neurol Sci 106:123–127
Tang WK, Chan SS, Chiu HF, Ungvari GS, Wong KS, Kwok TC, Mok V, Wong KT, Richards PS, Ahuja AT (2004) Impact of applying NINDS-AIREN criteria of probable vascular dementia to clinical and radiological characteristics of a stroke cohort with dementia. Cerebrovasc Dis 18:98–103
Tanoi Y, Okeda R, Budka H (2000) Binswanger’s encephalopathy: serial sections and morphometry of the cerebral arteries. Acta Neuropathol (Berl) 100:347–355
Tanskanen M, Lindsberg PJ, Tienari PJ, Polvikoski T, Sulkava R, Verkkoniemi A, Rastas S, Paetau A, Kiuru-Enari S (2005) Cerebral amyloid angiopathy in a 95+ cohort: complement activation and apolipoprotein E (APOE) genotype. Neuropathol Appl Neurobiol 31:589–599
Tatemichi TK, Desmond DW, Mayeux R, Paik M, Stern Y, Sano M, Remien RH, Williams JB, Mohr JP, Hauser WA et al (1992) Dementia after stroke: baseline frequency, risks, and clinical features in a hospitalized cohort. Neurology 42:1185–1193
Tatemichi TK, Desmond DW, Prohovnik I, Cross DT, Gropen TI, Mohr JP, Stern Y (1992) Confusion and memory loss from capsular genu infarction: a thalamocortical disconnection syndrome? Neurology 42:1966–1979
Tatemichi TK, Sacktor N, Mayeux R (1994) Dementia associated with cerebrovascular disease, other degenerative diseases, and metabolic disorders. In: Bick KL (ed) Alzheimer’s disease. Raven Press, New York, pp 123–166
Tatemichi TK, Desmond DW, Prohovnik I (1995) Strategic infarcts in vascular dementia. A clinical and brain imaging experience. Arzneimittelforschung 45:371–385
Taylor DH, Doraiswamy PM, Sloan FA (2004) Rates of diagnosis of vascular dementia over 10 years in the United States Medicare claims records (Abstr). Neurobiol Aging 52:S484.
Thal DR, Ghebremedhin E, Rub U, Yamaguchi H, Tredici KD, Braak H (2002) Two types of sporadic cerebral amyloid angiopathy. J Neuropathol Exp Neurol 61:282–293
Thal DR, Rub U, Orantes M, Braak H (2002) Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 58:1791–1800
Thal DR, Ghebremedhin E, Orantes M, Wiestler OD (2003) Small vessel lesions related to cerebral amyloid angiopathy (CAA) and arteriosclerosis/lipohylinosis (AS/LH) are associated with Alzheimer disease (abstr.). Brain Pathol 13(Suppl 1):S7–S8
Thomas A, Ballard C, Kenny RA, O’Brien J, Oakley A, Kalaria R (2005) Correlation of entorhinal amyloid with memory in Alzheimer’s and vascular but not Lewy body dementia. Dement Geriatr Cogn Disord 19:57–60
Thomas AJ, O’Brien JT, Davis S, Ballard C, Barber R, Kalaria RN, Perry RH (2002) Ischemic basis for deep white matter hyperintensities in major depression: a neuropathological study. Arch Gen Psychiatry 59:785–792
Tohgi H, Abe T, Kimura M, Saheki M, Takahashi S (1996) Cerebrospinal fluid acetylcholine and choline in vascular dementia of Binswanger and multiple small infarct types as compared with Alzheimer-type dementia. J Neural Transm 103:1211–1220
Tomimoto H, Akiguchi I, Wakita H, Kimura J (1994) [Changes in glial cells in Binswanger-type infarction]. No To Shinkei 46:771–779
Tomimoto H, Akiguchi I, Akiyama H, Ikeda K, Wakita H, Lin JX, Budka H (1999) Vascular changes in white matter lesions of Alzheimer’s disease. Acta Neuropathol (Berl) 97:629–634
Tomlinson BE, Blessed G, Roth M (1970) Observations on the brains of demented old people. J Neurol Sci 11:205–242
Torvik A (1984) The pathogenesis of watershed infarcts in the brain. Stroke 15:221–223
Tournier-Lasserve E, Iba-Zizen MT, Romero N, Bousser MG (1991) Autosomal dominant syndrome with strokelike episodes and leukoencephalopathy. Stroke 22:1297–1302
Trembath D, Ervin JF, Broom L, Szymanski M, Welsh-Bohmer K, Pieper C, Hulette CM (2007) The distribution of cerebrovascular amyloid in Alzheimer's disease varies with APOE genotype. Acta Neuropathol (Berl) DOI 10.1007/s00401-006-0162-9
Trojanowski JQ, Dickson D (2001) Update on the neuropathological diagnosis of frontotemporal dementias. J Neuropathol Exp Neurol 60:1123–1126
Tsuang D, Simpson KL, Li G, Barnhart RL, Edland SD, Bowen J, McCormick W, Teri L, Nochlin D, Larson EB, Thompson ML, Leverenz JB (2005) Evaluation of selection bias in an incident-based dementia autopsy case series. Alzheimer Dis Assoc Disord 19:67–73
Tullberg M, Fletcher E, DeCarli C, Mungas D, Reed BR, Harvey DJ, Weiner MW, Chui HC, Jagust WJ (2004) White matter lesions impair frontal lobe function regardless of their location. Neurology 63:246–253
Tuszynski MH, Petito CK, Levy DE (1989) Risk factors and clinical manifestations of pathologically verified lacunar infarctions. Stroke 20:990–999
Ueda K, Kawano H, Hasuo Y, Fujishima M (1992) Prevalence and etiology of dementia in a Japanese community. Stroke 23:798–803
Ulrich J, Probst A, Wuest M (1986) The brain diseases causing senile dementia. A morphological study on 54 consecutive autopsy cases. J Neurol 233:118–122
Uspenskaia O, Liebetrau M, Herms J, Danek A, Hamann GF (2004) Aging is associated with increased collagen type IV accumulation in the basal lamina of human cerebral microvessels. BMC Neurosci 5:37
Van de Nes JAP, Zimmer R, Janzen RWC, Turowski B, Hürtle E, Schlote W (2003) Mini-infarct encephalopathy associated with uncommon microvessel convolute formation presenting with presenile dementia. Clin Neuropathol 22:101–109
van den Heuvel DM, ten Dam VH, de Craen AJ, Admiraal-Behloul F, Olofsen H, Bollen EL, Jolles J, Murray HM, Blauw GJ, Westendorp RG, van Buchem MA (2006) Increase in periventricular white matter hyperintensities parallels decline in mental processing speed in a non-demented elderly population. J Neurol Neurosurg Psychiatry 77:149–153
Van der Eecken H (1959) Anastomoses between the leptomeningeal arteries of the brain. Charles C. Thomas, Springfield
van der Flier WM, van Straaten EC, Barkhof F, Ferro JM, Pantoni L, Basile AM, Inzitari D, Erkinjuntti T, Wahlund LO, Rostrup E, Schmidt R, Fazekas F, Scheltens P (2005) Medial temporal lobe atrophy and white matter hyperintensities are associated with mild cognitive deficits in non-disabled elderly people: the LADIS study. J Neurol Neurosurg Psychiatry 76:1497–1500
van der Flier WM, van Straaten EC, Barkhof F, Verdelho A, Madureira S, Pantoni L, Inzitari D, Erkinjuntti T, Crisby M, Waldemar G, Schmidt R, Fazekas F, Scheltens P (2005) Small vessel disease and general cognitive function in nondisabled elderly: the LADIS study. Stroke 36:2116–2120
van Straaten EC, Scheltens P, Knol DL, van Buchem MA, van Dijk EJ, Hofman PA, Karas G, Kjartansson O, de Leeuw FE, Prins ND, Schmidt R, Visser MC, Weinstein HC, Barkhof F (2003) Operational definitions for the NINDS-AIREN criteria for vascular dementia: an interobserver study. Stroke 34:1907–1912
van Swieten JC, Geyskes GG, Derix MM, Peeck BM, Ramos LM, van Latum JC, van Gijn J (1991) Hypertension in the elderly is associated with white matter lesions and cognitive decline. Ann Neurol 30:825–830
van Swieten JC, van den Hout JH, van Ketel BA, Hijdra A, Wokke JH, van Gijn J (1991) Periventricular lesions in the white matter on magnetic resonance imaging in the elderly. A morphometric correlation with arteriolosclerosis and dilated perivascular spaces. Brain 114(Pt 2):761–774
Vanderploe RD, Yuspeh RL, Schinka JA (2001) Differential episodic and semantic memory performance in Alzheimer’s disease and vascular dementias. J Int Neuropsychol Soc 7:563–573
Vermeer SE, Prins ND, den Heijer T, Hofman A, Koudstaal PJ, Breteler MM (2003) Silent brain infarcts and the risk of dementia and cognitive decline. N Engl J Med 348:1215–1222
Vidal R, Frangione B, Rostagno A, Mead S, Revesz T, Plant G, Ghiso J (1999) A stop-codon mutation in the BRI gene associated with familial British dementia. Nature 399:776–781
Vinters HV, Gilbert JJ (1983) Cerebral amyloid angiopathy: incidence and complications in the aging brain. II. The distribution of amyloid vascular changes. Stroke 14:924–928
Vinters HV, Ellis WG, Zarow C, Zaias BW, Jagust WJ, Mack WJ, Chui HC (2000) Neuropathologic substrates of ischemic vascular dementia. J Neuropathol Exp Neurol 59:931–945
Vinters HV, Farag ES (2003) Amyloidosis of cerebral arteries. In: Meldrum H (ed) Ischemic stroke: advances in neurology, vol 92. Lippincott Williams & Wilkins, Philadelphia, pp 105
Viswanathan A, Gray F, Bousser MG, Baudrimont M, Chabriat H (2006) Cortical neuronal apoptosis in CADASIL. Stroke 37:2690–2695
Vonsattel JP, Myers RH, Hedley-Whyte ET, Ropper AH, Bird ED, Richardson EP Jr (1991) Cerebral amyloid angiopathy without and with cerebral hemorrhages: a comparative histological study. Ann Neurol 30:637–649
Wade JP, Mirsen TR, Hachinski VC, Fisman M, Lau C, Merskey H (1987) The clinical diagnosis of Alzheimer’s disease. Arch Neurol 44:24–29
Wahlund LO, Barkhof F, Fazekas F, Bronge L, Augustin M, Sjogren M, Wallin A, Ader H, Leys D, Pantoni L, Pasquier F, Erkinjuntti T, Scheltens P (2001) A new rating scale for age-related white matter changes applicable to MRI and CT. Stroke 32:1318–1322
Wakita H, Tomimoto H, Akiguchi I, Matsuo A, Lin JX, Ihara M, McGeer PL (2002) Axonal damage and demyelination in the white matter after chronic cerebral hypoperfusion in the rat. Brain Res 924:63–70
Wang C, Stebbins GT, Nyenhuis DL, deToledo-Morrell L, Freels S, Gencheva E, Pedelty L, Sripathirathan K, Moseley ME, Turner DA, Gabrieli JD, Gorelick PB (2006) Longitudinal changes in white matter following ischemic stroke: a three-year follow-up study. Neurobiol Aging 27:1827–1833
Ward N, Brown MM (2003) Cerebral blood flow and metabolism in vascular dementia. In: Bowler JV, Hachinski V (eds) Vascular cognitive impairment. Preventable dementia. Oxford University Press, London, pp 192–207
Ward NS, Brown MM (1990) Leukoaraiosis. In: Cummings JL (ed) Subcortical dementia. Oxford University Press, Oxford, pp 47–68
Wardlaw JM, Dennis MS, Warlow CP, Sandercock PA (2001) Imaging appearance of the symptomatic perforating artery in patients with lacunar infarction: occlusion or other vascular pathology? Ann Neurol 50:208–215
Wardlaw JM (2005) What causes lacunar stroke? J Neurol Neurosurg Psychiatry 76:617–619
Wattendorff AR, Frangione B, Luyendijk W, Bots GT (1995) Hereditary cerebral haemorrhage with amyloidosis, Dutch type (HCHWA-D): clinicopathological studies. J Neurol Neurosurg Psychiatry 58:699–705
Waxman SG (2002) Molecular mechanisms of subcortical versus cortical infarction. In: Bogousslavsky J (ed) Subcortical stroke. Oxford University Press, Oxford, pp 67–83
Weller RO, Kida S, Zhang ET (1992) Pathways of fluid drainage from the brain—morphological aspects and immunological significance in rat and man. Brain Pathol 2:277–284
Weller RO, Massey A, Newman TA, Hutchings M, Kuo YM, Roher AE (1998) Cerebral amyloid angiopathy: amyloid beta accumulates in putative interstitial fluid drainage pathways in Alzheimer’s disease. Am J Pathol 153:725–733
Wen HM, Mok VC, Fan YH, Lam WW, Tang WK, Wong A, Huang RX, Wong KS (2004) Effect of white matter changes on cognitive impairment in patients with lacunar infarcts. Stroke 35:1826–1830
Wen HM, Baum L, Cheung WS, Mok V, Lam WW, Tomlinson B, Wong KS, Ng HK (2006) Apolipoprotein E epsilon4 allele is associated with the volume of white matter changes in patients with lacunar infarcts. Eur J Neurol 13:1216–1220
Wentzel C, Rockwood K, MacKnight C, Hachinski V, Hogan DB, Feldman H, Ostbye T, Wolfson C, Gauthier S, Verreault R, McDowell I (2001) Progression of impairment in patients with vascular cognitive impairment without dementia. Neurology 57:714–716
Wetterling T, Kanitz RD, Borgis KJ (1996) Comparison of different diagnostic criteria for vascular dementia (ADDTC, DSM-IV, ICD-10, NINDS-AIREN). Stroke 27:30–36
White L, Petrovich H, Hardman J (2002) Cerebrovascular pathology and dementia in autopsied Honolulu-Asia Aging Study partizipants. Ann NY Acad Sci 977:9–23
Whitman GT, Tang Y, Lin A, Baloh RW (2001) A prospective study of cerebral white matter abnormalities in older people with gait dysfunction. Neurology 57:990–994
Winblad B, Wallace W, Hardy J, Fowler C, Bucht G, Alafuzoff I, Adolfson R (1986) Neurochemical, genetic and clinical aspects of Alzheimer’s disease. In: Bergener M, Ermini M, Stähelin HB (eds) Dimensions in aging. Academic, London, pp 183–203
Wisniewski HM, Frackowiak J, Zoltowska A, Kim KS (1994) Vascular ß-amyloid in Alzheimer´s disease angiopathy is produced by proliferating and degenerating smooth muscle cells. Int J Exp Clin Invest 8–16
Wiszniewska M, Devuyst G, Bogousslavsky J, Ghika J, van Melle G (2000) What is the significance of leukoaraiosis in patients with acute ischemic stroke? Arch Neurol 57:967–973
Wolfe N, Linn R, Babikian VL, Knoefel JE, Albert ML (1990) Frontal systems impairment following multiple lacunar infarcts. Arch Neurol 47:129–132
Woodward M, Mackenzie IR, Feldman H (2006) High prevalence of multiple brain pathologies in dementia. Alzheimers Dement 2(Suppl 1):S426
World Health Organization (1993) ICD-10 classification of mental and behavioural disorders: diagnostic criteria for research. WHO, Geneva
Wu CC, Mungas D, Petkov CI, Eberling JL, Zrelak PA, Buonocore MH, Brunberg JA, Haan MN, Jagust WJ (2002) Brain structure and cognition in a community sample of elderly Latinos. Neurology 59:383–391
Yamanouchi H (1991) Loss of white matter oligodendrocytes and astrocytes in progressive subcortical vascular encephalopathy of Binswanger type. Acta Neurol Scand 83:301–305
Yamauchi H, Fukuyama H, Nagahama Y, Shiozaki T, Nishizawa S, Konishi J, Shio H, Kimura J (1999) Brain arteriolosclerosis and hemodynamic disturbance may induce leukoaraiosis. Neurology 53:1833–1838
Ylikoski A, Erkinjuntti T, Raininko R, Sarna S, Sulkava R, Tilvis R (1995) White matter hyperintensities on MRI in the neurologically nondiseased elderly. Analysis of cohorts of consecutive subjects aged 55 to 85 years living at home. Stroke 26:1171–1177
Yoshikawa T, Murase K, Oku N, Kitagawa K, Imaizumi M, Takasawa M, Nishikawa T, Matsumoto M, Hatazawa J, Hori M (2003) Statistical image analysis of cerebral blood flow in vascular dementia with small-vessel disease. J Nucl Med 44:505–511
Yoshikawa T, Murase K, Oku N, Kitagawa K, Imaizumi M, Takasawa M, Rishu P, Hashikawa K, Nishikawa T, Hori M, Matsumoto M (2003) Quantification of the heterogeneity of cerebral blood flow in vascular dementia. J Neurol 250:194–200
Yoshimura M, Yamanouchi H, Kuzuhara S, Mori H, Sugiura S, Mizutani T, Shimada H, Tomonaga M, Toyokura Y (1992) Dementia in cerebral amyloid angiopathy: a clinicopathological study. J Neurol 239:441–450
Yoshita M, Fletcher E, Harvey D, Ortega M, Martinez O, Mungas DM, Reed BR, DeCarli CS (2006) Extent and distribution of white matter hyperintensities in normal aging, MCI, and AD. Neurology 67: 2192–2198
Yoshitake T, Kiyohara Y, Kato I, Ohmura T, Iwamoto H, Nakayama K, Ohmori S, Nomiyama K, Kawano H, Ueda K et al (1995) Incidence and risk factors of vascular dementia and Alzheimer’s disease in a defined elderly Japanese population: the Hisayama Study. Neurology 45:1161–1168
Yousry TA, Seelos K, Mayer M, Bruning R, Uttner I, Dichgans M, Mammi S, Straube A, Mai N, Filippi M (1999) Characteristic MR lesion pattern and correlation of T1 and T2 lesion volume with neurologic and neuropsychological findings in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). AJNR Am J Neuroradiol 20:91–100
Zambenedetti P, Schmitt HP, Zatta P (2002) Metallothionein I–II immunocytochemical reactivity in Binswanger’s encephalopathy. J Alzheimers Dis 4:459–466
Zarow C, Vinters HV, Ellis WG, Weiner MW, Mungas D, White L, Chui HC (2005) Correlates of hippocampal neuron number in Alzheimer’s disease and ischemic vascular dementia. Ann Neurol 57:896–903
Zekry D, Duyckaerts C, Moulias R, Belmin J, Geoffre C, Herrmann F, Hauw JJ (2002) Degenerative and vascular lesions of the brain have synergistic effects in dementia of the elderly. Acta Neuropathol Berl 103:481–487
Zekry D, Duyckaerts C, Belmin J, Geoffre C, Herrmann F, Moulias R, Hauw JJ (2003) The vascular lesions in vascular and mixed dementia: the weight of functional neuroanatomy. Neurobiol Aging 24:213–219
Zekry D, Duyckaerts C, Belmin J, Geoffre C, Moulias R, Hauw JJ (2003) Microvascular changes induced by cerebral amyloid angiopathy in the elderly: relationship with dementia. Acta Neuropathol 106:367–373
Zhan SS, Beyreuther K, Schmitt HP (1994) Synaptophysin immunoreactivity of the cortical neuropil in vascular dementia of Binswanger type compared with the dementia of Alzheimer type and nondemented controls. Dementia 5:79–87
Zhang WW, Badonic T, Hoog A, Jiang MH, Ma KC, Nie JX, Olsson Y, Sourander P (1994) Structural and vasoactive factors influencing intracerebral arterioles in cases of vascular dementia and other cerebrovascular disease: a review. Immunohistochemical studies on expression of collagens, basal lamina components and endothelin-1. Dementia 5:153–162
Zhou DH, Wang JY, Li J, Deng J, Gao C, Chen M (2004) Study on frequency and predictors of dementia after ischemic stroke. The Chongqing Stroke Study. J Neurol 251:421–427
Chui HC, Zarow C, Mack WJ, Ellis WG, Zheng L, Jagust WJ, Mungas D, Reed BR, Kramer JH, Decarli CC, Weiner MW, Vinters HV (2006) Cognitive impact of subcortical vascular and Alzheimer’s disease pathology. Ann Neurol 60:677–687
Tomimoto H, Lin J, Ihara M, Ohtani R, Matsuo A, Miki Y (2007) Subinsular vascular lesions: an analysis of 119 consecutive autopsied brains. Eur J Neurol 14:95–101
Acknowledgments
The study was supported by the Society for the Support of Research in Experimental Neurology, Vienna, Austria. The author thanks the medical staff of many Vienna hospitals for the clinical and autopsy reports and brain material. Special thanks are due to Mrs. V. Rappelsberger for excellent laboratory work and Mr. E. Mitter-Ferstl, PhD, for secretarial and computer work.
Author information
Authors and Affiliations
Corresponding author
Additional information
After a lecture at the XI International Congress of Neuropathology, San Francisco, CA, on 11 September 2006.
Addendum
In a recent clinicopathological evaluation of 79 autopsy cases derived from a prospective longitudinal study of subcortical ischemic vascular disease (SIVD) and Alzheimer disease (AD), Chui et al. [555] found significant cerebrovascular lesions (CVL) in 30%, AD pathology in 54%, and hippocampal sclerosis (HS) in 18%. Statistical assessment showed that all three pathology variables contributed independently to cognitive status, but only the neuritic Braak scores contributed significantly to cognitive impairment, indicating that advancing AD pathology overwhelms the effects of the two other factors that, in the absence of considerable AD, contribute to mild cognitive impairment. A recent histologic-neuroimaging study in two patients with SIVD showed correspondence between subinsular T2 hyperintensive lesions and demyelination, gliosis, and dilated perivascular spaces [556].
Rights and permissions
About this article
Cite this article
Jellinger, K.A. The enigma of vascular cognitive disorder and vascular dementia. Acta Neuropathol 113, 349–388 (2007). https://doi.org/10.1007/s00401-006-0185-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-006-0185-2