
Abstract
We tested the hypothesis that pressure overload exacerbates oxidative stress associated with augmented mitochondrial permeability transition (MPT) pore opening and cell death in ischemic-reperfused hearts. Pressure overload decreased the level of reduced glutathione but increased nitrotyrosine and 8-hydroxydeoxyguanosine levels in ischemic-reperfused hearts. The activity of catalase, but not superoxide dismutase (SOD), was lower in ischemic-reperfused hearts perfused at higher pressure. Mitochondria from ischemic-reperfused hearts subjected to higher perfusion pressure displayed significantly greater [3H]-2-deoxyglucose-6-P entrapment suggestive of greater MPT pore opening and consistent with greater necrosis and apoptosis. Tempol (SOD mimetic) reduced infarct size in both groups but it remained greater in the higher pressure group. By contrast, uric acid (peroxynitrite scavenger) markedly reduced infarct size at higher pressure, effectively eliminating the differential between the two groups. Inhibition of xanthine oxidase, with allopurinol, reduced infarct size but did not eliminate the differential between the two groups. However, amobarbital (inhibitor of mitochondrial complex I) or apocynin [inhibitor of NAD(P)H oxidase] reduced infarct size at both pressures and also abrogated the differential between the two groups. Consistent with the effect of apocynin, pressure-overloaded hearts displayed significantly higher NAD(P)H oxidase activity. Furthermore, pressure-overloaded hearts displayed increased nitric oxide synthase activity which, along with increased propensity to superoxide generation, may underlie uric acid-induced cardioprotection. In conclusion, increased oxidative and nitrosative stress, coupled with lack of augmented SOD and catalase activities, contributes importantly to the exacerbating impact of pressure overload on MPT pore opening and cell death in ischemic-reperfused hearts.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Reactive oxygen species (ROS) play important roles in various normal cellular processes as well as pathologies. Indeed, excessive ROS generation is a critical event contributing to cell death in ischemic-reperfused hearts [10, 12, 16, 27, 35, 39]. The relative importance of ROS during ischemia and reperfusion has been the subject of intense investigation. Some studies suggest that during the ischemic phase, low levels of ROS are generated which can damage the electron transport chain thereby causing inefficient transfer of electrons with consequent increase in ROS generation. This aspect becomes particularly important with the availability of oxygen during early reperfusion of the myocardium thereby contributing to a large burst of ROS which plays a pivotal role in the genesis of reperfusion-induced injury [7, 12, 16, 21, 27, 28, 31, 39]. Important myocardial sources of ROS (e.g., superoxide) during reperfusion include the mitochondrial respiratory chain distal to complex I (NADH dehydrogenase), xanthine oxidase and NAD(P)H oxidase [27, 39]. It is noteworthy that although the myocardium possesses endogenous antioxidant defenses, such as the superoxide dismutase (SOD) and catalase, these mechanisms are usually overwhelmed following ischemia and reperfusion. In turn, these conditions are conducive to the interaction of superoxide with nitric oxide to produce the potent oxidant, peroxynitrite [31]. Consequently, exacerbated oxidative and nitrosative stress serves as a major trigger for the mitochondrial permeability transition (MPT) pore opening and subsequent cell death primarily via necrosis [4–7, 12, 14, 16, 21, 27, 28, 31, 39].
Our previous demonstration of load-dependency of myocardial ischemia reperfusion (IR) injury indicates that pressure overload favors pathways that promote cell death [24–26]. However, the association between pressure overload and ROS generation in relation to myocardial IR injury has not been explored. Since increased ROS generation is a key determinant of cell death following myocardial IR insult [6, 7, 10, 12, 14, 16, 21, 27, 28, 31, 39], we tested the hypotheses that (a) pressure overload exacerbates oxidative stress and (or) reduces the ability to scavenge ROS, (b) pressure overload-induced exacerbation of oxidative stress promotes greater MPT pore opening thereby worsening cell death via both necrosis and apoptosis and (c) inhibitors of ROS generation or the availability of ROS scavengers causes greater cardioprotection in hearts subjected to higher pressure. Accordingly, we determined the levels of several indicators of oxidative stress in association with activities of scavenging enzymes, catalase and SOD, in ischemic-reperfused rat hearts subjected to low and high perfusion pressures. Assessment of MPT pore opening in the isolated heart was achieved using mitochondrial entrapment of [3H]-2-deoxyglucose-6-P ([3H]-2DOG-6P) protocol [2, 15], while a flow cytometry-based assay was used for determination of apoptosis and necrosis. In addition, we examined the impact of pharmacological agents that either scavenge ROS (tempol and uric acid) [17, 22, 34] or inhibit their generation via complex I (amobarbital) [8, 32], xanthine oxidase (allopurinol) [39] and NAD(P)H oxidase (apocynin) [21] on infarct size in hearts subjected to low and high perfusion pressures.
Methods
Animals
Male Sprague–Dawley rats (9–11 weeks of age) were purchased from Harlan Laboratories (Indianapolis, IN). The use of animals for this study was approved by the Animal Care and Use Committee of the Medical College of Georgia.
Heart perfusion and determination of oxidative stress indicators
For all isolated heart perfusion experiments, the animals were heparinized (1,000 U/kg; i.p.), decapitated and hearts removed for perfusion on a Langendorff apparatus. The perfusion medium was standard Krebs–Henseleit (KH) buffer containing 11 mM glucose and equilibrated with 95% O2–5% CO2 at 37°C [24–26]. The perfusion pressure was set at either 80 or 160 cmH2O during the ischemia reperfusion protocol; these values correspond to about 60 and 120 mmHg, respectively [24–26].
The experimental protocols used in this investigation are summarized in Fig. 1. For determination of tissue levels and activities of indices of oxidative stress, hearts were subjected to the global model of IR insult. Accordingly, following 25 min of stabilization and 40 min of global ischemia, each heart was reperfused for 5 or 15 min followed by snap freezing in liquid nitrogen and kept at −80°C until assayed; hearts which were not subjected to the IR protocol served as normoxic controls. Tissue samples were then pulverized and used for assessment of reduced glutathione level and activities of SOD and catalase using commercially available kits (Cayman Chemical Company, Ann Arbor, Michigan). Tissue nitrotyrosine (as a stable footprint of oxidative stress) levels were determined using the slot blot analysis as described previously [23]. On the other hand, tissue 8-hydroxydeoxyguanonse (8-OHdG), as an index of oxidative DNA damage, was measured using an ELISA kit following manufacturer’s instruction (Northwest Life Science Specialties, LLC; Vancouver, WA) [23].

Diagram illustrating experimental protocols described in detail under Methods. [3H]-2DOG: [3H]-2-deoxyglucose
Myocardial NAD(P)H oxidase and nitric oxide synthase (NOS) activities were determined as described previously [20, 33]. Accordingly, the myocardial tissue was pulverized and placed in ice-cold homogenization buffer [vol/wt ratio 10; 50 mmol/L Tris–HCl (pH 7.4), 0.1 mmol/L EDTA, 0.1 mmol/L EGTA, 250 mmol/L sucrose and 10% glycerol] in the presence of protease inhibitors (1 mmol/L PMSF, 2 μmol/L leupeptin, 1 μmol/L pepstatin A, and 0.1% aprotinin). The tissue was homogenized on ice with a glass–Teflon homogenizer (15 strokes). Protein concentration was determined by standard Bradford assay (Bio-Rad). Myocardial NAD(P)H oxidase activity was measured by determining NAD(P)H-dependent superoxide production, in the presence and absence of tempol (10 mM), using the enhanced chemiluminescence technique as described previously [20]. On the other hand, total NOS activity was determined in the heart homogenates based on the rate of L-[3H] citrulline formation from L-[3H] arginine and defined as [3H] arginine to [3H] citrulline conversion inhibited by the nonselective NOS inhibitor, Nω-nitro-l-arginine (LNNA, 1 mmol/L). Aliquots of heart homogenates were incubated with [3H]arginine (10 μM final arginine, 71 Ci/mmol) in the presence of 1 mM NADPH, 30 nM calmodulin, 3 μM tetrahydrobiopterin, 2 mM CaCl2, 1 μM FAD, and 1 μM FMN in a final volume of 50 μl for 30 min at room temperature. The remainder of the assay was done as recently described [33].
Assessment of MPT pore opening
The induction of MPT pore opening, with ensuing cell death, occurs during the initial few minutes of reperfusion and the novel technique of mitochondrial entrapment of [3H]-2DOG-6P allows direct assessment of MPT pore opening and subsequent closure in the isolated perfused heart; [3H]-2DOG-6P is a metabolite of [3H]-2-deoxyglucose ([3H]-2DOG) which remains exclusively in the cytosol unless MPT pore opens [2, 15]. Accordingly, in the pre-ischemic stabilization phase, hearts were perfused with KH buffer containing 0.1 μCi/ml of [3H]-2DOG for 15 min followed by a 10-min washout of extracellular [3H]-2DOG. This was followed by 40 min of global ischemia and 15 min of reperfusion. Hearts which were not subjected to the IR protocol served as normoxic controls. Thereafter, hearts were minced and homogenized in buffer containing 250 mM sucrose, 10 mM Tris pH 7.4 and 1 mM EDTA using a dounce homogenizer; mitochondrial pellet was recovered following sequential centrifugation and assayed for [3H] and citrate synthase (Sigma-Aldrich); whole homogenate was also assayed for radioactivity. Mitochondrial entrapment of [3H]-2DOG-6P was expressed as (mitochondrial [3H] dpm per unit citrate synthase)/(total heart [3H] dpm/g wet wt.) × 105 [2, 15].
Assessment of cell necrosis and apoptosis
At the end of global IR protocol (40 min ischemia and 15 min reperfusion), hearts were perfused with KH buffer containing 100 units/ml of collagenase II (2 ml/min for 5 min; Sigma-Aldrich); hearts which were not subjected to the IR protocol served as normoxic controls. Additional hearts were treated with apocynin [100 μM as an NAD(P)H oxidase inhibitor] during the IR protocol to determine its effect on cell death via necrosis and apoptosis [21]. Thereafter, the hearts were transferred to KH buffer containing 400 units/ml of collagenase II and incubated at 37°C for 40 min [13]. Subsequently, hearts were made into a single-cell suspension and centrifuged at 1500 rpm for 10 min. This was followed by determination of cell necrosis by propidium iodide fluorescence and assessment of apoptosis by the terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) assay; these protocols were carried out using flow cytometry [FACSCalibur machine (BD Biosciences, Mountain View, CA)] and the APO-BRDU kit (Phoenix Flow System), following manufacturer’s protocols [3, 19].
Infarct size studies
For infarct size studies, the regional model (i.e., coronary ligation) of IR injury using the Langendorff perfused heart was employed [24–26]. In brief, after a 25-min stabilization period, each heart was subjected to 40 min of regional ischemia followed by 2 h of reperfusion (Fig. 1). Accordingly, the perfusion buffer contained either no drug (control) or one of the following agents: tempol (1 mM as a SOD mimetic) [17, 22], uric acid (100 μM as a peroxynitrite scavenger) [34], allopurinol (1 mM as a xanthine oxidase inhibitor) [39], amobarbital (2.5 mM as an inhibitor of NADH dehydrogenase distal to mitochondrial complex I) [8, 32] or apocynin (100 μM) [21]. Infarct size was expressed as the ratio of infarcted zones (lacking staining with tetrazolium) to risk zones (excluding zinc cadmium particles), which were determined by computerized planimetry [24–26]. Cardiac function was measured with a pressure transducer by inserting a 23 gauge needle through the ventricular wall; the pressure transducer was connected to a computerized heart performance analyzer (MicroMed, Louisville, KY); rate-pressure-product was calculated as an index of myocardial work [25, 26]. Also determined were indices of myocardial contractility and relaxation (maximum ± dP/dt) [25, 26]. Since amobarbital reduces myocardial function, data prior to its administration were recorded as baseline values.
Statistics
Data are expressed as mean ± SEM. Data were analyzed using the analysis of variance for comparison of multiple groups followed by Duncan’s post hoc test to establish significance (p < 0.05).
Results
Ischemia reperfusion insult was associated with decreased myocardial level of reduced glutathione but increased nitrotyrosine and 8-OHdG levels compared to those not subjected to the IR protocol (i.e., normoxic hearts; Fig. 2a–c). In hearts subjected to 40 min of global ischemia followed by 15 min of reperfusion, high perfusion pressure resulted in significantly lower levels of reduced glutathione but higher levels of nitrotyrosine and 8-OHdG relative to those perfused at lower pressure (Fig. 2a–c). In order to determine whether exacerbated oxidative stress in pressure-overloaded hearts is associated with decreased ability to scavenge superoxide and hydrogen peroxide, the activities of SOD and catalase were measured in hearts subjected to 40 min of global ischemia followed by 5 or 15 min of reperfusion. As shown in Fig. 3a, the activity of SOD was similar in hearts subjected to either low or high pressure. On the other hand, hearts subjected to the lower, but not higher, perfusion pressure displayed mild, albeit significant, increases in catalase activity following 5 and 15 min of reperfusion (Fig. 3b). As a result, a mild, albeit significant, differential in catalase activity was detectable between the 15-min reperfused hearts.

Bar graphs showing tissue levels of reduced glutathione (GSH; panel a), nitrotyrosine (panel b) and 8-hydroxydeoxyguanosine (8-OHdG; panel c) in normoxic control and ischemic-reperfused hearts that were subjected to low (80 cmH2O) and high (160 cmH2O) perfusion pressures. Nitrotyrosine level is expressed as the percent of the respective lower pressure group and representative bands are shown for each group. Data represent mean ± SEM of n = 5–7 hearts/group for normoxic hearts and n = 8–10 hearts/group for ischemic-reperfused hearts. *p < 0.05 compared to their counterparts at the lower perfusion pressure. #p < 0.05 compared to their normoxic counterparts

Bar graphs showing tissue activities of superoxide dismutase (SOD; panel a) and catalase (panel b) in normoxic control hearts (n = 8 hearts/group) or hearts subjected to ischemia (40 min) and reperfusion (either 5 or 15 min; n = 4 or 8 hearts/group, respectively) at low (80 cmH2O) and high (160 cmH2O) perfusion pressures. Data represent mean ± SEM. *p < 0.05 compared to their normoxic counterparts. #p < 0.05 compared to their lower pressure counterparts
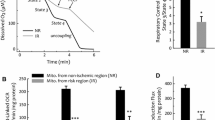
Oxidative stress is a major trigger for MPT pore opening and subsequent cell death via apoptosis and necrosis. Consistent with this notion, mitochondrial entrapment of [3H]-2DOG-6P was significantly increased in ischemic-reperfused than normoxic hearts; further, mitochondrial [3H]-2DOG-6P uptake was also significantly greater for ischemic-reperfused hearts at the higher pressure (Fig. 4). In order to establish the impact of greater induction of MPT pore opening in pressure-overloaded hearts on cell necrosis and apoptosis, a flow cytometry-based assay was employed. Accordingly, cell necrosis was determined by propidium iodide staining while apoptosis relied on labeling of 3-hydroxyl ends of single- or double-stranded DNA breaks. As shown in Fig. 5, an IR insult significantly increased necrotic and apoptotic cell death compared to normoxic condition. However, pressure overload markedly increased the contribution of both necrosis and apoptosis to cell death in IR hearts, a finding consistent with a significant increase in infarct size in hearts subjected to higher pressure as described below [24–26].

Bar graphs showing mitochondrial entrapment of [3H]-2DOG-6P (an index of MPT pore opening) in normoxic control (n = 4 hearts/group) and ischemic-reperfused hearts subjected to low (80 cmH2O; n = 4) and high (160 cmH2O; n = 6) perfusion pressure. Data represent mean ± SEM. *p < 0.05 compared to their counterparts at the lower perfusion pressure. #p < 0.05 compared to their normoxic counterparts

Flow cytometry analysis of apoptosis and necrosis in cells isolated from hearts subjected to low (80 cmH2O) and high (160 cmH2O) perfusion pressures. Panel a shows representative dot matrices while panel b shows histograms for apoptotic and necrotic cells for ischemic-reperfused hearts subjected to either low or high pressure. Also shown, in panel c, are percent cell death (both necrosis and apoptosis) for normoxic (n = 3 hearts/group), vehicle-treated (n = 5 hearts/group) and apocynin-treated (n = 4 hearts/group) ischemic-reperfused hearts at either perfusion pressure. Data are mean ± SEM. *p < 0.05 compared to their counterparts perfused at the lower pressure. # p < 0.05 compared to normoxic or apocynin-treated ischemic-reperfused hearts at the same pressure
We next determined the impact of several pharmacological agents that are known to either scavenge ROS or inhibit their generation via pathways serving as important sources of ROS production in ischemic-reperfused hearts. Consistent with previous studies [24–26], pressure overload was associated with significant increase in baseline rate-pressure-product (RPP), an index of myocardial work, in all groups (Fig. 6). However, treatment with tempol, allopurinol or apocynin was associated with variable but significant reductions in RPP at either low or high pressure; on the other hand, uric acid did not affect RPP at either pressure (Fig. 6).

Bar graphs showing baseline rate-pressure-product (RPP; an index of myocardial work) of untreated control hearts (n = 6–8 hearts/group) and hearts treated with either tempol (1 mM; n = 6 hearts/group), uric acid (100 μM; n = 5–6 hearts/group), allopurinol (1 mM; n = 5 hearts/group), amobarbital (2.5 mM; n = 5–6 hearts/group) or apocynin (100 μM; n = 6 hearts/group) at low (80 cmH2O) and high (160 cmH2O) perfusion pressures, as described in “Methods” and summarized in Fig. 1. Data represent mean ± SEM. *p < 0.05 compared to their counterparts at the lower pressure. #p < 0.05 compared to their control counterparts subjected to the same pressure
As shown in Fig. 7a, pressure overload caused significant worsening of infarct size in vehicle-treated control hearts. Treatment with tempol (SOD mimetic) resulted in a similar and significant decrease in infarct size at either high or low perfusion pressure. However, a significant differential in infarct size persisted between hearts subjected to higher and lower pressures. By contrast, treatment with uric acid (scavenger of peroxynitrite) was associated with a significant reduction in infarct size of hearts that were subjected to the higher pressure. Consequently, the pressure-induced differential in infarct size was nearly eliminated in hearts treated with uric acid. On the other hand, treatment with allopurinol caused similar and significant reductions of infarct size in hearts subjected to either high or low perfusion pressure, resulting in a significant differential in infarct size between the two groups. However, perfusion of hearts with amobarbital or apocynin reduced infarct size at either perfusion pressure, with abrogation of pressure-related differential in infarct size (Fig. 7, panel a). Subsequent flow cytometry-based studies showed that treatment with apocynin markedly reduced both necrosis and apoptosis in ischemic-reperfused hearts (Fig. 5). It is noteworthy that the IR insult was associated with significant reductions in functional recovery of the pressure-overloaded hearts as determined by indices of myocardial contractility and relaxation (maximum ± dP/dt; range of 37–43 vs. 8–10% for lower vs. higher pressure groups, respectively; Fig. 7, panels b, c). Treatment with pharmacological agents used in this study were associated with greater recovery of pressure-overloaded hearts [range of 20–30% (contractility) and relaxation (20–25%)] thereby diminishing the pressure-related differential of respective drug-treated groups (Fig. 7, panels b, c; p > 0.05).

Panel a shows infarct size, expressed as percent of risk zone, of untreated control and drug-treated hearts subjected to low (80 cmH2O) and high (160 cmH2O) perfusion pressures, as described in Fig. 6. On the other hand, panels b and c show percent recovery (from baseline values) of myocardial contractility (maximum +dP/dt) and relaxation (maximum −dP/dt) of experimental groups following 40 min of ischemia and 2 h of reperfusion. Data represent mean ± SEM. *p < 0.05 compared to their counterparts at the lower pressure. # p < 0.05 compared to their control counterparts at the same pressure
To further explore the impact of pressure overload on NAD(P)H oxidase, we determined the activity of this enzyme in hearts that were subjected to 40 min of global ischemia and 15 min of reperfusion. As shown in Fig. 8, tissue generation of superoxide was significantly higher in hearts subjected to higher pressure. Inclusion of tempol in the assay medium caused marked reductions in both of these values and also abrogated the differential in superoxide generation between the hearts exposed to higher and lower pressures (Fig. 8).

Bar graphs showing myocardial NAD(P)H-dependent superoxide generation, in the presence and absence of tempol (10 mM), of hearts subjected to low (80 cmH2O) and high (160 cmH2O) perfusion pressures as described in “Methods” and summarized in Fig. 1. Data represent mean ± SEM of 6 hearts/group/condition. *p < 0.05 compared to their counterparts at the lower pressure. #p < 0.05 compared to their counterparts in the absence of tempol
Finally, in light of increased nitrotyrosine levels and the marked cardioprotective effect of uric acid in pressure-overloaded ischemic-reperfused hearts, NOS activity was measured in freeze-clamped hearts that were subjected to 40 min of ischemia and 15 min of reperfusion. As shown in Fig. 9, ischemic-reperfused hearts perfused at the higher pressure showed significant increase in NOS activity compared to their lower pressure counterparts. Pressure overload also tended to increase myocardial NOS activity in normoxic hearts although the change was not significant (p = 0.0635).

Bar graphs showing myocardial nitric oxide synthase (NOS) activity of normoxic and ischemic-reperfused hearts subjected to low (80 cmH2O) and high (160 cmH2O) perfusion pressures as described in Fig. 1. Data are mean ± SEM of 5–6 hearts/group/condition. *p < 0.05 compared to their ischemic-reperfused counterparts or the normoxic controls at the lower pressure
Discussion
The main finding of the present study is that pressure overload is associated with exacerbated oxidative and nitrosative stress, greater MPT pore opening, apoptosis and necrosis in ischemic-reperfused hearts. The pressure overload-induced exacerbation of infarct size was abrogated by inhibition of mitochondrial complex I or NAD(P)H oxidase or by scavenging of peroxynitrite. Collectively, the results indicate that pressure overload is an important determinant of ROS generation, and subsequent cell death, in hearts subjected to an IR insult.
Commonly used indicators of oxidative stress include reduced glutathione, nitrotyrosine and 8-OHdG [23, 28, 31]. Thus, the decreased level of reduced glutathione but increased nitrotyrosine and 8-OHdG levels is consistent with the notion of greater oxidative and nitrosative stress burden of pressure-overloaded ischemic-reperfused hearts. Interestingly, however, neither SOD nor catalase activity was greater in pressure-overloaded hearts; on the contrary, pressure-overloaded hearts displayed a mild but significant reduction in catalase activity. Clearly, the imbalance between ROS generation and their scavenging underlies exacerbated oxidative stress in pressure-overloaded hearts.
One noted consequence of oxidative and nitrosative stress is opening of the MPT pore and subsequent cell death by apoptosis and necrosis [5, 6, 12, 16]. Indeed, the novel technique of [3H]-2DOG-6P entrapment allows determination of MPT pore opening in the isolated hearts [2, 15]. Accordingly, hearts perfused at a higher pressure displayed greater mitochondrial entrapment of radioactivity than those perfused at a lower pressure. This finding is consistent with previous indirect evidence for greater MPT pore induction in pressure loaded hearts [24–26]. First, cyclosporine A, an inhibitor of the MPT pore, causes greater cardioprotection in hearts subjected to the higher pressure [25, 26]. Second, inhibitors of glycogen synthase kinase 3β (GSK-3β) exert greater infarct-sparing effects in pressure-overloaded hearts [26]; GSK-3β has emerged as a major regulator of the MPT pore [12, 16, 27]. Third, pressure overload is associated with reduced level of the (ser-9) phosphorylated form of GSK-3β which is considered to be inactive and cardioprotective [24]. Although necrosis is the major mode of cell death in the ischemic-reperfused heart [16], the contribution of both necrosis and apoptosis to cell death markedly increases at the higher pressure. It is noteworthy that apoptotic and necrotic cell death was markedly lower in normoxic hearts and apocynin-treated ischemic-reperfused hearts at either pressure thereby demonstrating the great potential of flow cytometry for studies focused on IR injury. Collectively, the data point to the pivotal role for perfusion pressure as a major determinant of cell death via both apoptosis and necrosis. Thus, subsequent studies focused on assessment of the relative contribution of major pathways that regulate ROS generation in the ischemic-reperfused hearts subjected to pressure overload.
Previous studies have led to the recognition that mitochondrial damage does occur during the ischemic phase of IR injury and that the mitochondria sustain additional damage during reperfusion [1, 18]. The importance of ischemic mitochondrial damage is illustrated by the demonstration that amobarbital treatment 1 min before the ischemic phase causes a more profound reduction in infarct size of isolated rat heart compared to its administration during the first 5 min of reperfusion [8, 32]. We subjected the hearts to amobarbital both immediately before the ischemic phase and during early reperfusion in an attempt to maximize its protective effects. Although this protocol resulted in significant infarct-sparing effect in hearts subjected to both low and high perfusion pressures, the reduction in infarct size was greater for hearts subjected to the higher pressure, suggesting a pressure-related pivotal role for mitochondrial ROS generation via complex I. However, it is noteworthy that amobarbital exerts other effects which can potentially benefit the ischemic heart upon reperfusion [8, 32]. These include increasing efficiency of oxidative phosphorylation thereby favoring high-energy phosphate production [8] and enhanced glycolysis and persistent intracellular acidosis thereby decreasing MPT pore opening and myocardial injury [32].
Another major finding of this study is the demonstration that apocynin-induced inhibition of NAD(P)H oxidase also abrogated pressure-related differential in infarct size. Apocynin inhibits the binding of the cytosolic subunits of NAD(P)H oxidase to the membrane-bound p22phox/Nox subunits thereby preventing its activation; the importance of NAD(P)H oxidase as a major contributor to superoxide generation in the heart is well-recognized [31]. Our observations with apocynin suggest that pressure overload causes greater activation of NAD(P)H oxidase thereby augmenting superoxide production. This notion is supported by the demonstration of increased NAD(P)H oxidase activity in ischemic-reperfused hearts subjected to high pressure. These findings are consistent with previous reports indicating that mechanical stress increases superoxide production, likely through paracrine- and autocrine-induced release of angiotensin II and subsequent activation of angiotensin II receptor subtype 1 (AT1R) [7, 25, 29]. It is noteworthy that mechanical stress is also shown to activate AT1R independent of angiotensin II release [38]. Since activation of AT1R is linked to increased NAD(P)H oxidase activity, this provides a plausible explanation for both increased activity of NAD(P)H oxidase activity and greater apocynin-induced infarct-sparing effect in hearts subjected to pressure overload. This observation is supported by our previous demonstration that treatment with the AT1R inhibitor, candesartan, reduced infarct size in a pattern very similar to apocynin [25]. Collectively, these results provide strong support for the hypothesis that pressure overload augments the AII/AT1R/NAD(P)H oxidase axis thereby exacerbating oxidative stress and consequent worsening of IR injury.
Another source of ROS generation is the enzyme xanthine oxidase which acts upon hypoxanthine and xanthine, products of ATP metabolism. Thus, substrate availability (e.g., secondary to ischemia reperfusion insult) is a critical determinant for the contribution of xanthine oxidase to ROS generation [39]. Moreover, ischemia promotes conversion of xanthine dehydrogenase to xanthine oxidase thereby increasing the contribution of this pathway to ROS generation [39]. Consistent with previous reports that inhibition of xanthine oxidase reduces ROS generation and protects against IR injury, allopurinol treatment caused significant reduction in infarct size in hearts subjected to either high or low pressure. Interestingly, however, the treatment did not abrogate infarct size differential between the two groups. One likely explanation may relate to the localization of xanthine oxidase in the endothelium and the requirement of transport of adenosine and inosine from the cardiomyocyte to endothelial cells by the nucleotide transport system [39]. Indeed, it has been shown that inhibition of either adenosine deaminase (i.e., enzyme converting adenosine to inosine) or the nucleotide transporter protein effectively blocks free radical generation [11, 39]. Therefore, the lack of greater efficacy of allopurinol in pressure-overloaded hearts may relate to interference with processes that are prerequisite for substrate availability to xanthine oxidase. On the other hand, unlike the cardiomyocytes, the contribution of endothelial ROS generation by xanthine oxidase may not be differentially affected by pressure overload.
We also examined whether scavenging of superoxide and peroxynitrite would exert differential effects in pressure-overloaded hearts. Consistent with previous reports [22], the SOD mimetic tempol reduced infarct size in hearts subjected to either low or high pressure although a significant differential remained between the two groups. Interestingly, however, uric acid, which is a potent scavenger of peroxynitrite [34], caused minimal effect on infarct size at the lower pressure but markedly reduced infarct size of hearts subjected to the higher pressure, effectively abrogating infarct size differential between the two groups. Several forms of SOD are known to exist; MnSOD detoxifies superoxide generated by mitochondria while Cu/ZnSOD and ecSOD scavenge cytoplasmic and extracellularly generated superoxide. The availability of superoxide for peroxynitrite generation depends on the activities of the different forms of SOD [31]. It is suggested that when intracellular generation of nitric oxide approaches the levels of superoxide, more than half of the superoxide will react with nitric oxide to produce the potent oxidant peroxynitrite [31]. Interestingly, however, neither the activity of SOD nor that of catalase was greater in pressure-overloaded hearts; on the contrary, pressure-overloaded hearts displayed a mild but significant reduction in catalase activity. Thus, it is likely that greater generation of superoxide, coupled with the lack of upregulation of scavenging enzymes, and increased nitric oxide synthase activity in pressure-overloaded ischemic-reperfused hearts contributes to greater generation of peroxynitrite thereby providing a plausible explanation for the relatively greater efficacy of uric acid to reduce infarct size. While nitric oxide is generally cardioprotective [30], permissive conditions (e.g., pressure overload) which promote peroxynitrite formation can elicit deleterious consequences for the myocardium [28, 31, 39].
Our collective observations indicate that pressure overload exacerbates oxidative and nitrosative stress in the ischemic-reperfused heart. Interestingly, ROS production in cardiomyocytes [e.g., through NAD(P)H oxidase] can act to further exacerbate oxidative stress in a process known as ROS-induced-ROS generation and release. This phenomenon is likely related to mitochondrial depolarization and subsequent mitochondrial ROS release through the mitochondrial permeability transition (and the mitochondrial inner membrane anion channel) [7, 12]. The generated ROS can be released into the cytosol and trigger ROS-induced-ROS generation and release in neighboring mitochondria thereby establishing a positive feedback mechanism culminating in exacerbated injury [12, 37]. Thus, the increased ROS production in the pressure-overloaded hearts could be both a cause and an effect of MPT pore opening.
Another noted finding of this study relates to the reduction in baseline RPP in hearts that were treated with pharmacological agents that modulate ROS levels, an effect more prominent for tempol, allopurinol and apocynin. It is important to note that while excessive ROS generation (e.g., following an IR insult) is detrimental, low levels of ROS are implicated in various physiological processes [12, 39]. Indeed, increasing evidence implicate ROS as an important determinant of myocardial contractility. For example, increased superoxide generation via NAD(P)H oxidase is believed to contribute to the positive inotropic effects of angiotensin II and endothelin-1 [9]. While the molecular mechanisms mediating the effect of superoxide on cardiac contractility remain to be established, ROS are known to modulate the activity of a number of ion channels and transporters involved in cardiomyocyte calcium regulation [36]. Our observation that baseline RPP in the isolated heart was most affected by agents that either scavenge superoxide (tempol) or prevent its generation (apocynin and to a lesser degree allopurinol) is in general agreement with increasing evidence implicating superoxide in the regulation of calcium and myocardial function [9, 36]. It is noteworthy that myocardial functional recovery was generally improved in drug-treated pressure-overloaded hearts although the effect was mild-moderate. The lack of a marked beneficial impact on functional recovery with agents that cause marked infarct-sparing effects is consistent with previous observations [25, 26]. Likely explanations may relate to differences in mechanisms regulating cell death and functional recovery, the use of regional model of IR injury and the relatively long durations of ischemia (40 min) and reperfusion (2 h).
In conclusion, mitochondrial complex I and NAD(P)H oxidase contribute importantly to exacerbated oxidative stress in pressure-overloaded ischemic-reperfused hearts. In addition, our previous studies established that calcium overload contributes to the detrimental impact of pressure overload on infarct size [25]. In turn, both oxidative stress and calcium overload are major triggers for MPT pore opening and subsequent cell death via apoptosis and necrosis, aspects that were unraveled in the present study.
References
Ambrosio G, Zweier JL, Duilio C, Kuppusamy P, Santoro G, Elia PP, Tritto I, Cirillo P, Condorelli M, Chiariello M, Flaherty JT (1993) Evidence that mitochondrial respiration is a source of potentially toxic oxygen free radicals in intact rabbit hearts subjected to ischemia and reflow. J Biol Chem 268: 18532-18541. Available at http://www.jbc.org/
Ananthakrishnan R, Kaneko M, Hwang YC, Quadri N, Gomez T, Li Q, Caspersen C, Ramasamy R (2009) Aldose reductase mediates myocardial ischemia-reperfusion injury in part by opening mitochondrial permeability transition pore. Am J Physiol Heart Circ Physiol 296:H333–H341. doi:10.1152/ajpheart.01012.2008
Baban B, Chandler PR, Sharma MD, Pihkala J, Koni PA, Munn DH, Mellor AL (2009) IDO activates regulatory T cells and blocks their conversion into Th17-like T cells. J Immunol 183:2475–2483. doi:10.4049/jimmunol.0900986
Baines CP (2009) The mitochondrial permeability transition pore and ischemia-reperfusion injury. Basic Res Cardiol 104:181–188. doi:10.1007/s00395-009-0004-8
Boengler K, Stahlhofen S, van de Sand A, Gres P, Ruiz-Meana M, Garcia-Dorado D, Heusch G, Schulz R (2009) Presence of connexin 43 in subsarcolemmal, but not in interfibrillar cardiomyocyte mitochondria. Basic Res Cardiol 104:141–147. doi:10.1007/s00395-009-0007-5
Borutaite V, Morkuniene R, Brown GC (1999) Release of cytochrome c from heart mitochondria is induced by high Ca2+ and peroxynitrite and is responsible for Ca(2+)-induced inhibition of substrate oxidation. Biochim Biophys Acta 1453(1):41–48. doi:10.1016/S0925-4439(98)00082-9
Brandes RP (2005) Triggering mitochondrial radical release: a new function for NADPH oxidases. Hypertension 45:847–848. doi:10.1161/01.HYP.0000165019.32059.b2
Chen Q, Moghaddas S, Hoppel CL, Lesnefsky EJ (2006) Reversible blockade of electron transport during ischemia protects mitochondria and decreases myocardial injury following reperfusion. J Pharmacol Exp Ther 319:1405–1412. doi:10.1124/jpet.106.110262
De Giusti VC, Correa MV, Villa-Abrille MC, Beltrano C, Yeves AM, Chiappe de Cingolani GE, Cingolani HE, Aiello EA (2008) The positive inotropic effect of endothelin-1 is mediated by mitochondrial reactive oxygen species. Life Sci 83:264–271. doi:10.1016/j.lfs.2008.06.008
Di Lisa F, Klaudercic N, Carpi A, Menabo R, Giorgio M (2009) Mitochondrial pathways for ROS formation and myocardial injury: the relevance of p66(Shc) and monoamine oxidase. Basic Res Cardiol 104:131–139. doi:10.1007/s00395-009-0008-4
Guarnieri C, Flamigni F, Caldarera CM (1980) Role of oxygen in the cellular damage induced by reoxygenation of hypoxic hearts. J Mol Cell Cardiol 12:797–808. doi:10.1016/0022-2828(80)90081-4
Gustafsson AB, Gottlieb RA (2008) Heart mitochondria: gates of life and death. Cardiovasc Res 77:334–343. doi:10.1093/cvr/cvm005
Hadzimichalis NM, Baliga SS, Golfetti R, Jaques KM, Firestein BL, Merrill GF (2007) Acetaminophen-mediated cardioprotection via inhibition of the mitochondrial permeability transition pore-induced apoptotic pathway. Am J Physiol Heart Circ Physiol 293:H3348–H3355. doi:10.1152/ajpheart.00947.2007
Heusch G, Boengler K, Schulz R (2010) Inhibition of mitochondrial permeability transition pore opening: the holy grail of cardioprotection. Basic Res Cardiol 105:151–154. doi:10.1007/s00395-009-0080-9
Javadov SA, Clarke S, Das M, Griffiths EJ, Lim KH, Halestrap AP (2003) Ischaemic preconditioning inhibits opening of mitochondrial permeability transition pores in the reperfused rat heart. J Physiol 549:513–524. doi:10.1113/jphysiol.2003.034231
Javadov S, Karmazyn M (2007) Mitochondrial permeability transition pore opening as an endpoint to initiate cell death and as a putative target for cardioprotection. Cell Physiol Biochem 20:11–22. doi:10.1159/000103747
Kutala VK, Khan M, Mandal R, Potaraju V, Colantuono G, Kumbala D, Kuppuamy P (2006) Prevention of postischemic myocardial reperfusion injury by the combined treatment of NCX-4016 and tempol. J Cardiovasc Pharmacol 48:79–87. doi:10.1097/01.fjc.0000242050.16790.65
Lesnefsky E, Chen Q, Moghaddas S, Hassan MO, Tandler B, Hoppel CL (2004) Blockade of electron transport during ischemia protects cardiac mitochondria. J Biol Chem 279:47961–47967. doi:10.1074/jbc.M409720200
Lin KM, Lin B, Lian IY, Mestril R, Scheffler IE, Dillmann WH (2001) Combined and individual mitochondrial HSP60 and HSP10 expression in cardiac myocytes protects mitochondrial function and prevents apoptotic cell death induced by simulated ischemia-reoxygenation. Circulation 103:1787–1792. Available at http://circ.ahajournals.org/
Loomis ED, Sullivan JC, Osmond DA, Pollock DM, Pollock JS (2005) Endothelin mediates superoxide production and vasoconstriction through activation of NADPH oxidase and uncoupled nitric-oxide synthase in the rat aorta. J Pharmacol Exp Ther 315:1058–1064. doi:10.1124/jpet.105.091728
Matsushima S, Kinugawa S, Yokota T, Inoue N, Ohta Y, Hamaguchi S, Tsutsui H (2009) Increased myocardial NAD(P)H oxidase-derived superoxide causes the exacerbation of postinfarct heart failure in type 2 diabetes. Am J Physiol Heart Circ Physiol 297:H409–H416. doi:10.1152/ajpheart.01332.2008
McDonald MC, Zacharowski K, Bowes J, Cuzzocrea S, Thiemermann C (1999) Tempol reduces infarct size in rodent models of regional myocardial ischemia and reperfusion. Free Radic Biol Med 27:493–503. doi:10.1016/S0891-5849(99)00100-8
Mozaffari MS, Abdelsayed R, Liu JY, Wimborne H, El-Remessy A, El-Marakby A (2009) Effects of chromium picolinate on glycemic control and kidney of the obese Zucker rat. Nutr Metab (Lond) 6:51. doi:10.1186/1743-7075-6-51
Mozaffari MS, Liu JY, Schaffer SW (2010) Effect of pressure overload on cardioprotection via PI3K-Akt: comparison of postconditioning, insulin, and pressure unloading. Am J Hypertens 23:668–674. doi:10.1038/ajh.2010.43
Mozaffari MS, Patel C, Schaffer SW (2006) Mechanisms underlying afterload-induced exacerbation of myocardial infarct size: role of T-type Ca2+ channel. Hypertension 47:912–919. doi:10.1161/01.HYP.0000209940.65941.46
Mozaffari MS, Schaffer SW (2008) Effect of pressure overload on cardioprotection of mitochondrial KATP channels and GSK-3beta: interaction with the MPT pore. Am J Hypertens 21:570–575. doi:10.1038/ajh.2008.25
Murphy E, Steenbergen C (2008) Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev 88:581–609. doi:10.1152/physrev.00024.2007
Paravicini TM, Touyz RM (2008) NADPH oxidases, reactive oxygen species, and hypertension: clinical implications and therapeutic possibilities. Diabetes Care 31:S170–S180. doi:10.2337/dc08-s247
Pimentel DR, Amin JK, Xiao L, Miller T, Vierek J, Oliver-Krasinski J, Baliga R, Wang J, Siwik DA, Singh K, Pagano P, Colucci WS, Sawyer DB (2001) Reactive oxygen species mediate amplitude-dependent hypertrophic and apoptotic responses to mechanical stretch in cardiac myocytes. Circ Res 89:453–460. doi:10.1161/hh1701.096615
Shiva S, Gladwin MT (2009) Nitrite mediates cytoprotection after ischemia/reperfusion by modulating mitochondrial function. Basic Res Cardiol 104:113–119. doi:10.1007/s00395-009-0009-3
Sorescu D, Griendling KK (2002) Reactive oxygen species, mitochondria, and NAD(P)H oxidases in the development and progression of heart failure. Congest Heart Fail 8:132–140. doi:10.1111/j.1527-5299.2002.00717.x
Stewart S, Lesnefsky E, Chen Q (2009) Reversible blockade of electron transport with amobarbital at the onset of reperfusion attenuates cardiac injury. Transl Res 153:224–231. doi:10.1016/j.trsl.2009.02.003
Sullivan JC, Pardieck JL, Hyndman KA, Pollock JS (2010) Renal NOS activity, expression and localization in male and female spontaneously hypertensive rats. Am J Physiol Regul Integr Comp Physiol 298(1):R61–R69. doi:10.1152/ajpregu.00526.2009
Wang HC, Zhang HF, Guo WY, Su H, Zhang KR, Li QX, Yan W, Ma XL, Lopez BL, Christopher TA, Gao F (2006) Hypoxic postconditioning enhances the survival and inhibits apoptosis of cardiomyocytes following reoxygenation: role of peroxynitrite formation. Apoptosis 11:1453–1460. doi:10.1007/s10495-006-7786-z
Wojtovich AP, Brookes PS (2009) The complex II inhibitor atpenin A5 protects against cardiac ischemia-reperfusion injury via activation of mitochondrial KATP channels. Basic Res Cardiol 104:121–129. doi:10.1007/s00395-009-0001-y
Zhang YH, Dingle L, Hall R, Casadei B (2009) The role of nitric oxide and reactive oxygen species in the positive inotropic response to mechanical stretch in the mammalian myocardium. Biochim Biophys Acta 1787:811–817. doi:10.1016/j.bbabio.2009.03.020
Zorov DB, Juhaszova M, Sollott SJ (2006) Mitochondrial ROS-induced-ROS release: an update and review. Biochim Biophys Acta 1757:509–517. doi:10.1016/j.bbabio.2006.04.029
Zou Y, Akazawa H, Qin Y, Sano M, Takano H, Minamino T, Makita N, Iwanaga K, Zhu W, Kudoh S, Toko H, Tamura K, Kihara M, Nagai T, Fukamizu A, Umemura S, Iiri T, Fujita T, Komuro I (2004) Mechanical stress activates angiotensin II type 1 receptor without the involvement of angiotensin II. Nat Cell Biol 6:499–506. doi:10.1038/ncb1137
Zweier JL, Talukder MA (2006) The role of oxidants and free radicals in reperfusion injury. Cardiovasc Res 70:181–190. doi:10.1016/j.cardiores.2006.02.025
Acknowledgments
This study was supported by a grant from the American Heart Association, Southeast Affiliate (0755627B; MSM).
Conflict of interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mozaffari, M.S., Baban, B., Liu, J.Y. et al. Mitochondrial complex I and NAD(P)H oxidase are major sources of exacerbated oxidative stress in pressure-overloaded ischemic-reperfused hearts. Basic Res Cardiol 106, 287–297 (2011). https://doi.org/10.1007/s00395-011-0150-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00395-011-0150-7