
Abstract
Although mitochondria are considered the most relevant site for the formation of reactive oxygen species (ROS) in cardiac myocytes, a major and unsolved issue is where ROS are generated in mitochondria. Respiratory chain is generally indicated as a main site for ROS formation. However, other mitochondrial components are likely to contribute to ROS generation. Recent reports highlight the relevance of monoamine oxidases (MAO) and p66Shc. The importance of these systems in the irreversibility of ischemic heart injury will be discussed along with the cardioprotective effects elicited by both MAO inhibition and p66Shc knockout. Finally, recent evidence will be reviewed that highlight the relevance of mitochondrial ROS formation also in myocardial failure and atherosclerosis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The complete reduction of O2 into two molecules of H2O requires four electrons. This process occurs sequentially since O2 accepts only one electron at a time. Consequently, O2 reduction inevitably implies the formation of partially reduced intermediates. In particular, the addition of the first electron yields superoxide anion (O2 · –) that becomes hydrogen peroxide when one additional electron is added. A further single electron reduction produces the hydroxyl radical (·OH) that accepting another electron becomes H2O. O2 · – and ·OH are oxygen radicals (i.e., containing unpaired electrons) and represent quite reactive species. H2O2 is less reactive per se, but, according to the Fenton reaction, in the presence of Fe2+ or Cu+ generates ·OH. These partially reduced forms of oxygen are commonly referred to as reactive oxygen species (ROS), which also include the highly reactive singlet oxygen (1O2). This excited form of oxygen is produced by the Haber-Weiss reaction (i.e., the non-enzymatic dismutation of O2 · –) or high energy irradiation (i.e., UV light).
ROS formation and toxicity are counterbalanced by a complex defense system. The most efficacious strategy is the enzymatic removal of ROS that is catalyzed by superoxide dismutases (SODs) and peroxidases [48, 49]. Through SOD reaction O2 · – is transformed into H2O2 that is then reduced into water by peroxidases including catalase and glutathione peroxidases. Reduction of oxidized molecules can be also catalyzed by thioredoxin and peroxyredoxin [101]. The enzymatic defenses are paralleled by non-enzymatic mechanisms that rely upon antioxidants such as vitamins A, E and C, ubiquinone, urate, lipoic acid, and glutathione [50].
The imbalance between formation and removal of ROS is termed oxidative stress and plays a major role in all cardiac diseases. In this respect, major attention has been focused on the relationship between oxidative stress and ischemia/repefusion injury [15, 22, 50, 73]. Nevertheless, an increased formation of ROS is suggested to be involved in heart failure [24, 50, 59, 89] as well as in atherosclerosis [6, 35, 40, 56, 66].
Besides altering every cell component, oxidative stress increases the occurrence of cell death. To this end, a relevant consequence of ROS accumulation is the increased susceptibility to opening of the mitochondrial permeability transition pore (PTP) [10] that is recognized as a major determinant of myocyte injury during post-ischemic reperfusion [4, 38]. PTP opening is especially sensitive to oxidative stress since it is favored by decreases in NADPH(H+)/NADP+ and -SH/-S–S ratios [32, 33]. Recent evidence suggests that PTP opening and ROS formation are linked in a vicious cycle. In fact, besides being a likely consequence of oxidative stress, PTP opening has been shown to increase mitochondrial ROS formation in cardiac myocytes [54]. In addition, the reduced susceptibility to PTP opening elicited by ischemic preconditioning appears to be associated with a decrease in mitochondrial oxidative stress [27]. On the other hand, it must be pointed out that ROS can also inhibit PTP opening. This appears to be the case with low doses of H2O2 [31] and singlet oxygen [90].
Although major emphasis has been put on the pathological consequences resulting from ROS-induced derangements of every macromolecule, ROS also contribute to several physiological processes [50]. The myriad of roles, at times contrasting, are likely to be related to the involvement of ROS, especially H2O2, in signaling pathways [52, 60]. Signaling modulation by ROS is mostly due to oxidation of cysteinyl residues resulting in covalent modifications of the affected proteins. These changes are potentially reversible due to the operation of several enzymes such as protein disulfide isomerase, glutaredoxin, and thioredoxin that catalyze the reduction of the oxidized cysteines [11, 60]. ROS modulate signaling pathways through three major modalities:
-
(i)
activation of protein kinases. Besides the well-established modulation of extracellular signal-regulated kinase (ERK), Jun N-terminal kinase (JNK), apoptosis signal-regulating kinase 1 (ASK-1) and protein kinase C (PKC) [5, 65, 60], recent elegant evidence has been reported that ROS also modulate the myocardial activity of cyclic AMP-dependent protein kinase (PKA) [17], cyclic GMP- dependent protein kinase (PKG) [20] and Ca2+/calmodulin-dependent protein kinase II (CaMKII) [43];
-
(ii)
H2O2 formation downstream of receptors activated by cytokines and growth factors, including insulin [12, 50, 52];
-
(iii)
inhibition of protein phosphatases, such as protein tyrosine phosphatase 1B and PTEN [46, 93, 60].
Regarding the protective mechanisms related to ischemic preconditioning, the role of ROS, reactive nitrogen species (RNS), and their downstream cellular targets is somewhat controversial (see [44] for extensive review). However, ROS/RNS appear to play a critical role in the signal transduction pathway leading to PKC activation.
Mitochondrial ROS formation
ROS are produced at various intracellular sites, yet it is generally accepted that in cardiac myocytes the largest amount of ROS are formed within mitochondria [5, 39, 53, 72, 97]. Besides impairment of energy metabolism and ionic homeostasis, formation of ROS [21, 80, 97] represents an additional process through which mitochondrial dysfunction accelerates, or even determines, the evolution of cell injury toward necrosis or apoptosis [9, 37, 58, 62, 76].
The large majority of oxygen delivered to mitochondria is fully reduced to water at the level of Complex IV. In this terminal step of the mitochondrial respiratory chain, oxygen is reduced sequentially, yet reaction intermediates remain bound to Complex IV, so that only the final product, i.e., water, is released. Nevertheless, electrons flowing through the respiratory chain can be donated to oxygen at other sites, but in these cases the reduction is not complete, resulting in the release of partially reduced forms, especially superoxide anion. In fact, O2 · – is formed at the level of Complex I and III and is then rapidly dismutated into H2O2 by SOD [49]. Besides the classical notion of the mitochondrial localization of MnSOD or SOD-2, also CuZnSOD (SOD-1), commonly referred to as the cytosolic isoform, is present in the intermembrane space of mitochondria [78].
The relevance of SOD as an antioxidant defense has been demonstrated by genetic approaches. Mice lacking SOD-2 display widespread organ damage associated with severe mitochondrial dysfunctions [68], and heterozygous deficiency of this enzyme impairs postischemic recovery of the heart [2]. On the other hand, SOD-2 overexpression elicits cardioprotection against the ischemia/reperfusion injury [25]. In addition, the absence of SOD-2 increases mtDNA damage and accelerates atherosclerosis in apoE knockout mice [77] providing support to the tight link between mitochondrial oxidative stress and atherogenesis [66].
The mitochondrial formation of ROS might be modulated by NO· [86, 91] as a consequence of the inhibition of cytochrome oxidase [8, 18, 28, 57, 97]. This reversible process can be transformed into irreversible alterations of respiratory chain when NO· formation is sustained. Indeed, NO· reacting with O2 · – generates peroxynitrite, which can produce the irreversible nitration of proteins [7]. Interestingly, a proteomic study showed that one-third of the proteins nitrated during inflammatory challenge are of mitochondrial origin [3].
It must be pointed out that ROS are also produced within mitochondria at sites other than the inner mitochondrial membrane [36, 39], such as monoamine oxidase (MAO) and p66Shc. These additional mitochondrial processes produce significant amounts of ROS. For instance, in brain mitochondria the highest rate of H2O2 formation from respiratory chain, as observed in the presence of the Complex III inhibitor antimycin A, is 48-fold lower than that originating from MAO activity [21]. Therefore, generation of ROS, especially H2O2, far from being just an unfortunate side effect of respiration, is catalyzed by specific enzymes, such as MAO and p66Shc. It is tempting to speculate that under physiological conditions the inner mitochondrial membrane could scavenge ROS produced at other mitochondrial or cellular sites. Then, the increase in ROS formation detected under pathological conditions might result, at least in part, from the loss of scavenging properties of the inner mitochondrial membrane due to its dysfunction.
The following paragraphs will focus on the relevant contribution of MAO and p66Shc to both mitochondrial ROS formation and cell injury.
p66Shc and cardiovascular pathology
Role of p66Shc in mitochondrial ROS generation
p66Shc is a vertebrate splice variant of p52Shc and p46Shc, two cytoplasmic adaptor proteins involved in the propagation of intracellular signals from activated tyrosine kinases to Ras [82]. The Shc acronym indicates proteins sharing a C-terminal SH2 domain adjacent to a collagen homology (CH) region [71]. p66Shc has the same modular structure as p52Shc/p46Shc (SH2-CH-PTB) and contains a unique N-terminal region (CH 2); however, it is not involved in Ras regulation, but rather functions in the intracellular pathway(s) that regulates ROS metabolism and apoptosis [70]. Within its additional CH region p66Shc becomes phosphorylated on Ser36 upon UV irradiation or treatment with oxidants linking p66Shc with redox signaling. Indeed, p66Shc has been shown to play a relevant role in a wide range of pathological alterations related to oxidative stress [30, 71]. A great deal of interest was attracted to p66Shc by the initial report that its deletion results in a 30% prolongation of lifespan [70]. Subsequently, studies carried out in a wide range of experimental models demonstrated quite clearly that ROS formation is reduced in cells lacking p66Shc, and that systemic and intracellular markers of oxidative stress are diminished in p66 Shc–/– mice [51, 79, 96].
A decrease in ROS levels might result from either a reduced formation and/or an increased removal. Initially, the reduced oxidative stress associated with p66Shc absence was attributed to a possible increase in ROS catabolism related to the Forkhead transcription factor FKHR-L1 that induces the expression of several antioxidant enzymes [75, 87]. Indeed, FKHR-L1 is phosphorylated and inactivated as a downstream target of Ser36-phosphorylated p66Shc, whereas FKHR-L1 activity is remarkably increased in cells devoid of p66Shc [75]. However, more recently, it became clear that ROS formation is directly enhanced by p66Shc that localizing to mitochondria catalyzes an alternative redox reaction [51]. In particular, electrochemical experiments demonstrated that the amino terminal portion of p66Shc contains a redox active sequence able to couple the reduction of molecular oxygen to H2O2 with the oxidation of cytochrome c [51]. This reaction is crucial for p66Shc apoptotic function that is abolished by mutating critical residues (E132, E133 and W134) in its redox center. Notably, these mutations impair the reaction of p66Shc with cytochrome c without affecting other properties (i.e., binding to and phosphorylation of the tyrosine kinase receptor and other substrates, including Grb2) [51].
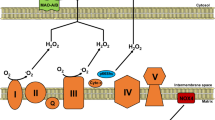
More recent work suggests that PKC β phosphorylation of p66Shc on Ser36 could cause its translocation to mitochondria [84]. Therefore, the increase in mitochondrial ROS formation caused by p66Shc appears to amplify PKC β signaling triggered by an initial oxidative stress (Fig. 1).

Mitochondrial sources of reactive oxygen species (ROS). Under physiological conditions (left panel), p66Shc is localized in the cytosol while ROS production by MAO is neutralized by antioxidant defenses. However, pathological stimuli (such as ischemia/reperfusion injury, right panel) may trigger signaling pathways that mediate p66Shc translocation into the mitochondria or release of catecholamines from endogenous stores rendering them available for catabolism by MAO. Both these events result in increased ROS production that may overwhelm cellular antioxidant defenses leading to contractile dysfunction, PTP opening, cell death, and heart failure. OMM outer mitochondrial membrane, IMM inner mitochondrial membrane, cyt c cytochrome c, MAO monoamine oxidase, PTP permeability transition pore, AO defenses antioxidant defenses
Since p66 Shc deletion is beneficial in a wide range of pathological conditions, its expression might appear as an unfortunate oversight of Mother Nature. Although the physiological roles of this protein are far from being conclusively elucidated, besides the possible contribution to heart development, recent studies relate p66Shc-induced ROS formation to both adipogenesis and immune response [12, 45].
p66Shc in cardiac and vascular pathologies
A large body of evidence supports the relevance of p66Shc in cardiovascular pathophysiology. In adult cardiac myocytes p66Shc is barely detectable. However, in dog hearts its expression was found to increase progressively during the transition toward decompensated hypertrophy induced by ventricular pacing [24]. Studies in dog heart could hardly elucidate the causative link between p66Shc-induced oxidative stress and myocardial injury that was established by investigating the deleterious effects of angiotensin II. In fact, mice devoid of p66Shc displayed a significant reduction in both the occurrence of apoptosis and the degree of hypertrophy induced by a sub-pressor dose of angiotensin II [55]. While supporting the bidirectional link between oxidative stress and p66Shc expression observed in other experimental models [83], these findings suggest the occurrence of a vicious cycle whereby an initial slight formation of ROS that might not affect myocardial function and viability is amplified by the increased expression of p66Shc leading to a ROS overload that causes cell death and contractile impairment. Interestingly, it has been proposed that this alteration could be part of the return to a fetal phenotype observed in diseased hearts [16]. Indeed, during heart development the expression of Shc proteins is increased [47]. This might be aimed at potentiating apoptosis associated with proliferative processes required for myocardial growth. Supporting this view, p66Shc knockout was found to be associated with myocardial hyperplasia, although contractile function is apparently similar to that of wild type littermates [55]. Oxidative stress and ROS formation appear to be relevant also for cells other than differentiated cardiomyocytes. In fact, the lack of p66Shc was shown to protect against diabetic cardiomyopathy by preventing the senescence of cardiac progenitor cells that hampers cardiac and vascular cell turnover [89].
Besides heart failure, oxidative stress related to p66Shc is likely to exacerbate ischemic injury. In this respect, the only available study was carried out in skeletal muscles using the protocol of hindlimb ischemia [100]. Upon reperfusion the extent of cell death was fivefold lower in p66 Shc–/– mice as compared to wild type littermates. In addition, the absence of p66Shc resulted in a significant decrease of oxidative stress in both ischemic muscles and isolated endothelial cells subjected to simulated ischemia. The beneficial action of p66Shc down-regulation against ischemia/reperfusion damage is also supported by the observation that protection elicited by preconditioning in neuronal SH-SY5Y cells is associated with a decreased expression of p66Shc that appears to be linked to preconditioning-induced increase in NO availability [1].
The link between p66Shc and vascular pathology was originally highlighted by studying the effects of hypercholesterolemia [74]. As compared to wild-type littermates hypercholesterolemic p66 Shc–/– mice displayed reduced levels of isoprostane and oxidized LDL, a decreased amount of foam cells, and a reduction in the extent of both apoptosis, and early atherogenic lesions. These initial findings prompted several other studies, especially in the field of diabetic vasculopathy. For instance, evidence was obtained that p66 Shc–/– mice are protected against glomerulopathy as shown by the maintenance of renal structure and function along with a marked reduction in oxidative stress [69]. Unfortunately, at present the translation of these experimental findings into the clinical practice is limited by the lack of drugs that might prevent the ROS forming activity of p66Shc.
Monoamine oxidase and oxidative stress
Biochemical features
Monoamine oxidase is a flavoenzyme located within the outer mitochondrial membrane, responsible for the oxidative deamination of neurotransmitters and dietary amines. It exists in two isoforms, MAO-A and B that differ for substrate specificity and inhibitor sensitivity [42]. In both isoforms FAD is covalently bound to a cysteine residue [41]. In peripheral tissues, MAO is involved in the oxidative catabolism of amines from the blood and in preventing the entry of dietary amines into the circulation. In the central and peripheral nervous system, intraneuronal MAO-A and B protect neurons from exogenous amines, terminate the actions of amine neurotransmitters, and regulate the contents of intracellular amine stores [99].
MAO-A catalyzes preferentially the oxidative deamination of norepinephrine (NE), and serotonin (5-HT) and is inhibited by low concentrations of clorgyline. In contrast, MAO-B has a higher affinity for phenylethylamine and benzylamine, and is inhibited by selegiline [99]. Both isoforms catalyze the deamination of dopamine, tyramine, octopamine, and tryptamine and are inhibited by pargyline. A wide range of MAO inhibitors is available today and these are proving to have a therapeutic value in several pathologies, including affective disorders, neurodegenerative diseases, stroke, and aging [88, 99]. They can be classified into three groups: (i) irreversible and non selective inhibitors, such as phenelzine and tranylcypromine; (ii) irreversible and selective inhibitors, such as selegiline for MAO-B and clorgyline for MAO-A; (iii) reversible and selective inhibitors, such as moclobemide for MAO-A and lazabemide for MAO-B.
MAO catalyze the following reaction:
Kinetic studies have shown that the binding of the amine group to the enzyme precedes the binding of oxygen [95]. In a first moment, the reduction of the cofactor FAD yields an aldehyde intermediate and ammonia, while in a second moment the oxidized form of the prosthetic group is restored with the concomitant production of hydrogen peroxide.
The aldehyde intermediates are rapidly metabolized to the corresponding acid by the action of aldehyde dehydrogenase (ALDH). A failure of this latter enzyme might increase the deleterious aspects of MAO activity generating potentially harmful aldehyde compounds that could exacerbate the damage produced by MAO-induced H2O2 formation. Indeed, the decrease in ALDH activity appears to be involved in both oxidative stress and nitrate tolerance [34, 98], whereas an increased activity of ALDH has been reported to result in a decreased injury of the ischemic heart [26].
Deletion of MAO-A and MAO-B genes has proven the important role of these enzymes in neurotransmitter metabolism and behavior. MAO-A –/– mice display elevated brain levels of serotonin, norepinephrine, and to a lesser extent, dopamine [23], whereas only 2-phenylethylamine is increased in MAO-B knockout mice [99]. Both MAO-A and B knockout mice show increased reactivity to stress, similar to that observed after administration of non-selective MAO inhibitors. However, these studies, and the deletion of both MAO-A and B in a rare form of human Norrie disease, indicate that MAO is not essential for survival [63]. Gene deletion has shown that MAO-A activity is important during development. A compulsive-aggressive behavior results from lack of MAO-A function in humans [19] and mice [92]. This effect, which might reflect the importance of serotonin during development, can be mimicked by the administration of MAO-A inhibitor clorgyline during the early postnatal period.
Monoamine oxidases distribution has been particularly studied in the brain, where MAO-A has been prevalently found in noradrenergic neurons whereas MAO-B has been detected in serotoninergic and histaminergic neurons and in glial cells [64]. In peripheral tissues, MAO-A has been found in placenta, liver, intestine, and thyroid gland, while platelets, liver, and kidney contain mainly MAO-B. Human cardiomyocytes contain both enzymes, although MAO-A is the predominant isoform [94].
MAO inhibition: from neurological disorders to cardiovascular diseases
The roles of MAO in terminating the actions of neurotransmitters and dietary amines in central and peripheral nervous system, as well as in extraneuronal tissues, have been extensively studied. In contrast, less attention has been given to the products of MAO activity. Monoamine catabolism results in the formation of aldehydes, ammonia and H2O2.
MAO is involved in numerous pathologies, in particular in neuronal and psychiatric disorders, as demonstrated by the beneficial effects elicited by MAO inhibitors. The therapeutic potential of MAO inhibition has been discovered in the early 1950s, when antituberculosis treatment with iproniazid was shown to improve the mood while reducing MAO activity [85, 88].
MAO-B appears to be involved in the loss of dopaminergic neurons that occurs in Parkinson’s disease, most likely due to the increased dopamine catabolism, resulting in elevated production of ROS responsible for the oxidative damage at the level of nigrostriatal neurons. Indeed, MAO-B inhibition has been proven to afford neuroprotection [99]. An increase in MAO-B activity in brain is also associated with diseases such as Alzheimer’s or Huntington’s disease. Depression, panic attacks and personality disorders are also associated with changes in dopaminergic, noradrenergic, and serotoninergic neurotransmission, which are regulated by both isoforms of MAO [99].
Besides their implication in neurodegenerative diseases, MAO isoforms, especially MAO-A, have been shown to play a relevant role in myocardial injury caused by post-ischemic reperfusion [13], and preliminary evidence suggests that MAO-A contributes also to the maladaptive evolution of myocardial hypertrophy into failure [61] (Fig. 1). In particular, MAO-A has been demonstrated to be an important source of ROS in receptor-independent apoptotic effects of serotonin in isolated cardiomyocytes and post ischemic myocardial injury [13, 14]. In fact, MAO-dependent ROS increase appears to be relevant for serotonin-induced myocyte hypertrophy in vitro [14]. In addition, MAO-A can promote cell apoptosis through ROS-dependent sphingosine kinase inhibition with accumulation of ceramide [81]. As far as vasculature is concerned, MAO-A mediated ROS production has been shown to induce mitogenic signaling in smooth muscle cells in a process that might involve the activation of metalloproteinase MMP-2, likely contributing to vascular wall remodeling [29]. Interestingly, MAO-A activity has been reported to increase with aging [67]. It is tempting to speculate that the consequent increase in H2O2 formation might contribute to aging-associated pathologies, such as congestive heart failure and vascular alterations.
Considering the important role of MAO as a source of H2O2 that has been described both in the brain as well as in the heart following I/R injury, MAO inhibition is likely to represent an important tool for both the study and the treatment of vascular pathologies that share oxidative stress as a common denominator.
At present the relationships between p66Shc and MAO are still to be clarified. They might represent two independent sources of mitochondrial ROS. On the other hand, while allosteric or covalent changes do not appear to be involved in MAO activity, H2O2 produced by these flavoenzymes could induce conformational changes in p66Shc, facilitating its phosphorylation and/or activation as a ROS producing enzyme. This latter possibility appears to be supported by preliminary evidence from our laboratory (unpublished data). In fact, while MAO inhibition and p66Shc deletion resulted in comparable degrees of protection against post-ischemic reperfusion, clorgyline did not elicit additional protection in p66 Shc–/– mice.
In conclusion, besides opening new possibilities for therapeutical interventions against cardiovascular disorders, studies on p66Shc and MAO clearly indicate that mitochondrial ROS formation is not only an accidental by-product of the respiratory chain, but also that most of the intracellular oxidative stress originate in mitochondria.
References
Andoh T, Lee SY, Chiueh CC (2000) Preconditioning regulation of bcl-2 and p66Shc by human NOS1 enhances tolerance to oxidative stress. FASEB J 14:2144–2146
Asimakis GK, Lick S, Patterson C (2002) Postischemic recovery of contractile function is impaired in SOD2(+/–) but not SOD1(+/–) mouse hearts. Circulation 105:981–986
Aulak KS, Miyagi M, Yan L, West KA, Massillon D, Crabb JW, Stuehr DJ (2001) Proteomic method identifies proteins nitrated in vivo during inflammatory challenge. Proc Natl Acad Sci USA 98:12056–12061
Baines CP (2009) The mitochondrial permeability transition pore and ischemia-reperfusion injury. Basic Res Cardiol (in press)
Balaban RS, Nemoto S, Finkel T (2005) Mitochondria, oxidants, and aging. Cell 120:483–495
Bauersachs J, Widder JD (2008) Endothelial dysfunction in heart failure. Pharmacol Rep 60:119–126
Beckman JS, Koppenol WH (1996) Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol 271:C1424–C1437
Beltran B, Mathur A, Duchen MR, Erusalimsky JD, Moncada S (2000) The effect of nitric oxide on cell respiration: a key to understanding its role in cell survival or death. Proc Natl Acad Sci USA 97:14602–14607
Bernardi P, Petronilli V, Di Lisa F, Forte M (2001) A mitochondrial perspective on cell death. Trends Biochem Sci 26:112–117
Bernardi P, Krauskopf A, Basso E, Petronilli V, Blalchy-Dyson E, Di Lisa F, Forte MA (2006) The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J 273:2077–2099
Berndt C, Lillig CH, Holmgren A (2007) Thiol-based mechanisms of the thioredoxin and glutaredoxin systems: implications for diseases in the cardiovascular system. Am J Physiol Heart Circ Physiol 292:H1227–H1236
Berniakovich I, Trinei M, Stendardo M, Migliaccio E, Minucci S, Bernardi P, Pelicci PG, Giorgio M (2008) p66Shc-Generated oxidative signal promotes fat accumulation. J Biol Chem 283:34283–34293
Bianchi P, Kunduzova O, Masini E, Cambon C, Bani D, Raimondi L, Seguelas MH, Nistri S, Colucci W, Leducq N, Parini A (2005) Oxidative stress by monoamine oxidase mediates receptor-independent cardiomyocyte apoptosis by serotonin and postischemic myocardial injury. Circulation 112:3297–3305
Bianchi P, Pimentel DR, Murphy MP, Colucci WS, Parini A (2005) A new hypertrophic mechanism of serotonin in cardiac myocytes: receptor-independent ROS generation. FASEB J 19:641–643
Bolli R, Marban E (1999) Molecular and cellular mechanisms of myocardial stunning. Physiol Rev 79:609–634
Booz GW (2005) Growing old, angiotensin II, cardiac hypertrophy, and death: making the connection with p66Shc. Hypertension 46:259–260
Brennan JP, Bardswell SC, Burgoyne JR, Fuller W, Schroder E, Wait R, Begum S, Kentish JC, Eaton P (2006) Oxidant-induced activation of type I protein kinase A is mediated by RI subunit interprotein disulfide bond formation. J Biol Chem 281:21827–21836
Brown GC, Cooper CE (1994) Nanomolar concentrations of nitric oxide reversibly inhibit synaptosomal respiration by competing with oxygen at cytochrome oxidase. FEBS Lett 356:295–298
Brunner HG, Nelen M, Breakefield XO, Ropers HH, van Oost BA (1993) Abnormal behavior associated with a point mutation in the structural gene for monoamine oxidase A. Science 262:578–580
Burgoyne JR, Madhani M, Cuello F, Charles RL, Brennan JP, Schroder E, Browning DD, Eaton P (2007) Cysteine redox sensor in PKGIa enables oxidant-induced activation. Science 317:1393–1397
Cadenas E, Davies KJ (2000) Mitochondrial free radical generation, oxidative stress, and aging. Free Radic Biol Med 29:222–230
Canton M, Neverova I, Menabò R, Van Eyk JE, Di Lisa F (2004) Evidence of myofibrillar protein oxidation induced by postischemic reperfusion in isolated rat hearts. Am J Physiol Heart Circ Physiol 286:H870–H877
Cases O, Seif I, Grimsby J, Gaspar P, Chen K, Pournin S, Muller U, Aguet M, Babinet C, Shih JC (1995) Aggressive behavior and altered amounts of brain serotonin and norepinephrine in mice lacking MAO-A. Science 268:1763–1766
Cesselli D, Jakoniuk I, Barlucchi L, Beltrami AP, Hintze TH, Nadal-Ginard B, Kajstura J, Leri A, Anversa P (2001) Oxidative stress-mediated cardiac cell death is a major determinant of ventricular dysfunction and failure in dog dilated cardiomyopathy. Circ Res 89:279–286
Chen Z, Siu B, Ho YS, Vincent R, Chua CC, Hamdy RC, Chua BH (1998) Overexpression of MnSOD protects against myocardial ischemia/reperfusion injury in transgenic mice. J Mol Cell Cardiol 30:2281–2289
Chen CH, Budas GR, Churchill EN, Disatnik MH, Hurley TD, Mochly-Rosen D (2008) Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science 321:1493–1495
Clarke SJ, Khaliulin I, Das M, Parker JE, Heesom KJ, Halestrap AP (2008) Inhibition of mitochondrial permeability transition pore opening by ischemic preconditioning is probably mediated by reduction of oxidative stress rather than mitochondrial protein phosphorylation. Circ Res 102:1082–1090
Cleeter MW, Cooper JM, rley-Usmar VM, Moncada S, Schapira AH (1994) Reversible inhibition of cytochrome c oxidase, the terminal enzyme of the mitochondrial respiratory chain, by nitric oxide. Implications for neurodegenerative diseases. FEBS Lett 345:50–54
Coatrieux C, Sanson M, Negre-Salvayre A, Parini A, Hannun Y, Itohara S, Salvayre R, Auge N (2007) MAO-A-induced mitogenic signaling is mediated by reactive oxygen species, MMP-2, and the sphingolipid pathway. Free Radic Biol Med 43:80–89
Cosentino F, Francia P, Camici GG, Pelicci PG, Luscher TF, Volpe M (2008) Final common molecular pathways of aging and cardiovascular disease: role of the p66Shc protein. Arterioscler Thromb Vasc Biol 28:622–628
Costa AD, Quinlan CL, Andrukhiv A, West IC, Jaburek M, Garlid KD (2006) The direct physiological effects of mitoK(ATP) opening on heart mitochondria. Am J Physiol Heart Circ Physiol 290:H406–H415
Costantini P, Chernyak BV, Petronilli V, Bernardi P (1995) Selective inhibition of the mitochondrial permeability transition pore at the oxidation-reduction sensitive dithiol by monobromobimane. FEBS Lett 362:239–242
Costantini P, Chernyak BV, Petronilli V, Bernardi P (1996) Modulation of the mitochondrial permeability transition pore by pyridine nucleotides and dithiol oxidation at two separate sites. J Biol Chem 271:6746–6751
Daiber A, Wenzel P, Oelze M, Schuhmacher S, Jansen T, Munzel T (2008) Mitochondrial aldehyde dehydrogenase (ALDH-2)-Maker of and marker for nitrate tolerance in response to nitroglycerin treatment. Chem Biol Interact (in press)
Davidson SM, Duchen MR (2007) Endothelial mitochondria: contributing to vascular function and disease. Circ Res 100:1128–1141
Di Lisa F, Bernardi P (2005) Mitochondrial function and myocardial aging. A critical analysis of the role of permeability transition. Cardiovasc Res 66:222–232
Di Lisa F, Menabò R, Canton M, Petronilli V (1998) The role of mitochondria in the salvage and the injury of the ischemic myocardium. Biochim Biophys Acta 1366:69–78
Di Lisa F, Canton M, Menabò R, Dodoni G, Bernardi P (2003) Mitochondria and reperfusion injury. The role of permeability transition. Basic Res Cardiol 98:235–241
Droge W (2002) Free radicals in the physiological control of cell function. Physiol Rev 82:47–95
Dworakowski R, Alom-Ruiz SP, Shah AM (2008) NADPH oxidase-derived reactive oxygen species in the regulation of endothelial phenotype. Pharmacol Rep 60:21–28
Edmondson DE, Binda C, Mattevi A (2004) The FAD binding sites of human monoamine oxidases A and B. Neurotoxicology 25:63–72
Edmondson DE, Mattevi A, Binda C, Li M, Hubalek F (2004) Structure and mechanism of monoamine oxidase. Curr Med Chem 11:1983–1993
Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, Oddis CV, Bartlett RK, Lowe JS, O’Donnell SE, ykin-Burns N, Zimmerman MC, Zimmerman K, Ham AJ, Weiss RM, Spitz DR, Shea MA, Colbran RJ, Mohler PJ, Anderson ME (2008) A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 133:462–474
Ferdinandy P, Schulz R, Baxter GF (2007) Interaction of cardiovascular risk factors with myocardial ischemia/reperfusion injury, preconditioning, and postconditioning. Pharmacol Rev 59:418–458
Finetti F, Pellegrini M, Ulivieri C, Savino MT, Paccagnini E, Ginanneschi C, Lanfrancone L, Pelicci PG, Baldari CT (2008) The proapoptotic and antimitogenic protein p66Shc acts as a negative regulator of lymphocyte activation and autoimmunity. Blood 111:5017–5027
Finkel T (2000) Redox-dependent signal transduction. FEBS Lett 476:52–54
Fiorina P, Corradi D, Pinelli S, Maestri R, Lagrasta C, Buscaglia M, Davalli A, Folli F, Astorri E (2004) Apoptotic/mytogenic pathways during human heart development. Int J Cardiol 96:409–417
Flohe L, Ursini F (2008) Peroxidase: a term of many meanings. Antioxid Redox Signal 10:1485–1490
Fridovich I (1995) Superoxide radical and superoxide dismutases. Annu Rev Biochem 64:97–112
Giordano FJ (2005) Oxygen, oxidative stress, hypoxia and heart failure. J Clin Invest 115:500–508
Giorgio M, Migliaccio E, Orsini F, Paolucci D, Moroni M, Contursi C, Pelliccia G, Luzi L, Minucci S, Marcaccio M, Pinton P, Rizzuto R, Bernardi P, Paolucci F, Pelicci PG (2005) Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell 122:221–233
Giorgio M, Trinei M, Migliaccio E, Pelicci PG (2007) Hydrogen peroxide: a metabolic by-product or a common mediator of ageing signals? Nat Rev Mol Cell Biol 8:722–728
Glab M, Lojek A, Wrzosek A, Dolowy K, Szewczyk A (2006) Endothelial mitochondria as a possible target for potassium channel modulators. Pharmacol Rep 58(Suppl):89–95
Gordon LI, Burke MA, Singh AT, Prachand S, Lieberman ED, Sun L, Naik TJ, Naga Prasad SV, Ardehali H (2008) Blockade of the erbB2 receptor induces cardiomyocyte death through mitochondrial- and reactive oxygen species-dependent pathways. J Biol Chem (in press)
Graiani G, Lagrasta C, Migliaccio E, Spillmann F, Meloni M, Madeddu P, Quaini F, Padura IM, Lanfrancone L, Pelicci P, Emanueli C (2005) Genetic deletion of the p66Shc adaptor protein protects from angiotensin II-induced myocardial damage. Hypertension 46:433–440
Gutierrez J, Ballinger SW, Darley-Usmar VM, Landar A (2006) Free radicals, mitochondria, and oxidized lipids: the emerging role in signal transduction in vascular cells. Circ Res 99:924–932
Haynes V, Elfering SL, Squires RJ, Traaseth N, Solien J, Ettl A, Giulivi C (2003) Mitochondrial nitric-oxide synthase: role in pathophysiology. IUBMB Life 55:599–603
Hengartner MO (2000) The biochemistry of apoptosis. Nature 407:770–776
Ide T, Tsutsui H, Kinugawa S, Suematsu N, Hayashidani S, Ichikawa K, Utsumi H, Machida Y, Egashira K, Takeshita A (2000) Direct evidence for increased hydroxyl radicals originating from superoxide in the failing myocardium. Circ Res 86:152–157
Janssen-Heininger YM, Mossman BT, Heintz NH, Forman HJ, Kalyanaraman B, Finkel T, Stamler JS, Rhee SG, van d, V (2008) Redox-based regulation of signal transduction: principles, pitfalls, and promises. Free Radic Biol Med 45:1–17
Kaludercic N, Feng N, Nagayama T, Bedja D, Carpi A, Vecoli C, Cormaci G, Gabrielson K, Kass D, Paolocci N, Di Lisa F (2007) Monoamine oxidase A is upregulated in cardiac hypertrophy and is a major determinant of the transition from compensation to failure. Circ Res 101(11):7 (abstract)
Lemasters JJ, Qian T, Bradham CA, Brenner DA, Cascio WE, Trost LC, Nishimura Y, Nieminen AL, Herman B (1999) Mitochondrial dysfunction in the pathogenesis of necrotic and apoptotic cell death. J Bioenerg Biomembr 31:305–319
Lenders JW, Eisenhofer G, Abeling NG, Berger W, Murphy DL, Konings CH, Wagemakers LM, Kopin IJ, Karoum F, van Gennip AH, Brunner HG (1996) Specific genetic deficiencies of the A and B isoenzymes of monoamine oxidase are characterized by distinct neurochemical and clinical phenotypes. J Clin Invest 97:1010–1019
Levitt P, Pintar JE, Breakefield XO (1982) Immunocytochemical demonstration of monoamine oxidase B in brain astrocytes and serotonergic neurons. Proc Natl Acad Sci USA 79:6385–6389
Liu Y, Yang XM, Iliodromitis EK, Kremastinos DT, Dost T, Cohen MV, Downey JM (2008) Redox signaling at reperfusion is required for protection from ischemic preconditioning but not from a direct PKC activator. Basic Res Cardiol 103:54–59
Madamanchi NR, Runge MS (2007) Mitochondrial dysfunction in atherosclerosis. Circ Res 100:460–473
Maurel A, Hernandez C, Kunduzova O, Bompart G, Cambon C, Parini A, Frances B (2003) Age-dependent increase in hydrogen peroxide production by cardiac monoamine oxidase A in rats. Am J Physiol Heart Circ Physiol 284:H1460–H1467
Melov S, Coskun PE, Wallace DC (1999) Mouse models of mitochondrial disease, oxidative stress, and senescence. Mutat Res 434:233–242
Menini S, Amadio L, Oddi G, Ricci C, Pesce C, Pugliese F, Giorgio M, Migliaccio E, Pelicci P, Iacobini C, Pugliese G (2006) Deletion of p66Shc longevity gene protects against experimental diabetic glomerulopathy by preventing diabetes-induced oxidative stress. Diabetes 55:1642–1650
Migliaccio E, Giorgio M, Mele S, Pelicci G, Reboldi P, Pandolfi PP, Lanfrancone L, Pelicci PG (1999) The p66Shc adaptor protein controls oxidative stress response and life span in mammals. Nature 402:309–313
Migliaccio E, Giorgio M, Pelicci PG (2006) Apoptosis and aging: role of p66Shc redox protein. Antioxid Redox Signal 8:600–608
Murphy MP (2009) How mitochondria produce reactive oxygen species. Biochem J (in press)
Murphy E, Steenbergen C (2008) Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev 88:581–609
Napoli C, Martin-Padura I, de Nigris F, Giorgio M, Mansueto G, Somma P, Condorelli M, Sica G, De Rosa G, Pelicci P (2003) Deletion of the p66Shc longevity gene reduces systemic and tissue oxidative stress, vascular cell apoptosis, and early atherogenesis in mice fed a high-fat diet. Proc Natl Acad Sci USA 100:2112–2116
Nemoto S, Finkel T (2002) Redox regulation of forkhead proteins through a p66Shc-dependent signaling pathway. Science 295:2450–2452
Newmeyer DD, Ferguson-Miller S (2003) Mitochondria: releasing power for life and unleashing the machineries of death. Cell 112:481–490
Ohashi M, Runge MS, Faraci FM, Heistad DD (2006) MnSOD deficiency increases endothelial dysfunction in ApoE-deficient mice. Arterioscler Thromb Vasc Biol 26:2331–2336
Okado-Matsumoto A, Fridovich I (2001) Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu, Zn-SOD in mitochondria. J Biol Chem 276:38388–38393
Orsini F, Migliaccio E, Moroni M, Contursi C, Raker VA, Piccini D, Martin-Padura I, Pelliccia G, Trinei M, Bono M, Puri C, Tacchetti C, Ferrini M, Mannucci R, Nicoletti I, Lanfrancone L, Giorgio M, Pelicci PG (2004) The life span determinant p66Shc localizes to mitochondria where it associates with mitochondrial heat shock protein 70 and regulates trans-membrane potential. J Biol Chem 279:25689–25695
Papa S, Skulachev VP (1997) Reactive oxygen species, mitochondria, apoptosis and aging. Mol Cell Biochem 174:305–319
Pchejetski D, Kunduzova O, Dayon A, Calise D, Seguelas MH, Leducq N, Seif I, Parini A, Cuvillier O (2007) Oxidative stress-dependent sphingosine kinase-1 inhibition mediates monoamine oxidase A-associated cardiac cell apoptosis. Circ Res 100:41–49
Pelicci G, Lanfrancone L, Grignani F, McGlade J, Cavallo F, Forni G, Nicoletti I, Grignani F, Pawson T, Pelicci PG (1992) A novel transforming protein (SHC) with an SH2 domain is implicated in mitogenic signal transduction. Cell 70:93–104
Pellegrini M, Pacini S, Baldari CT (2005) p66Shc: the apoptotic side of Shc proteins. Apoptosis 10:13–18
Pinton P, Rimessi A, Marchi S, Orsini F, Migliaccio E, Giorgio M, Contursi C, Minucci S, Mantovani F, Wieckowski MR, Del SG, Pelicci PG, Rizzuto R (2007) Protein kinase C beta and prolyl isomerase 1 regulate mitochondrial effects of the life-span determinant p66Shc. Science 315:659–663
Pletscher A (1991) The discovery of antidepressants: a winding path. Experientia 47:4–8
Poderoso JJ, Carreras MC, Lisdero C, Riobo N, Schopfer F, Boveris A (1996) Nitric oxide inhibits electron transfer and increases superoxide radical production in rat heart mitochondria and submitochondrial particles. Arch Biochem Biophys 328:85–92
Purdom S, Chen QM (2003) p66(Shc): at the crossroad of oxidative stress and the genetics of aging. Trends Mol Med 9:206–210
Riederer P, Lachenmayer L, Laux G (2004) Clinical applications of MAO-inhibitors. Curr Med Chem 11:2033–2043
Rota M, LeCapitaine N, Hosoda T, Boni A, De AA, Padin-Iruegas ME, Esposito G, Vitale S, Urbanek K, Casarsa C, Giorgio M, Luscher TF, Pelicci PG, Anversa P, Leri A, Kajstura J (2006) Diabetes promotes cardiac stem cell aging and heart failure, which are prevented by deletion of the p66Shc gene. Circ Res 99:42–52
Salet C, Moreno G, Ricchelli F, Bernardi P (1997) Singlet oxygen produced by photodynamic action causes inactivation of the mitochondrial permeability transition pore. J Biol Chem 272:21938–21943
Sarkela TM, Berthiaume J, Elfering S, Gybina AA, Giulivi C (2001) The modulation of oxygen radical production by nitric oxide in mitochondria. J Biol Chem 276:6945–6949
Shih JC (2004) Cloning, after cloning, knock-out mice, and physiological functions of MAO A and B. Neurotoxicology 25:21–30
Siddall HK, Warrell CE, Yellon DM, Mocanu MM (2008) Ischemia-reperfusion injury and cardioprotection: investigating PTEN, the phosphatase that negatively regulates PI3 K, using a congenital model of PTEN haploinsufficiency. Basic Res Cardiol 103:560–568
Sivasubramaniam SD, Finch CC, Rodriguez MJ, Mahy N, Billett EE (2003) A comparative study of the expression of monoamine oxidase-A and -B mRNA and protein in non-CNS human tissues. Cell Tissue Res 313:291–300
Tipton KF, Boyce S, O’Sullivan J, Davey GP, Healy J (2004) Monoamine oxidases: certainties and uncertainties. Curr Med Chem 11:1965–1982
Trinei M, Giorgio M, Cicalese A, Barozzi S, Ventura A, Migliaccio E, Milia E, Padura IM, Raker VA, Maccarana M, Petronilli V, Minucci S, Bernardi P, Lanfrancone L, Pelicci PG (2002) A p53–p66Shc signalling pathway controls intracellular redox status, levels of oxidation-damaged DNA and oxidative stress-induced apoptosis. Oncogene 21:3872–3878
Turrens JF (2003) Mitochondrial formation of reactive oxygen species. J Physiol 552:335–344
Wenzel P, Muller J, Zurmeyer S, Schuhmacher S, Schulz E, Oelze M, Pautz A, Kawamoto T, Wojnowski L, Kleinert H, Munzel T, Daiber A (2008) ALDH-2 deficiency increases cardiovascular oxidative stress–evidence for indirect antioxidative properties. Biochem Biophys Res Commun 367:137–143
Youdim MB, Edmondson D, Tipton KF (2006) The therapeutic potential of monoamine oxidase inhibitors. Nat Rev Neurosci 7:295–309
Zaccagnini G, Martelli F, Fasanaro P, Magenta A, Gaetano C, Di Carlo A, Biglioli P, Giorgio M, Martin-Padura I, Pelicci PG, Capogrossi MC (2004) p66ShcA modulates tissue response to hindlimb ischemia. Circulation 109:2917–2923
Zhao W, Fan GC, Zhang ZG, Bandyopadhyay A, Zhou X, Kranias EG (2008) Protection of peroxiredoxin II on oxidative stress-induced cardiomyocyte death and apoptosis. Basic Res Cardiol (in press)
Acknowledgments
This work has been supported by University of Padova (Post-Doctoral Fellowship to NK), MIUR and CNR.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Di Lisa, F., Kaludercic, N., Carpi, A. et al. Mitochondrial pathways for ROS formation and myocardial injury: the relevance of p66Shc and monoamine oxidase. Basic Res Cardiol 104, 131–139 (2009). https://doi.org/10.1007/s00395-009-0008-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00395-009-0008-4