
Abstract
Introduction
Atypical teratoid/rhabdoid tumor (AT/RT) is an aggressive embryonal tumor of the central nervous system with a generally dismal prognosis, especially in patients younger than 12 months.
Discussion
We here describe the unusual case of an infant with AT/RT with long-term survival despite low-cumulative dose chemotherapy after subtotal resection. Due to a poor neurological situation and an unfavorable oncological prognosis, therapy was halted after two partial surgical resections and four of the nine chemotherapy courses recommended by the European Rhabdoid Registry, without the patient receiving either radiotherapy or high-dose chemotherapy. The patient is alive without evidence of disease 52 months after diagnosis.
Conclusion
This case report highlights that independent prognostic factors are urgently needed for optimizing treatment stratification and preventing overtreatment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Atypical teratoid/rhabdoid tumors (AT/RTs) of the brain occur predominantly in infants and are central nervous system manifestations of the malignant rhabdoid tumor family. Common to this family are inactivating mutations of the gene encoding the SMARCB1 (formerly Ini1) protein, a tumor suppressor and core subunit of the SWI/SNF chromatin-remodeling complex. SMARCB1 inactivation results in the formation of malignant rhabdoid tumors, localized either in the kidney, soft tissues or the brain [1, 2].
AT/RT is an extremely aggressive malignancy with dismal prognosis. Data from 56 patients registered in the German HIT database revealed a 3-year overall and event-free survival of 22 and 13 %, respectively [3]. The 5-year overall survival in AT/RT patients from the Austrian Brain Tumor Registry was 39.5 % [4], while the 5-year overall and event-free survival of the 22 patients treated only at the Medical University of Vienna was 56.3 and 52.9 %, respectively. A provocative 100 % overall survival was reported for a single cohort of 9 patients treated according to the MUV-ATRT regimen at the Medical University of Vienna (median follow-up of 76 months) [5]. A similar approach employing high-dose chemotherapy with methotrexate induction has yielded poor results [6]. Treatment approaches are multimodal, combining surgical resection with systemic and intrathecal chemotherapy and adding focal or craniospinal irradiation. Recent data from the European Rhabdoid Registry (EU-RHAB) suggested that infants benefit from radiotherapy with tolerable acute side effects [7]. High-dose chemotherapy combined with autologous stem cell transplantation has been reported to improve survival [5, 8–11]. The efficacy of current treatment regimens, however, remains limited in infants and patients with metastatic disease.
Here, we report the unusual case of an infant with AT/RT with long-term survival despite young age at presentation (<12 months), large unresectable tumor, and low-cumulative dose chemotherapy.
Case presentation
An 11-month-old female presented with a 4-week history of lethargy and vomiting. She was admitted to a community hospital for acute loss of vigilance, spasticity of the lower extremity, and strabismus. Magnetic resonance imaging (MRI) demonstrated an inhomogeneously enhancing, 6 × 4.6 × 5 cm tumor filling the fourth ventricle, compressing the dorsal brainstem, and causing occlusive hydrocephalus (Fig. 1a, b). An incomplete tumor resection was performed. She remained in a comatose state during the 7 days following surgery and was transferred to our hospital with a Glasgow Coma Scale of 3. MRI showed a rostral herniation through the tentorial incisures. Tumor progression and local hemorrhage made a second surgery necessary, which partially resected the tumor and removed the hematoma. The subsequent postoperative course was complicated by tachycardia, arterial hypertension and hyperthermia. Neurologically, she demonstrated the clinical picture of a deep mesencephalic locked-in syndrome.

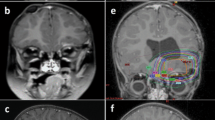
Cranial MRI showed an inhomogeneously enhancing, 6 × 4.6 × 5 cm tumor filling the fourth ventricle with compression of the dorsal brainstem and occlusive hydrocephalus (a, b). Control MRI demonstrated no evidence of tumor 41 months after diagnosis (c, d)
The highly cellular tumor consisted of small round blue cells exhibiting primitive features and elevated proliferation, as evidenced by the overabundance of mitotic cells (Fig. 2a, b). Tumor cells lacked nuclear SMARCB1 expression (Fig. 2c), indicating an AT/RT, which was confirmed by the national reference center. Sanger sequencing and multiplex ligation-dependent probe amplification from tumor tissue revealed a hemizygous nonsense mutation (c.978C>G) and reduced SMARCB1 gene dosage (Fig. 3a). No SMARCB1 germline mutation was detected (Fig. 3b).

Histology showed the highly cellular tumor consisting of small round blue cells (a) characterized by a high proliferation rate, as evidenced by immunohistochemical staining for Ki-67 (b). The nuclear expression of SMARCB1 was absent, as evidenced by immunohistochemical staining (c)

Salsa multiplex ligation-dependent probe amplification P258-C1 (MRC-Holland, Amsterdam, The Netherlands) was used to analyze the SMARCB1 region (chromosomes 22q11.21–22q12.2) in tumor cells (a) and leukocytes (b). Bars in light blue represent control tissue and bars in green patient’s tissue. Bars in dark blue indicate the gene dose difference between control and patient’s tissues. The gene dose of SMARCB1 including all exons (exons 1–9) and of the surrounding genes was reduced by 50 % in tumor cells (a). The SMARCB1 gene dose in patient’s leukocytes was not reduced (b)
Postoperative staging excluded metastases. The patient underwent four courses of intravenous chemotherapy (Table 1) with intraventricular methotrexate, according to recommendations of the European Rhabdoid Registry. MRI at completion of the fourth chemotherapy course documented a complete remission of the postoperative residual tumor; however, the clinical condition of mesencephalic locked-in syndrome continued. Chemotherapy was halted based on the poor neurological situation and unfavorable oncological prognosis, and the patient was transferred to a hospice.
Against all expectations, the neurological condition improved steadily and small advances in development were observed. MRI continued to confirm complete tumor remission 3 months after intravenous chemotherapy was stopped. Oral chemotherapy with cyclophosphamide, idarubicin and etoposide was initiated and carried out for 6 months. The patient is now 5 years and 3 months of age and alive without evidence of disease 4 years and 4 months after diagnosis. She crawls, can pull herself up to a standing position, and stands when supported. She says some single words.
Discussion
Case reports of long-term survival in patients treated for AT/RT have been published [12–14]; however, no case presented to date in the literature described a patient with the two most unfavorable prognostic factors, diagnosis in infancy and a large unresectable tumor, and with discontinuation of treatment at an early point. Here, we describe a patient diagnosed and treated for AT/RT and who remains alive 52 months after diagnosis without evidence of tumor recurrence. Therapy was halted after two partial surgical resections and four of the nine chemotherapy courses recommended by the European Rhabdoid Registry EU-RHAB, without the patient receiving either radiotherapy or high-dose chemotherapy.
The presented case raises several interesting questions concerning the appropriate treatment of AT/RT. According to international treatment protocols, the highly aggressive AT/RT has to be treated as intensively as possible. Gross total tumor resection followed by intravenous and intrathecal chemotherapy combined with radiotherapy is the recommended therapy for AT/RT. The therapeutic benefit of combined high-dose chemotherapy with autologous stem cell transplantation is still unclear. Some reports reasoned that selected patients might benefit from high-dose chemotherapy [5, 8–11]. However, the morphologic diagnosis of AT/RT includes tumors that can be controlled using less intensive treatment, as evidenced by our case. To date, there are no reliable parameters for identifying patients with a favorable outcome. Data from the German HIT database showed that a median age of less than 1.2 years correlated with a worse 3-year overall survival (37 ± 10 vs. 5 ± 5 %) and 3-year event-free survival (21 ± 8 vs. 5 ± 5 %) [3]. A multivariate analysis of French patients supported these results by identifying an age of less than 2 years as a negative prognostic factor for survival (hazard ratio = 3.1 ± 1.8; p = 0.01) [15]. Inactivating mutations of the tumor suppressor gene SMARCB1 are associated with fatal outcome [16, 17]. Despite the presence of common prognostic factors for unfavorable outcome, the patient presented here survived for a long period of time without undergoing complete chemotherapy, irradiation, or high-dose chemotherapy and remains in complete remission. The largest integrated molecular and clinicopathological analysis of AT/RT in children identified two subgroups with differential enrichment of genetic pathways. The two groups could be reliably differentiated by immunostaining of ASCL1, a regulator of the NOTCH signaling. Group 1 (ASCL1 positive) was correlated with a supratentorial location and a more favorable 5-year overall survival than ASCL1-negative group 2 (35 ± 22 vs. 20 ± 14 %; p = 0.033). Further, in patients who were treated with chemotherapy without irradiation, group 1 was associated with a superior 5-year overall survival (34 ± 27 vs. 9 ± 11 %; p = 0.001) [18]. There is a definite need for a prospective validation of these results and proposed risk stratification.
To conclude, the achievement of long-term survival in an infant patient with low-cumulative dose chemotherapy shows that among the patients with a highly aggressive AT/RT, there are selected patients with a less poor prognosis. For optimizing treatment stratification and preventing overtreatment, independent prognostic factors have to be identified.
References
Hollmann TJ, Hornick JL (2011) INI1-deficient tumors: diagnostic features and molecular genetics. Am J Surg Pathol 35(10):e47–e63
Wilson BG, Roberts CW (2011) SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer 11(7):481–492
von Hoff K, Hinkes B, Dannenmann-Stern E, von Bueren AO, Warmuth-Metz M, Soerensen N et al (2011) Frequency, risk-factors and survival of children with atypical teratoid rhabdoid tumors (AT/RT) of the CNS diagnosed between 1988 and 2004, and registered to the German HIT database. Pediatr Blood Cancer 57(6):978–985
Woehrer A, Slavc I, Waldhoer T, Heinzl H, Zielonke N, Czech T et al (2010) Incidence of atypical teratoid/rhabdoid tumors in children: a population-based study by the Austrian Brain Tumor Registry, 1996-2006. Cancer 116(24):5725–5732
Slavc I, Chocholous M, Leiss U, Haberler C, Peyrl A, Azizi AA et al (2014) Atypical teratoid rhabdoid tumor: improved long-term survival with an intensive multimodal therapy and delayed radiotherapy. The Medical University of Vienna Experience 1992–2012. Cancer Med 3(1):91–100
Zaky W, Dhall G, Ji L, Haley K, Allen J, Atlas M et al (2014) Intensive induction chemotherapy followed by myeloablative chemotherapy with autologous hematopoietic progenitor cell rescue for young children newly-diagnosed with central nervous system atypical teratoid/rhabdoid tumors: the Head Start III experience. Pediatr Blood Cancer 61(1):95–101
Seeringer A, Bartelheim K, Kerl K, Hasselblatt M, Leuschner I, Rutkowski S et al (2014) Feasibility of intensive multimodal therapy in infants affected by rhabdoid tumors—experience of the EU-RHAB registry. Klin Padiatr 226(3):143–148
Benesch M, Bartelheim K, Fleischhack G, Gruhn B, Schlegel PG, Witt O et al (2014) High-dose chemotherapy (HDCT) with auto-SCT in children with atypical teratoid/rhabdoid tumors (AT/RT): a report from the European Rhabdoid Registry (EU-RHAB). Bone Marrow Transplant 49(3):370–375
Chi SN, Zimmerman MA, Yao X, Cohen KJ, Burger P, Biegel JA et al (2009) Intensive multimodality treatment for children with newly diagnosed CNS atypical teratoid rhabdoid tumor. J Clin Oncol 27(3):385–389
Finkelstein-Shechter T, Gassas A, Mabbott D, Huang A, Bartels U, Tabori U et al (2010) Atypical teratoid or rhabdoid tumors: improved outcome with high-dose chemotherapy. J Pediatr Hematol Oncol 32(5):e182–e186
Park ES, Sung KW, Baek HJ, Park KD, Park HJ, Won SC et al (2012) Tandem high-dose chemotherapy and autologous stem cell transplantation in young children with atypical teratoid/rhabdoid tumor of the central nervous system. J Korean Med Sci 27(2):135–140
Bouvier C, De Paula AM, Fernandez C, Quilichini B, Scavarda D, Gentet JC et al (2008) Atypical teratoid/rhabdoid tumour: 7-year event-free survival with gross total resection and radiotherapy in a 7-year-old boy. Childs Nerv Syst 24(1):143–147
Gidwani P, Levy A, Goodrich J, Weidenheim K, Kolb EA (2008) Successful outcome with tandem myeloablative chemotherapy and autologous peripheral blood stem cell transplants in a patient with atypical teratoid/rhabdoid tumor of the central nervous system. J Neurooncol 88(2):211–215
Modena P, Sardi I, Brenca M, Giunti L, Buccoliero AM, Pollo B et al (2013) Case report: long-term survival of an infant syndromic patient affected by atypical teratoid-rhabdoid tumor. BMC Cancer 13:100
Dufour C, Beaugrand A, Le Deley MC, Bourdeaut F, Andre N, Leblond P et al (2012) Clinicopathologic prognostic factors in childhood atypical teratoid and rhabdoid tumor of the central nervous system: a multicenter study. Cancer 118(15):3812–3821
Bourdeaut F, Lequin D, Brugieres L, Reynaud S, Dufour C, Doz F et al (2011) Frequent hSNF5/INI1 germline mutations in patients with rhabdoid tumor. Clin Cancer Res 17(1):31–38
Sevenet N, Sheridan E, Amram D, Schneider P, Handgretinger R, Delattre O (1999) Constitutional mutations of the hSNF5/INI1 gene predispose to a variety of cancers. Am J Hum Genet 65(5):1342–1348
Torchia J, Picard D, Lafay-Cousin L, Hawkins CE, Kim SK, Letourneau L et al (2015) Molecular subgroups of atypical teratoid rhabdoid tumours in children: an integrated genomic and clinicopathological analysis. Lancet Oncol 16(5):569–582
Acknowledgments
The authors thank K. Astrahantseff for proofreading the manuscript and V. Klepatschow for figure preparation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Arnhold, V., Oyen, F., Schneppenheim, R. et al. Long-term survival of an infant with an atypical teratoid/rhabdoid tumor following subtotal resection and low-cumulative dose chemotherapy: a case report. Childs Nerv Syst 32, 1157–1161 (2016). https://doi.org/10.1007/s00381-015-2999-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-015-2999-5