
Abstract
Atypical teratoid rhabdoid tumors (ATRT) are highly malignant tumors of the central nervous system with a peak incidence in children less than 3 years of age. Despite multimodal therapy including surgery, radiation and chemotherapy, the prognosis remains dismal. No specific treatment guidelines are defined for ATRTs but a gross total resection and radiation therapy (RT) appear to improve overall outcome. In children less than 3 years of age, the prognosis is dismal due in part to the reluctance to utilize RT given its severe neurological sequelae. To avoid RT in this age group, intensification of chemotherapy has been tried and has shown to improve outcome. Myeloablative chemotherapy followed by autologous stem cell re-infusion has been used as a modality to intensify therapy but there are no reports of use of tandem myeloablative regimens and autologous stem cell re-infusions for treatment of ATRT. We herein report the case of a 4-month-old boy with ATRT with partial resection of his tumor who achieved complete remission using tandem high-dose therapy followed by autologous peripheral blood stem cell re-infusions despite having biopsy proven disease at the time of starting the tandem regimens. This was achieved without the use of RT as a treatment modality.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Central nervous system (CNS) tumors are the second most common pediatric tumors with an overall survival of 70% [1]. Rhabdoid tumors of the CNS, also known as atypical teratoid rhabdoid tumors (ATRT), comprise of approximately 2–3% of pediatric brain tumors with a peak incidence in children less than 3 years of age [2]. They are associated with characteristic genetic abnormalities, which include either monosomy 22 or deletion involving the hSNF5/INI1 gene located on 22q11.2 thus leading to the absence of INI1 protein which is a hallmark of ATRT [3, 4]. ATRTs are highly malignant tumors with a median survival of less than 1 year after diagnosis. Due to the small numbers and relatively recent recognition of these tumors as a distinct entity, there are no standards of treatment defined yet. Two prognostic factors that affect overall outcome are: (1) the extent of tumor resection and (2) early radiation therapy (RT) [2]. Given the young age of many patients with ATRT, the short and long term effects of RT are prohibitively severe. Therefore, chemotherapy is the treatment of choice after surgery for patients less than 3 years old. Intensification of chemotherapy appears to improve outcome [5–7]. The use of myeloablative chemotherapy followed by autologous stem cell rescue as a modality to intensify therapy has been reported but there are no reports of the use of tandem transplants for treatment of ATRTs. In this report, we present a young patient diagnosed with ATRT who achieved long-term disease free survival, despite an incomplete resection of his tumor and without using RT, by intensive chemotherapy followed by tandem high-dose myeloablative chemotherapy regimens and autologous stem cell rescues.
Clinical report
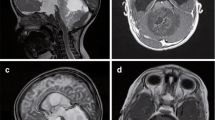
A 4-month-old boy presented to the emergency room with altered mental status and right gaze preference. A computed tomography scan of the head showed a 6 cm lobulated lesion in the right cerebral hemisphere with mass effect, subfalcine herniation, transtentorial herniation & compression of left lateral ventricle. A subsequent brain magnetic resonance imaging (MRI) confirmed the right parieto-temporal cerebral tumor with additional mass effect on the brainstem (Fig. 1a). MRI of the spine did not show gross tumor and spinal fluid analysis was negative for malignant cells. He underwent an emergent craniotomy for partial resection of the tumor. Neuropathologic study showed a malignant neoplasm composed of large cells with eccentric eosinophilic cytoplasm, large pleomorphic nuclei and prominent nucleoli (Fig. 2a). Ill-defined cytoplasmic inclusions were positive for cytokeratin and vimentin (Fig. 2b). Immunohistochemical staining for INI1 protein was negative, confirming the diagnosis of ATRT. Cytogenetic analysis of the tumor cells revealed deletion of hSNF5/INI1 gene located on 22q11.2. He received five cycles of chemotherapy including cisplatin, vincristine, cyclophosphamide, etoposide and high-dose methotrexate as per Headstart II protocol [5] (Table 1), which he tolerated well. His tumor regressed with each course of chemotherapy. Peripheral blood stem cells were harvested after the second cycle of chemotherapy in anticipation of consolidating therapy with myeloablative regimen and autologous stem cell rescue and 23 million CD34+ cells/kilogram (kg) body weight were obtained. Subsequently, he underwent a second surgical exploration after the fifth cycle in an attempt to achieve a gross total resection but due to the close proximity of the tumor to the midbrain, it was not accomplished. Repeat pathological analyses of the tissue revealed viable tumor consistent with ATRT. He received two more courses of the same chemotherapy and his tumor continued to shrink on MRI evaluation (Fig. 1b). He was then evaluated for RT but due to his age (12 months), tumor location and the size of the original tumor volume, expected toxicity, even with intensity-modulated radiation therapy, was deemed unacceptable. Thus, he received high-dose myeloablative chemotherapy including carboplatin, etoposide and thiotepa as per the Headstart II protocol (Table 1) with autologous stem cell rescue [5]. He received 10 million CD34+ cells/kg body weight. His neutrophil count was greater than 1,000/μl without cytokine support by day 14 post-transplant and his platelet count was greater than 75,000/μl without transfusion support by day 30 post-transplant. A subsequent brain MRI showed a persistent enhancing nodule with a mild decrease in tumor size (Fig. 1c). Given his continued response to chemotherapy and excellent transplant course, he underwent a second course of high-dose chemotherapy at 14 months of age with busulfan, melphalan, and thiotepa, (Table 1) followed by autologous stem cell reinfusion of 8 million CD34+ cells/kg. His neutrophil count recovered by 20 days post-transplant and the platelet count recovered by 50 days post-transplant. No significant infectious complications or organ dysfunction occurred with either course of high-dose chemotherapy. A subsequent MRI done 2 months post-transplant showed no evidence of disease (Fig. 1d). Since then, he has been serially followed at our institution and has remained disease free for 2 years post therapy. He has received intensive physical, occupational and speech therapy and currently is able to walk, say a few words and follows simple instructions. His current chronological age is 3 years and developmental age is 2 years with signs of progressive catch-up growth.

MRI brain with gadolinium: (a) At diagnosis showing a large enhancing right parietal tumor (arrow) with significant mass effect; (b) after five courses of chemotherapy showing residual enhancing tumor; (c) before second course of myeloablative chemotherapy and stem cell transplant showing residual enhancing nodule in the tumor bed; (d) 2 months post therapy showing complete remission

(a) A section stained with hematoxylin and eosin shows a hypercellular tumor composed of cells wiith round to oval vesicular nuclei with occasional prominent nucleoli. A few cells have ill-defined pink inclusions in their cytoplasm (arrows). (b) immunostaining using the CAM5.2 cytokeratin antibody shows occasional cells with well-defined brown inclusions. Original magnification ×400
Discussion
We here report the case of a 4-month-old boy with a CNS ATRT who achieved complete remission of his disease following tandem myeloablative regimens and autologous stem cell re-infusions. This positive outcome was significant for the following reasons: (1) partial resection of the primary tumor, (2) avoidance of RT and (3) presence of viable disease before myeloablative therapy and radiographic evidence of residual disease in between the two myeloablative regimens.
Rhabdoid tumors are highly malignant neoplasms that occur primarily in the CNS and the kidneys. Other sites include liver, pelvis, heart and soft tissues. CNS tumors also referred to as ATRTs, can be both supratentorial and infratentorial in location [3]. They present in children less than 3 years of age and carry a dismal prognosis. They are not highly chemosensitive and most patients succumb to the disease within 1 year of diagnosis despite multimodal therapy. Although there are no standards of treatment defined for ATRTs, retrospective review of the literature suggests that the ability to achieve a complete resection is an important prognostic factor [6, 8]. Patients who achieve a complete resection and/or patients who receive RT appear to have improved overall survival. However, extensive radiation therapy or aggressive surgical resections may have a devastating effect in young patients. In general, rhabdoid tumors are considered to be relatively chemo-resistant, but there have been multiple reports of using intensive IRS-III type chemotherapy with some improvement in overall survival [6–8]. Thus intensifying chemotherapy including high-dose alkylator based therapy seems to be a rational therapeutic approach at this point in time.
High-dose myeloablative chemotherapy with autologous stem cell rescue has been used as a therapeutic approach and our review of the literature suggests that the majority of the long-term survivors either had a gross total resection or RT as accompanying therapies (Table 2). We found three reported cases of prolonged disease free survival in patients with ATRT who had a subtotal resection followed by chemotherapy, myeloablative therapy, and autologous stem cell transplant without the use of RT (Table 2). While high-dose myeloablative chemotherapy is not novel for ATRT, our report is novel because we are reporting the first result with tandem high-dose chemotherapy and that high-dose chemotherapy was effective in treating residual disease. The use of tandem high-dose therapy and autologous stem cell rescues has not been reported for treatment of CNS ATRTs. Childrens Cancer Group (CCG) conducted a pilot study (CCG 99703) from1998 to 2004 evaluating the feasibility of triple high-dose chemotherapy including carboplatin and escalating doses of thiotepa followed by autologous stem cell rescues in infants with malignant brain tumors, including some patients with ATRT. The results of this study have not yet been reported. Katzenstein et al. described safe administration of tandem myeloablative chemotherapy including cyclophosphamide, etoposide, carboplatin and melphalan, and autologous stem cell rescue in a 21-month-old patient with metastatic rhabdoid tumor of the liver. The patient had progressive disease and eventually died 9 months after diagnosis [14]. We chose busulfan, melphalan, and thiotepa for the second course of myeloablative chemotherapy instead of the same agents as the first course to potentially overcome resistance to the previously used agents. This combination has been previously used in conjunction with autologous stem cell rescue in patients with medulloblastomas and supratentorial primitive neuroectodermal tumors [15].
In this patient with evidence of residual but chemoresponsive disease, the strategy of tandem high-dose chemotherapy aided in rendering the patient disease-free. To our knowledge this is the first report of successful administration of tandem high-dose therapy with autologous stem cell rescues in a patient with local but partially resected tumor without the use of RT. The role of high-dose myeloablative chemotherapy and autologous stem cell rescue remains to be determined but in a chemo-sensitive tumor, this might be a strategy used to intensify therapy. Tandem high-dose chemotherapy and stem cell rescues might further help in achieving this goal and should be explored further.
References
Jemal A, Clegg LX, Ward E et al (2004) Annual report to the nation on the status of cancer, 1975–2001, with a special feature regarding survival. Cancer 101:3–27
Squire SE, Chan MD, Marcus KJ (2007) Atypical teratoid/rhabdoid tumor: the controversy behind radiation therapy. J Neurooncol 81(1):97–111
Reddy AT (2005) Atypical teratoid/rhabdoid tumors of the central nervous system. J Neurooncol 75(3):309–313
Judkins AR, Mauger J, Bielgel JA et al (2004) Immunohistochemical analysis of hSNF5/INI1 in pediatric CNS neoplasms. Am J Surg Pathol 28(5):644–650
Chi SN, Gardener SL, Levy AS et al (2004) Feasibility and response to induction chemotherapy intensified with high dose methotrexate for young children with newly diagnosed high-risk disseminated medulloblastoma. J Clin Oncol 22(24):4881–4887
Zimmerman MA, Goumnerova LC, Proctor M et al (2005) Continuous remission of newly diagnosed and relapsed central nervous system atypical teratoid/rhabdoid tumor. J Neurooncol 72(1):77–84
Olson TA, Bayar E, Kosnik E et al (1995) Successful treatment of disseminated central nervous system malignant rhabdoid tumor. J Pediatr Hematol Oncol 17(1):71–75
Tekautz TM, Fuller CE, Blaney S et al (2005) Atypical teratoid/rhabdoid tumors (ATRT): improved survival in children 3 years of age and older with radiation therapy and high-dose alkylator based chemotherapy. J Clin Oncol 23(7):1491–1499
Hilden JM, Watterson J, Longee DC et al (1998) Central nervous system atypical teratoid/rhabdoid tumor: response to intensive therapy and review of the literature. J Neuro Oncol 40(3):265–275
Beschorner R, Mittlebronn M, Koerbel A et al (2006) Atypical teratoid-rhabdoid tumor spreading along the trigeminal nerve. Pediatr Neurosurg 42(4):258–263
Fujita M, Sato M, Nakamura M et al (2005) Multicentric atypical teratoid/rhabdoid tumors occurring in the eye and fourth ventricle of an infant. J Neurosurg 102(3 suppl):299–302
Ronghe MD, Moss TH, Lowis SP (2004) Treatment of CNS malignant rhabdoid tumors. Pediatr Blood Cancer 42(3):254–260
Hilden JM, Meerbaum S, Burger P et al (2004) Central nervous system atypical teratoid/rhabdoid tumor: results of therapy in children enrolled in a registry. J Clin Oncol 22(14):2877–2884
Katzenstein HM, Kletzel M, Reynolds M et al (2003) Metastatic malignant rhabdoid tumor of the liver treated with tandem high-dose therapy and autologous peripheral blood stem cell rescue. Med Pediatr Oncol 40(3):199–201
Perez-Martinez A, Lassaletta A, Gonzalez-Vincent M et al (2005) High-dose chemotherapy with autologous stem cell rescue for children with high risk and recurrent medulloblastoma and supratentorial primitive neuroectodermal tumors. J Neurooncol 71(1):33–38
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gidwani, P., Levy, A., Goodrich, J. et al. Successful outcome with tandem myeloablative chemotherapy and autologous peripheral blood stem cell transplants in a patient with atypical teratoid/rhabdoid tumor of the central nervous system. J Neurooncol 88, 211–215 (2008). https://doi.org/10.1007/s11060-008-9553-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-008-9553-1