
Abstract
Purposes
The purpose of this study was to retrospectively study embryonal tumors with multilayered rosettes (ETMR), a rare new entity that gathers ETAN-TR (embryonal tumor with abundant neuropil and true rosettes), ependymoblastomas, and medulloepitheliomas, in order to improve their descriptions and try to better define therapeutic modalities.
Methods
Patients with ETMR, ETAN-TR, ependymoblastoma, and medulloepithelioma treated in SFCE centres (Société Française de lutte contre les Cancers et les leucémies de l'Enfant et de l'adolescent) since 2000 were collected. Data were retrieved from clinical charts.
Results
Thirty-eight patients were included in the analysis. Seventeen had an ETAN-TR, 13 had a medulloepithelioma, and 8 had an ETMR. No ependymoblastoma was included. The median age at diagnosis was 31 months (range, 2.8–141 months). The predominant tumor location was supratentorial (66 %); 18.4 % patients had metastatic lesion.
LIN28A expression was positive in 11/11 patients. Amplification of the locus 19q13.42 was positive in 10/12 patients. Thirty patients were treated according to the primitive neuroectodermal tumors of high risk (PNET-HR) protocol. The median time of follow-up was 0.9 years (range 0.1 to 15.3 years). The 1-year event-free survival (EFS) and overall survival (OS) were, respectively, 36 % CI 95 % (23–55) and 45 % CI 95 % (31–64). On multivariate analysis, complete surgical resection, radiotherapy, and high-dose chemotherapy were associated with a better overall survival with a relative risk of, respectively, 7.9 CI 95 % (2.6–23.5) p < 0.0002, 41.8 CI 95 % (9.4–186) p < 0.0001, and 3.5 CI 95 % (1.3–9.5) p = 0.012.
Conclusion
Prognosis of ETMR remains dismal despite multimodal therapy. LIN28A immunostaining and 19q13.42 amplification should be systematically done to secure the diagnosis. Complete surgical resection, radiotherapy, and high-dose chemotherapy are associated with better outcome.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Embryonal tumors with neuropil and true rosettes abundant (ETAN-TR), ependymoblastomas, and medulloepitheliomas are three distinct pathologies which actually represent different manifestations of the same histological and biological entity [1, 2]. The term embryonal tumors with multilayered rosettes (ETMR) was proposed in 2010 as a unifying entity by Paulus and Kleihues [3] as the histological central element found in these tumors was the presence of multilayered rosettes. Moreover, ETMR seem to have a specific molecular signature with a positivity of LIN28A on immuno-histochemistry and amplification of the locus 19q13.42 in FISH analysis [4, 5]. ETMR belongs to the group of the primitive neuroectodermal tumors of the central nervous system (PNET) according to the World Health Organization (WHO) classification [6].
ETMR are quite rare entities; their prevalence is not accurately estimated. They are likely under-diagnosed with only a few cases reported in the literature since 2000 [2, 3, 7, 8]. They occur mostly in children aged less than 2 years old. In the literature, the localization of the majority of the primitive tumor is supratentorial with signs of increased intracranial pressure. Treatment of ETMR usually starts with maximal surgical resection. The role of adjuvant treatment with either chemotherapy and/or radiotherapy has not yet been well established for this disease.
To improve our knowledge about ETMR, we collected cases treated in SFCE centers (Société Française des Cancers et leucémies de l’Enfant et de l’adolescent) from January 2000 to May 2014 in order to retrospectively study clinical, radiological, biological, and prognostic descriptions to improve their descriptions and try to better define therapeutic modalities.
Patients and methods
Study design, patients, data collection
Patients aged less than 18 years with newly diagnosed ETMR who were treated in France between January 2000 and 1 May 2014 were identified retrospectively by consulting local oncologists’ and pathologists’ databases in SFCE centers using the following diagnosis: ETMR, ETAN-TR, ependymoblastoma, and medulloepithelioma. A standardized data sheet was sent to pediatric oncologists who were members of the SFCE to collect clinical data. The questionnaire included information about sex, age at time of presentation, duration of the symptoms at time of presentation, location of tumor, extent of resection (complete resection, partial resection, biopsy), pathological biological and molecular diagnosis, adjuvant therapies, and follow-up. At diagnosis, stage of disease was evaluated by initial cranial and spinal MRI and CSF cytology. The extent of initial resection was assessed by MRI or computed tomography performed within 72 h after surgery. Metastatic disease was assessed using the Chang staging system. Tumors were subjected to a central histopathological review by members of the national pediatric brain tumors board (GENOP). When it was feasible, immuno-histochemistry analysis applying a LIN28A polyclonal antibody and array-based comparative genomic hybridization (CGHarray) was performed. The data were coded to ensure confidentiality. This study has been approved by the National Ethical Committee (CCTIRS).
Statistical analysis:
Relapses and deaths were considered as events. Event-free survival (EFS), overall survival (OS), and univariate analysis were estimated by the Kaplan-Meier method, and differences between groups were assessed by the log-rank test. Survival estimates referred to 1 and 3 years from diagnosis and the related 95 % confidence intervals (95 % CI) were calculated. Cox model was used to calculate the multivariate analysis by a stepwise selection. Descriptive statistics were reported as absolute frequencies and percentages for qualitative data, while median and range were used for continuous variables. When performing univariate analysis, those variables were used: age ≤4 years or >4 years, age <4 years or 4 to 5 years or 5 to 8 years or 8 to 11 years, histological diagnosis (ETMR, ETAN-TR, medulloepithelioma, ependymoblastoma), surgery, chemotherapy, radiotherapy, high-dose chemotherapy, and primitive neuroectodermal tumors of high risk (PNET-HR) (yes or no). Positive variables identified with univariate analysis were used in the multivaried analysis. All statistical tests were two sided and a “p” value <0.05 was considered significant. Statistical analysis was performed using R version 3.0.2.
Results:
Patients’ characteristics
Forty-two patients with an ETMR were identified in the SFCE centers, but 4 patients were excluded because data could not be retrieved or the diagnosis was uncertain. Thirty-eight patients were therefore analyzed. The median age at diagnosis was 31.1 months (range 2.8–141 months) and the sex ratio was 0.4 (male/female). Main clinical signs were a sided weakness (34 %) and increased intracranial pressure (53 %). Confusion, seizures, and cerebellar syndrome occurred in 26, 21, and 21 % of children, respectively. Torticollis occurred in 8 % of cases and visual impairment was found in 11 % of patients.
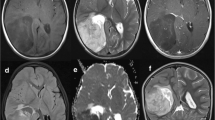
The predominant tumor location was supratentorial (66 %). Tumors were infratentorial in the others (26 %). One patient had a medullar lesion. Eighteen percent of patients had metastatic disease at initial presentation. In all cases, the metastasis was evidenced by MRI while metastatic cells were never found in the cerebrospinal fluid at the time of diagnosis.
Histopathological characteristics
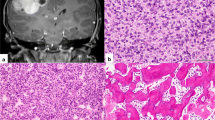
A histopathological review was performed in 100 % of the 38 patients. Noteworthy, 6 children had an uncorrect diagnosis (1 ependymoma, 5 PNET). Seventeen patients had ETAN-TR, 13 had medulloepithelioma, 8 had ETMR, and 0 had ependymoblastoma (Table 1). LIN28A expression was searched for 11 patients and was positive in 100 % of the cases. The amplification of the locus 19q13.42, searched for in 12 children, was present in 83 % of the patients. The gain of the chromosome 2 and other chromosome gains were searched for in 11 patients and were positive, respectively, in 27 and 54 % of the cases. Data are summarized in Table 2.
Treatment strategy and outcome
All children but 8 were treated according to the PNET-HR protocol. The strategy relies on the combination of surgery, conventional chemotherapy (2 courses of etoposide-carboplatin), followed by sequential high-dose chemotherapy (melphalan/cisplatinum/melphalan/cisplatinum/thiotepa) each course being followed by peripheral blood stem cell transplantation and reduced dose radiation therapy [9]. Details of all the 38 patients are provided in Table 3.
All children received adjuvant chemotherapy, after a complete resection for 12 patients, or a partial resection/biopsy for 18 patients. Out of the 30 patients who received etoposide/carboplatinum, 4 experienced disease progression (1 who had a complete resection and 3 who had a partial resection). In patients who underwent complete resection, 6 children received the complete PNET-HR protocol and 3 are alive with a follow-up of, respectively, 12, 8.6, and 3.6 years. Out of the patients who had a post-surgery residual disease (18 patients), only two completed the whole protocol PNET-HR. The remaining patients presented tumor progression and all patients but 2 died.
Among the patients who were not treated according the the PNET-HR protocol, one child with a localized ETMR underwent a complete removal of the tumor after 2 surgeries. CSF became positive after the second surgery. She was treated with temozolomide-irinotecan chemotherapy, followed by craniospinal radiotherapy and by a 1-year metronomic temozolomide-irinotecan maintenance. This child is alive and in CR with a follow-up of more than a year, without receiving high-dose chemotherapy. Three other children received chemotherapy with etoposide/carboplatin after a surgical resection (2 partial and 1 total) and radiotherapy after thiotepa. One of them received a maintenance treatment with temozolomide. Those children are alive with a follow-up of respectively 1, 1.3, and 5.8 years.
Of note, 6 tumors out of the 16 tumors of who received radiotherapy progressed and 10 children are alive. All patients alive and in complete remission underwent complete surgical resection and received radiotherapy.
The median time of follow-up for the whole population was 0.9 years (range 0.1 to 15.3 years). The 1-year event-free survival (EFS) and overall survival (OS) rate were 36 % CI 95 % (23–55) and 45 % CI 95 % (31–64), respectively (Figs. 1 and 2). The median EFS and OS rate were 5.4 and 10.7 months, respectively. The median time to first progression/relapsed was 4.3 months (range, 0.9–44.8 months). Twenty-seven relapses were observed, 74 % were localized, 22 % were localized and metastatic, and 4 % were only metastatic.

Overall survival

Event-free survival
Prognostic factors
The univariate analysis indicates that complete surgical resection, radiotherapy, and high-dose chemotherapy were significantly associated with a better OS (respectively, p < 0.003, p < 0.0001, and p < 0.012). Furthermore, according to the multivariate analysis, radiotherapy with a relative risk (RR) of 41.8 CI 95 % (9.4–186) p < 0.0001, surgical complete resection with a 7.9 CI 95 % (2.6–23.5) p < 0.0002, and high-dose chemotherapy with a RR 3.5 CI 95 % (1.3–9.5) p = 0.012 were prognostic factors associated with a better OS.
Discussion
ETMR is a new entity, which encompasses ETAN-TR, ependymoblastomas, medulloepitheliomas. Indeed, those tumors seem to present clinical, histopathological, and molecular similarities [2–4]. As those tumors are rare, have been only recently described and diagnosis difficult to establish; there is only a limited number of publications concerning ETMR [2, 3, 7, 8]. The series we report here represents the second largest series.
The patients’ characteristics we report here appear similar to those reported in previous studies [2, 10]. ETMR mainly occurs in young children [1, 2, 7], with a median of age at diagnosis of 31 months found in our cohort. The sex ratio in our series is 0.4. This ratio is in line with the ratio reported by Picard et al. [10], where a sex ratio of 0.6 were found in the 29 included patients. As previously reported, the most frequent clinical sign is an increased intracranial pressure, which is commonly found in cerebral tumor in children [11–13]. Clinical signs at diagnosis are similar for all ETMR histological variants [2]. Our results (Table 1) are consistent with these findings. Nevertheless, it should be noted that there is no ependymoblastoma in our series, probably due to the fact that the number of patients included in the cohort we report here is limited. A supratentorial location was found in the majority of the cases (66 %), as previously published [2]. We also report one patient with an ETMR localized to the spinal cord. This had never been reported before. Interestingly, metastasis was present at diagnosis in 18.9 % of the children as reported by Korshunov et al. [2]. Metastasis were only evidenced using MRI but were never found on CSF pathological analysis at diagnosis. Of note, one case had a CSF positivity after second surgery.
The diagnosis of ETMR appears to be challenging, as illustrated by the fact that among the histopathological review we report here, there was a change in diagnosis in 16 % cases (Table 2). The molecular diagnosis with LIN28A and the research of the gain of the chromosome 2 was performed in 11 patients. The amplification of the locus 19q13.42 was performed in 12 patients. LIN28A staining was positive in 100 % of children, and 83 % of children presented an amplification of the locus 19q13.42. Those results are in line with the data available in the literature and confirm that ETMR has a specific molecular signature. Therefore, immuno-histochemistry for LIN28A and CGHarray for amplification of the locus 19q13.42 should be done routinely to secure the diagnosis.
One of the interests of the series we report here relies on the fact that the majority of the patients were treated homogenously according to the PNET-HR protocol [9]. Despite a multimodal therapy given in 30 patients, progressions were observed in all patients with a residual tumor during induction standard therapy or sequential high-dose therapy, and 26 of them died. Noteworthy, most of the patients presented either a local or both a local and distant relapse highlighting the aggressiveness of ETMR.
In the literature, radiotherapy was the only prognostic factor in univariate analysis [11]. This is confirmed by our findings which show that using both univariate and multivariate analyses, radiotherapy is associated with a better outcome. To the best of our knowledge, there is no other data on the prognostic factors available in the medical literature. Noteworthy, radiotherapy is given at the end of the treatment after chemotherapy which might select patient and introduce a bias. In the present series, we also found that a complete surgery and high-dose chemotherapy were favorable prognostic factors. Indeed, out the 11 survivors, all but 2 children received high-dose chemotherapy. This is consistent with the experience of Dufour et al. [9] who reported the feasibility and effectiveness of high-dose chemotherapy (2 cycles of thiotepa) followed by conventional craniospinal radiotherapy with newly diagnosed high-risk medulloblastoma or supratentorial PNET. In their study, the 5-year overall survival was 85 % for the whole cohort. Unfortunately, the impact of high-dose chemotherapy on long-term survival is difficult to extrapolate from the literature on ETMR due to small patient numbers. But, such aggressive chemotherapy, as in other embryonal central nervous system, could compensate for the avoidance or dose reduction in prophylactic craniospinal irradiation [14]. Such a strategy might lead to acceptable outcomes both in terms of toxicity and survival [15].
Patients with ETMR have a poor prognosis with an EFS and OS of, respectively, 36 % CI 95 % (23–55) and 45 % CI 95 % (31–64). In comparison, Korshunov et al. found a worst OS (14 %). This may be due to their treatment strategy which relies on chemotherapy and high-dose chemotherapy after surgery [4] but not on radiotherapy. Radiotherapy may therefore be recommended in children with ETMR. However, as radiotherapy potentially impairs neurocognitive function, especially in young children [16], and as children with ETMR are for the most of all less than 4 years old, benefits and long-term toxicities have to be balanced.
Conclusion
ETMR are rare cerebral tumors of the young child. A biologic diagnosis with LIN28A immuno-histochemistry and the search for amplification of the locus 19q13.42 should be systematically done to ensure diagnosis. Prognosis remains dismal, but complete surgical removal seems to be an important part of the treatment. Radiotherapy may be another crucial point of the treatment but has to be balanced with neurocognitive toxicity that can be caused in young children. Chemotherapy and maintenance chemotherapy may be useful, but their respective roles remain to be determined.
References
Ceccom J, Bourdeaut F, Loukh N, Rigau V, Milin S, Takin R, et al. (2014) Embryonal tumor with multilayered rosettes: diagnostic tools update and review of the literature. Clin Neuropathol 33(1):15–22
Korshunov A, Sturm D, Ryzhova M, Hovestadt V, Gessi M, Jones DTW, et al. (2014) Embryonal tumor with abundant neuropil and true rosettes (ETANTR), ependymoblastoma, and medulloepithelioma share molecular similarity and comprise a single clinicopathological entity. Acta Neuropathol 128(2):279–289
Paulus W, Kleihues P (2010) Genetic profiling of CNS tumors extends histological classification. Acta Neuropathol 120(2):269–270
Korshunov A, Remke M, Gessi M, Ryzhova M, Hielscher T, Witt H, et al. (2010) Focal genomic amplification at 19q13.42 comprises a powerful diagnostic marker for embryonal tumors with ependymoblastic rosettes. Acta Neuropathol 120(2):253–260
Korshunov A, Ryzhova M, Jones DTW, Northcott PA, van Sluis P, Volckmann R, et al. (2012) LIN28A immunoreactivity is a potent diagnostic marker of embryonal tumor with multilayered rosettes (ETMR). Acta Neuropathol 124(6):875–881
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, et al. (2007) The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114(2):97–109
Eberhart CG, Brat DJ, Cohen KJ, Burger PC (2000) Pediatric neuroblastic brain tumors containing abundant neuropil and true rosettes. Pediatr Dev Pathol 3(4):346–352
Gessi M, Giangaspero F, Lauriola L, Gardiman M, Scheithauer BW, Halliday W, et al. (2009) Embryonal tumors with abundant neuropil and true rosettes: a distinctive CNS primitive neuroectodermal tumor. Am J Surg Pathol 33(2):211–217
Dufour C, Delisle M-B, Geoffray A, Laplanche A, Frappaz D, Icher C, et al. (2014) CT-003. Tandem high-dose chemotherapy with stem cell rescue followed by risk-adapted radiation in children with high-risk cerebral primitive neuroectodermal tumor: results of the prospective sfce-trial pnet hr + 5. Neuro-Oncology 16(suppl 1):i10–i13
Picard D, Miller S, Hawkins CE, Bouffet E, Rogers HA, Chan TSY, et al. (2012) Markers of survival and metastatic potential in childhood CNS primitive neuro-ectodermal brain tumours: an integrative genomic analysis. Lancet Oncol 13(8):838–848
Alexiou GA, Stefanaki K, Vartholomatos G, Sfakianos G, Prodromou N, Moschovi M (2013) Embryonal tumor with abundant neuropil and true rosettes: a systematic literature review and report of 2 new cases. J Child Neurol 28(12):1709–1715
Buccoliero AM, Castiglione F, Rossi Degl’Innocenti D, Franchi A, Paglierani M, Sanzo M, et al. (2010) Embryonal tumor with abundant neuropil and true rosettes: morphological, immunohistochemical, ultrastructural and molecular study of a case showing features of medulloepithelioma and areas of mesenchymal and epithelial differentiation. Neuropathology 30(1):84–91
Lafay-Cousin L, Hader W, Wei XC, Nordal R, Strother D, Hawkins C, et al. (2014) Post-chemotherapy maturation in supratentorial primitive neuroectodermal tumors. Brain Pathol 24(2):166–172
Odagiri K, Omura M, Hata M, Aida N, Niwa T, Goto H, et al. (2014) Treatment outcomes and late toxicities in patients with embryonal central nervous system tumors. Radiat Oncol 9:201
Kim SY, Sung KW, Hah JO, Yoo KH, Koo HH, Kang HJ, et al. (2010) Reduced-dose craniospinal radiotherapy followed by high-dose chemotherapy and autologous stem cell rescue for children with newly diagnosed high-risk medulloblastoma or supratentorial primitive neuroectodermal tumor. Korean J Hematol 45(2):120–126
Padovani L, André N, Constine LS, Muracciole X (2012) Neurocognitive function after radiotherapy for paediatric brain tumours. Nat Rev Neurol 8(10):578–588
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Horwitz, M., Dufour, C., Leblond, P. et al. Embryonal tumors with multilayered rosettes in children: the SFCE experience. Childs Nerv Syst 32, 299–305 (2016). https://doi.org/10.1007/s00381-015-2920-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-015-2920-2