
Abstract
Long-chain fatty acids (LCFAs) are a tremendous source of metabolic energy, an essential component of membranes, and important effector molecules that regulate a myriad of cellular processes. As an energy-rich nutrient source, the role of LCFAs in promoting bacterial survival and infectivity is well appreciated. LCFA degradation generates a large number of reduced cofactors that may confer redox stress; therefore, it is imperative to understand how bacteria deal with this paradoxical situation. Although the LCFA utilization pathway has been studied in great detail, especially in Escherichia coli, where the earliest studies date back to the 1960s, the interconnection of LCFA degradation with bacterial stress responses remained largely unexplored. Recent work in E. coli shows that LCFA degradation induces oxidative stress and also impedes oxidative protein folding. Importantly, both issues arise due to the insufficiency of ubiquinone, a lipid-soluble electron carrier in the electron transport chain. However, to maintain redox homeostasis, bacteria induce sophisticated cellular responses. Here, we review these findings in light of our current knowledge of the LCFA metabolic pathway, metabolism-induced oxidative stress, the process of oxidative protein folding, and stress combat mechanisms. We discuss probable mechanisms for the activation of defense players during LCFA metabolism and the likely feedback imparted by them. We suggest that besides defending against intrinsic stresses, LCFA-mediated upregulation of stress response pathways primes bacteria to adapt to harsh external environments. Collectively, the interplay between LCFA metabolism and stress responses is likely an important factor that underlies the success of LCFA-utilizing bacteria in the host.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Long-chain fatty acids (LCFAs) are amphiphilic molecules composed of a linear aliphatic chain of 12–20 carbon atoms and a terminal carboxyl group. Several bacteria acquire LCFAs from host tissues, which essentially have three fates inside the bacterial cell: degradation via β-oxidation, incorporation into membrane phospholipids, and recognition as signaling molecules. Thus, depending on their fate, LCFAs can provide metabolic energy, remodel bacterial membrane, and govern bacterial response to the environment. The effect of LCFAs on bacterial processes such as virulence, biofilm formation, and motility via their incorporation into the membrane and as signaling molecules has been reviewed recently (Kumar et al. 2020). Further, studies have associated membrane phospholipid composition with stress. In the bacterial strains where the ratio of unsaturated to saturated LCFAs in membrane phospholipids was altered due to a mutation in the enzyme involved in membrane incorporation of LCFAs or varied by either overproducing free fatty acids or overexpressing a fatty acid biosynthesis enzyme, the unsaturated LCFA content in membranes was found to be directly correlated with oxidative and membrane stress (Lennen et al. 2011; Oberg et al. 2013; Pradenas et al. 2012).
Studies have suggested that LCFAs also induce stress in bacteria when used as a nutrient source (Doi et al. 2014; Rodriguez et al. 2014). Several bacteria obtain metabolic energy from host-derived LCFAs, which contributes to their survival and virulence. For example, the LCFA degradation enzymes are induced in Mycobacterium tuberculosis and Pseudomonas aeruginosa during lung infection (Pan et al. 2020; Schnappinger et al. 2003; Son et al. 2007); the LCFA degradation enzymes are upregulated in Salmonella Typhimurium during infection, which likely contributes to the metabolism of pro-inflammatory host LCFAs and thereby suppression of the innate immune response (Mahan et al. 1995; Spector et al. 1999); in Vibrio cholerae, cholera toxin-dependent remodeling of the host metabolism causes lipolysis in target cells, accumulating LCFAs in the intestinal lumen, which are ultimately used by the pathogen for its enhanced growth (Rivera-Chavez and Mekalanos 2019); and mutants of M. tuberculosis, P. aeruginosa and S. Typhimurium defective in LCFA utilization exhibit reduced virulence (Fang et al. 2005; Kang et al. 2010; McKinney et al. 2000; Munoz-Elias and McKinney 2005). The widespread use of LCFAs as a nutrient source warrants a detailed understanding of the mechanisms by which LCFA utilization induces stress and the cellular responses employed by bacteria to mitigate them. Recent studies have investigated this issue primarily in Escherichia coli (Agrawal et al. 2017; Jaswal et al. 2020). In this review, we first describe the LCFA utilization pathway in E. coli and then discuss the recently uncovered association of LCFA degradation with stress response mechanisms.
LCFA degradation pathway in E. coli
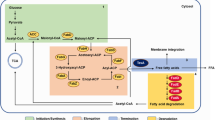
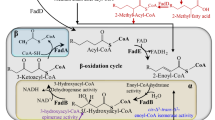
The LCFA utilization pathway has been extensively studied in E. coli, which can grow on this carbon source, both under aerobic and anaerobic conditions. LCFA metabolism is carried out by Fad (fatty acid degradation) proteins, which transport and activate LCFAs, and further, degrade these to acetyl-CoA via β-oxidation (Fig. 1). Briefly, the exogenous LCFAs are transported across the outer membrane by a β-barrel outer membrane protein, FadL. During aerobic metabolism, LCFAs are extracted from the inner membrane and activated to acyl-CoA thioesters by the inner membrane-associated acyl-CoA synthetase, FadD. Acyl-CoAs are further degraded in the cytoplasm via the various activities of β-oxidation enzymes FadE, FadB, and FadA. In each round of β-oxidation, two carbon atoms are released as acetyl-CoA, and the shortened acyl-CoA re-enters the degradation cycle. Acetyl-CoA enters the tricarboxylic acid (TCA) and glyoxylate cycles to generate metabolic precursors for growth (Fig. 1). LCFA metabolism is mainly regulated at a transcriptional level by three systems: (i) negative regulation by the transcriptional regulator, FadR, whose repression is relieved by binding of acyl-CoA, (ii) negative regulation by the ArcA–ArcB (anoxic redox control) two-component system, and (iii) positive regulation by the cAMP-CRP (cyclic AMP receptor protein-cyclic AMP) complex [reviewed in Clark and Cronan (2005), Cronan and Laporte (2006)].

The aerobic LCFA utilization pathway in E. coli. Exogenously provided LCFAs are transported across the outer membrane by FadL. LCFAs are extracted from the inner membrane and activated to acyl-CoA by FadD. Acyl-CoAs are degraded via the β-oxidation enzymes FadE, FadB, and FadA, to acetyl-CoA, which is further metabolized in the TCA and glyoxylate cycles. NADH and FADH2 generated in β-oxidation and TCA cycle are oxidized in the ETC by respiratory dehydrogenases, and the electrons are transferred to ubiquinone. Ubiquinol further gives electrons to the terminal oxidases. Arrows with e− labels indicate the direction of electron flow. The dotted arrow denotes that the players involved in FadE oxidation and transfer of electrons from FadE to the ETC are not known. CP cytoplasm, IM inner membrane, PP periplasm, OM outer membrane, Ub ubiquinone, UbH2 ubiquinol
Unlike glucose, a fermentable carbon source, which generates energy both in glycolysis and in the electron transport chain (ETC), LCFAs being non-fermentable carbon sources generate energy in the ETC (Berger 1973; Campbell et al. 2003; Clark and Cronan 2005; Romeo and Snoep 2005). The ETC components are present in the inner membrane of E. coli (Fig. 1). During aerobic metabolism of LCFAs, the reduced cofactors, NADH and FADH2, generated in β-oxidation and TCA cycle, are oxidized at the ETC by NADH dehydrogenases and succinate dehydrogenase, respectively, and the electrons are transferred to ubiquinone, a lipid-soluble electron carrier. Ubiquinol, the reduced form of ubiquinone, in turn, donates electrons to the terminal oxidases, which finally transfer electrons to molecular oxygen (O2) (Aussel et al. 2014b; Unden et al. 2014). The acyl-CoA dehydrogenase, FadE, which catalyzes the first step of β-oxidation, i.e., the conversion of acyl-CoA to enoyl-CoA together with the reduction of FAD to FADH2, has also been hypothesized to re-oxidize FADH2 by transferring electrons to the ETC (Fig. 1) (Campbell and Cronan 2002). During electron transfer through ETC, proton motive force is generated, which is then used by ATP synthase to drive ATP synthesis (Unden et al. 2014).
LCFA metabolism and stress responses
Since LCFA degradation generates a large number of reduced cofactors (Fig. 1), its utilization may confer redox stress in bacteria. Studies in a laboratory strain of E. coli, summarized below, showed that LCFA utilization generates elevated levels of reactive oxygen species (ROS) and causes problems in oxidative protein folding; however, bacteria induce cellular responses to counteract the detrimental effect of these stresses (Agrawal et al. 2017; Jaswal et al. 2020).
LCFA metabolism-associated oxidative stress and combat mechanisms
During metabolism, ROS are formed as an inevitable consequence of redox reactions. The ROS molecules, superoxide (O2−), hydrogen peroxide (H2O2), and hydroxyl radical (.OH), are produced by mechanisms that include electron leakage during oxidation-reduction cycles of ETC promoting the adventitious collision of free electrons with O2, extraction of electrons from metal centers of several metabolic enzymes by O2, and auto-oxidation of flavoproteins (Imlay 2003, 2013; Søballe and Poole 2000). E. coli grown aerobically in LCFAs generates higher levels of ROS compared to cells cultured in fermentable (glucose) or other non-fermentable (acetate and succinate) carbon sources (Agrawal et al. 2017; Doi et al. 2014). The fad mutants defective in different steps of LCFA transport and degradation are unable to produce ROS validating LCFA utilization as the reason for LCFA-induced oxidative stress (Agrawal et al. 2017). Importantly, the high NADH/NAD+ and FADH2/FAD ratios during LCFA metabolism increase electron flow in the ETC (Fig. 1) (Jaswal et al. 2020); this likely increases electron leakage and auto-oxidation of respiratory dehydrogenases. A predominant source of ROS during LCFA metabolism could be the flavoprotein involved in β-oxidation, i.e., the acyl-CoA dehydrogenase FadE. The auto-oxidation of reduced flavin bound to FadE might generate ROS (Agrawal et al. 2017).
The imbalance in ROS production and antioxidants leads to oxidative stress. The highly reactive ROS molecules oxidize macromolecular components resulting in DNA damage, lipid peroxidation, and disassembly of iron-sulfur clusters and formation of undesired disulfide bonds in proteins. For decades, the enzymes, catalases, peroxidases, and superoxide dismutases have been known as the major oxidative stress combat players in E. coli [reviewed in Chiang and Schellhorn (2012), Farr and Kogoma (1991), Imlay (2013)]. On the contrary, the role of ubiquinone as an antioxidant remained underappreciated.
In bacteria, the antioxidant function of ubiquinone was first suggested in a study by Søballe and Poole (2000), where an E. coli strain defective in ubiquinone biosynthesis was shown to exhibit several oxidative stress phenotypes in LB medium. However, the physiological condition under which ubiquinone plays a more prominent role in comparison to other oxidative stress response players had not been investigated. We identified ubiquinone as a key antioxidant during LCFA metabolism (Agrawal et al. 2017). The first hint came from the comparative analysis of the datasets from high-throughput genetic screens of the single-gene deletion library of E. coli on various carbon sources, which included non-fermentable carbon sources, acetate, succinate, and oleate (an LCFA; C18:1 cis-9). This large-scale analysis revealed that the requirement of aerobic ETC components for growth is inversely correlated with the ATP yield of non-fermentable carbon sources; their requirement is maximal in acetate, which has the poorest net ATP yield. However, the requirement of ubiquinone does not follow this trend; it is maximally required for growth in LCFAs. Following observations revealed that the increased requirement of ubiquinone in LCFAs is to mitigate oxidative stress: (i) the growth defect of ubiquinone biosynthesis mutants in oleate is partially recovered by chemical antioxidants, glutathione and thiourea, (ii) the exogenous supplementation of ubiquinone decreases ROS production in oleate-grown cells, and (iii) oleate utilization generates ROS and also results in ubiquinone accumulation; thus a feedback loop likely prevents excessive ROS formation during LCFA metabolism. Further, as long as ubiquinone is present in LCFA-grown cells, it does not allow ROS to build-up, thereby reducing dependence on other oxidative stress response players: (i) in oleate-utilizing cells, ROS levels increase in strains defective in ubiquinone biosynthesis but not in strains deleted for other oxidative stress combat players, and (ii) during oleate metabolism, enzymatic scavengers are induced only in a ubiquinone biosynthesis mutant. Collectively, our study established ubiquinone as the cell’s primary defense against LCFA-mediated oxidative stress (Agrawal et al. 2017).
There are at least two possible mechanisms by which ubiquinone might combat LCFA-induced ROS. Considering its electron shuttling role in the ETC, it is likely that the increased levels of ubiquinone in LCFA-utilizing cells enable the rapid transfer of a large flow of electrons derived from LCFA metabolism, decreasing the residence time of electrons at the site of ROS formation (Agrawal et al. 2017). Further, based on a previous finding that the terminal oxidase, Cyd, exhibits quinol peroxidase activity in vitro (Al-Attar et al. 2016), we can speculate that the increased accumulation of ubiquinol (reduced form of ubiquinone) during LCFA metabolism promotes the peroxidase activity of Cyd to scavenge ROS (Agrawal et al. 2017).
Besides upregulating antioxidant defense mechanisms to mitigate elevated ROS produced due to high NADH/NAD+ and FADH2/FAD ratios, it is plausible that bacteria induce anabolic pathways as a reductive sink to restore redox balance. In fact, the global transcriptome of M. tuberculosis cultured in a medium supplemented with a mixture of even-length LCFAs showed overexpression of WhiB3 and DosR, the two heme sensor proteins involved in maintaining intracellular redox balance, and of several genes involved in complex lipid biosynthesis, a process that consumes reduced cofactors (Rodriguez et al. 2014). Notably, the comparative analysis of the high-throughput genetic screens of the single-gene deletion library of E. coli on different carbon sources also revealed maximal enrichment of the anabolic pathway, gluconeogenesis, in LCFAs (Agrawal et al. 2017).
LCFA metabolism impedes oxidative protein folding and activates envelope stress response to restore homeostasis
Disulfide bond formation, an oxidative process that creates a covalent bond between the sulfur atoms of two cysteine residues, is required for the maturation and stability of many extracytoplasmic proteins in all domains of life. In Gram-negative bacteria, this process takes place in the oxidizing environment of the periplasm, an aqueous space surrounded by the outer and inner-membrane layers of the envelope. In E. coli, a periplasmic oxidoreductase, DsbA, forms disulfide bonds in substrate proteins. DsbB, an inner-membrane disulfide oxidoreductase, re-oxidizes DsbA and transfers electrons to quinones, ubiquinone and menaquinone, during aerobic and anaerobic metabolism, respectively (Fig. 2) (Landeta et al. 2018; Manta et al. 2019). The role of ETC in oxidative protein folding in E. coli has been demonstrated in several studies: (i) DsbA accumulates in a reduced form in mutants with a defective respiratory chain, i.e., in mutants defective in either heme or quinone biosynthesis (Kobayashi et al. 1997), (ii) disulfide bond formation is compromised in E. coli grown in a purely fermentative manner, a condition where the ETC is non-operational (Bader et al. 1999), (iii) in vitro reconstitution of the oxidative protein folding system showed the involvement of ubiquinone and terminal oxidases in re-oxidizing DsbA–DsbB (Bader et al. 1999), and (iv) mutants defective in ubiquinone biosynthesis exhibit thiol hypersensitivity (Zeng et al. 1998), a phenotype shared by dsb mutants (Bardwell et al. 1991, 1993; Missiakas et al. 1993).

The aerobic disulfide bond formation pathway in E. coli. DsbA oxidizes substrate proteins in the periplasm and becomes reduced. DsbB re-oxidizes DsbA by transferring electrons to ubiquinone. Ubiquinol further gives electrons to the terminal oxidases. Arrows with e− labels denote the direction of electron flow. CP cytoplasm, IM inner membrane, PP periplasm, OM outer membrane, Ub ubiquinone, UbH2 ubiquinol
An earlier study in E. coli, grown aerobically in glucose, reported ubiquinone to be present in ~ 15- to 20-fold excess over other ETC components (Cox et al. 1970). However, our observation that exogenous supplementation of ubiquinone decreases ROS production in oleate-grown cells strongly suggested ubiquinone to be limiting for its electron transfer function during LCFA metabolism (Agrawal et al. 2017). Given the convergence of metabolism and disulfide bond formation in the ETC, we investigated whether ubiquinone is also insufficient for oxidative protein folding in LCFA-utilizing cells (Jaswal et al. 2020). The various phenotypes exhibited by oleate-grown cells, i.e., decrease in the activity of alkaline phosphatase (a DsbA substrate), hypersensitivity to thiol agents, sensitivity to cadmium (binds with free thiols of proteins), and accumulation of the reduced form of DsbA and its substrate, DegP, convincingly established that disulfide bond formation is hampered during LCFA metabolism. Importantly, these hallmarks are prevented when ubiquinone is exogenously provided to LCFA-grown cells (Jaswal et al. 2020).
In E. coli, ~ 300 extracytoplasmic proteins are predicted to have disulfide bonds (Dutton et al. 2008). Of these, more than two dozen proteins involved in diverse biological processes are reported to be dependent on DsbA for correct folding (Delhaye et al. 2019; Kadokura et al. 2004; Manta et al. 2019). Thus, to ensure cellular homeostasis, it is important for bacteria to monitor the envelope redox status and mount an appropriate response when disturbances occur. During LCFA metabolism, DsbA accumulates in its reduced form only transiently (Jaswal et al. 2020), which suggested that defense mechanisms are upregulated to deal with the hypo-oxidizing environment of the envelope. Of the five dedicated envelope stress response (ESR) pathways (Bae, Cpx, Psp, Rcs, and σE), which sense damage in the envelope and change the transcriptome to mitigate stress (Mitchell and Silhavy 2019), we identified Cpx to be the major ESR system activated by LCFAs (Jaswal et al. 2020). Cpx is a two-component system comprised of an inner-membrane sensor histidine kinase, CpxA, and a cytoplasmic response regulator, CpxR. In the presence of envelope stress signals, CpxA autophosphorylates and transfers its phosphoryl group to CpxR, which then directs the transcription of its regulon members involved in combating stress (Grabowicz and Silhavy 2017; Raivio 2014; Raivio and Silhavy 1997). Importantly, Cpx induction in LCFA-grown cells is partially downregulated upon exogenous supplementation of ubiquinone, indicating that at least one of the signals for Cpx during LCFA metabolism is redox-dependent (Jaswal et al. 2020).
The outer membrane lipoprotein, NlpE, is a well-recognized signal for Cpx activation (Delhaye et al. 2019; Snyder et al. 1995). Recent observations that NlpE is a DsbA substrate, disruption of its C-terminal domain disulfide bond activates Cpx, and Cpx induction in ΔdsbA is NlpE-dependent, have suggested NlpE to be a sensor of oxidative protein folding defects (Delhaye et al. 2019). However, NlpE is not the molecular cue for Cpx during LCFA metabolism; Cpx is fully induced in ΔnlpE grown in oleate (Jaswal et al. 2020). A possible redox signal during LCFA metabolism is the periplasmic chaperone-protease DegP. The function of DegP is redox-dependent; its disulfide-bonded form is a chaperone, whereas the thiol form is a protease (Skorko-Glonek et al. 2008). One of the substrates of DegP protease is CpxP, a negative regulator of the Cpx response (Buelow and Raivio 2005; Isaac et al. 2005). Since DegP accumulates significantly in its thiol form in LCFA-utilizing cells (Jaswal et al. 2020), the reduced form of DegP may degrade CpxP to activate Cpx. Further, the other known inducers of Cpx, i.e., the inner-membrane respiratory complexes and lipoproteins other than NlpE (Guest et al. 2017; Miyadai et al. 2004), may constitute the redox-dependent or redox-independent signals for Cpx activation during growth in LCFAs. Clearly, detailed studies are needed to identify the Cpx-inducing signals in LCFA-utilizing cells and investigate their interplay that leads to robust Cpx activation.
Several mechanisms can be envisaged for the maintenance of envelope redox homeostasis by Cpx during LCFA metabolism: (i) Cpx response decreases envelope stress by repairing/degrading damaged proteins that accumulate due to inadequate disulfide bond formation, (ii) Cpx reduces the load on ETC by decreasing electron flow from LCFA metabolism, thereby increasing the availability of ubiquinone for disulfide bond formation, and (iii) Cpx facilitates electron transfer from disulfide bond-forming machinery by increasing the oxidizing power of ETC (Fig. 3). The following existing information on the Cpx pathway lends support to these suggested mechanisms: (i) Cpx upregulates several periplasmic chaperones, proteases and their modulators, and peptidyl-prolyl isomerases (Raivio et al. 2013), (ii) Cpx downregulates NADH dehydrogenase I and succinate dehydrogenase (Guest et al. 2017; Raivio et al. 2013). Further, in the ΔcydD strain, which has a hyper-oxidizing envelope (a situation inverse of LCFA metabolism), Cpx is downregulated, whereas fad genes are upregulated (Goldman et al. 1996; Holyoake et al. 2016; Jaswal et al. 2020; Messens et al. 2007), and (iii) the upregulation of ubiquinone in LCFA-utilizing cells is abrogated in a ΔcpxR strain (Jaswal et al. 2020).

Probable mechanisms by which Cpx restores disulfide bond formation in LCFA-grown cells. The CpxAR two-component system is comprised of an inner membrane histidine kinase, CpxA, and a cytoplasmic transcriptional regulator, CpxR. Under unstressed conditions, a periplasmic protein, CpxP, inhibits the phosphorylation of CpxA. In the presence of LCFA-generated stress signals, CpxA autophosphorylates and transfers its phosphoryl group to CpxR. The activation of the Cpx pathway decreases envelope stress by upregulating periplasmic chaperones, proteases and their modulators, and peptidyl-prolyl isomerases, which repair/remove damaged proteins. Besides, Cpx increases the oxidizing power for disulfide bond formation by upregulating ubiquinone and further increases ubiquinone availability for oxidative protein folding by downregulating the components of LCFA metabolism. CP cytoplasm, IM inner membrane, PP periplasm, OM outer membrane
Detailed studies are available on the regulation of LCFA metabolism by FadR and the ArcA–ArcB two-component system. Both FadR and the cytoplasmic response regulator ArcA repress fad genes, and ArcA additionally regulates ETC components (Bongaerts et al. 1995; Cho et al. 2006; Cotter and Gunsalus 1992; Feng and Cronan 2012; Fujita et al. 2007; Iuchi and Lin 1988; Kwon et al. 2005; Zhang and Javor 2003). Importantly, the activity of the inner-membrane sensor kinase ArcB is governed by the redox state of quinones (Alvarez et al. 2013; Georgellis et al. 2001). Since Cpx upregulates ubiquinone in LCFA-utilizing cells (Jaswal et al. 2020), it is possible that the effect of ArcA–ArcB on LCFA metabolism is modulated by Cpx. Understanding the feedback exerted by Cpx in LCFA-grown cells and its crosstalk with FadR and ArcA–ArcB under these metabolic conditions is an exciting area for future research.
Concluding remarks
Recent work on the interconnection between LCFA metabolism and stress responses in E. coli opens up several new areas of investigation. In the immediate future, studies will be required to identify the major site of ROS formation, understand the mechanism by which ubiquinone counteracts LCFA-induced oxidative stress, and identify the molecular signal for Cpx activation and investigate the nature of its feedback. Further, it will be important to examine whether bacterial pathogens use mechanisms equivalent to those identified in E. coli to defend against the harmful effects of LCFAs.
A pertinent question is why despite being predisposed to LCFA-induced stresses, bacteria readily use this nutrient source during infection. Does the use of LCFAs precondition bacteria to external stresses encountered in the host? In fact, a recent study demonstrated that hypoxia is less stressful for M. tuberculosis cultured in LCFAs, suggesting that inside the foamy macrophages, LCFA utilization enables the tubercle bacilli to survive environmental stresses (Del Portillo et al. 2018). During infection, bacteria often encounter ROS from the host immune system, such as the oxidative burst associated with neutrophils and phagocytes (Papp-Szabo et al. 1994; Rhen 2019). LCFA-induced ROS formation and the accumulation of ubiquinone might be a priming mechanism by which bacteria adapt to oxidative stress from the host. Notably, ubiquinone biosynthesis mutants of S. Typhimurium are impaired for intracellular proliferation in macrophages (Aussel et al. 2014a; Loiseau et al. 2017). Since during infection, S. Typhimurium uses LCFAs in macrophages (Fang et al. 2005; Mahan et al. 1995), it is plausible that LCFA-induced upregulation of ubiquinone serves to combat ROS produced by these immune cells.
The Cpx pathway governs many cellular processes in Gram-negative bacteria. Cpx affects biofilm formation, motility, and chemotaxis in E. coli and Salmonella Enteritidis by modulating the expression of structural, biogenesis, and regulatory components of flagella and curli (De Wulf et al. 1999; Dorel et al. 1999; Prigent-Combaret et al. 2001; Raivio 2014; Raivio et al. 2013; Shetty et al. 2019). Further, Cpx regulates the expression and assembly of cell-surface structures associated with virulence, such as pili of uropathogenic and enteropathogenic E. coli strains (Hernday et al. 2004; Hung et al. 2001; Nevesinjac and Raivio 2005), and controls the invasiveness of S. Typhimurium and Shigella sonnei in a pH-dependent manner by regulating the expression of type III secretion system (Humphreys et al. 2004; Nakayama and Watanabe 1995; Nakayama et al. 2003). The Cpx response is also associated with antibiotic resistance [reviewed in Raivio (2014)]. It is possible that LCFA-related problems in disulfide bond formation serve as a cue to induce Cpx to regulate cellular processes, which ultimately affect bacterial survival in the host.
Future research in the aforementioned directions will provide insights on how LCFA metabolism is integrated with stress responses and enable a deeper understanding of its impact on host-bacterial interactions.
Availability of data and materials
Not applicable.
Code availability
Not applicable.
References
Agrawal S, Jaswal K, Shiver AL, Balecha H, Patra T, Chaba R (2017) A genome-wide screen in Escherichia coli reveals that ubiquinone is a key antioxidant for metabolism of long-chain fatty acids. J Biol Chem 292:20086–20099. https://doi.org/10.1074/jbc.M117.806240
Al-Attar S, Yu Y, Pinkse M, Hoeser J, Friedrich T, Bald D, de Vries S (2016) Cytochrome bd displays significant quinol peroxidase activity. Sci Rep 6:27631. https://doi.org/10.1038/srep27631
Alvarez AF, Rodriguez C, Georgellis D (2013) Ubiquinone and menaquinone electron carriers represent the yin and yang in the redox regulation of the ArcB sensor kinase. J Bacteriol 195:3054–3061. https://doi.org/10.1128/JB.00406-13
Aussel L, Loiseau L, Hajj Chehade M, Pocachard B, Fontecave M, Pierrel F, Barras F (2014a) ubiJ, a new gene required for aerobic growth and proliferation in macrophage, is involved in coenzyme Q biosynthesis in Escherichia coli and Salmonella enterica serovar Typhimurium. J Bacteriol 196:70–79. https://doi.org/10.1128/JB.01065-13
Aussel L, Pierrel F, Loiseau L, Lombard M, Fontecave M, Barras F (2014b) Biosynthesis and physiology of coenzyme Q in bacteria. Biochem Biophys Acta 1837:1004–1011. https://doi.org/10.1016/j.bbabio.2014.01.015
Bader M, Muse W, Ballou DP, Gassner C, Bardwell JC (1999) Oxidative protein folding is driven by the electron transport system. Cell 98:217–227. https://doi.org/10.1016/s0092-8674(00)81016-8
Bardwell JC, McGovern K, Beckwith J (1991) Identification of a protein required for disulfide bond formation in vivo. Cell 67:581–589. https://doi.org/10.1016/0092-8674(91)90532-4
Bardwell JC, Lee JO, Jander G, Martin N, Belin D, Beckwith J (1993) A pathway for disulfide bond formation in vivo. Proc Natl Acad Sci USA 90:1038–1042. https://doi.org/10.1073/pnas.90.3.1038
Berger EA (1973) Different mechanisms of energy coupling for the active transport of proline and glutamine in Escherichia coli. Proc Natl Acad Sci USA 70:1514–1518. https://doi.org/10.1073/pnas.70.5.1514
Bongaerts J, Zoske S, Weidner U, Unden G (1995) Transcriptional regulation of the proton translocating NADH dehydrogenase genes (nuoA-N) of Escherichia coli by electron acceptors, electron donors and gene regulators. Mol Microbiol 16:521–534. https://doi.org/10.1111/j.1365-2958.1995.tb02416.x
Buelow DR, Raivio TL (2005) Cpx signal transduction is influenced by a conserved N-terminal domain in the novel inhibitor CpxP and the periplasmic protease DegP. J Bacteriol 187:6622–6630. https://doi.org/10.1128/JB.187.19.6622-6630.2005
Campbell JW, Cronan JE Jr (2002) The enigmatic Escherichia coli fadE gene is yafH. J Bacteriol 184:3759–3764. https://doi.org/10.1128/jb.184.13.3759-3764.2002
Campbell JW, Morgan-Kiss RM, Cronan JE Jr (2003) A new Escherichia coli metabolic competency: growth on fatty acids by a novel anaerobic beta-oxidation pathway. Mol Microbiol 47:793–805. https://doi.org/10.1046/j.1365-2958.2003.03341.x
Chiang SM, Schellhorn HE (2012) Regulators of oxidative stress response genes in Escherichia coli and their functional conservation in bacteria. Arch Biochem Biophys 525:161–169. https://doi.org/10.1016/j.abb.2012.02.007
Cho BK, Knight EM, Palsson BO (2006) Transcriptional regulation of the fad regulon genes of Escherichia coli by ArcA. Microbiology 152:2207–2219. https://doi.org/10.1099/mic.0.28912-0
Clark DP, Cronan JE (2005) Two-carbon compounds and fatty acids as carbon sources. EcoSal Plus. https://doi.org/10.1128/ecosalplus.3.4.4
Cotter PA, Gunsalus RP (1992) Contribution of the fnr and arcA gene products in coordinate regulation of cytochrome o and d oxidase (cyoABCDE and cydAB) genes in Escherichia coli. FEMS Microbiol Lett 70:31–36. https://doi.org/10.1016/0378-1097(92)90558-6
Cox GB, Newton NA, Gibson F, Snoswell AM, Hamilton JA (1970) The function of ubiquinone in Escherichia coli. Biochem J 117:551–562. https://doi.org/10.1042/bj1170551
Cronan JE, Laporte D (2006) Tricarboxylic acid cycle and glyoxylate bypass. In: Curtiss R III (ed) EcoSal—Escherichia coli and Salmonella: cellular and molecular biology. ASM Press, Washington, DC. https://doi.org/10.1128/ecosalplus.3.5.2
De Wulf P, Kwon O, Lin EC (1999) The CpxRA signal transduction system of Escherichia coli: growth-related autoactivation and control of unanticipated target operons. J Bacteriol 181:6772–6778. https://doi.org/10.1128/JB.181.21.6772-6778.1999
Del Portillo P et al (2018) Hypoxia is not a main stress when Mycobacterium tuberculosis is in a dormancy-like long-chain fatty acid environment. Front Cell Infect Microbiol 8:449. https://doi.org/10.3389/fcimb.2018.00449
Delhaye A, Laloux G, Collet JF (2019) The lipoprotein NlpE is a cpx sensor that serves as a sentinel for protein sorting and folding defects in the Escherichia coli envelope. J Bacteriol 201:e00611-00618. https://doi.org/10.1128/JB.00611-18
Doi H, Hoshino Y, Nakase K, Usuda Y (2014) Reduction of hydrogen peroxide stress derived from fatty acid β-oxidation improves fatty acid utilization in Escherichia coli. Appl Microbiol Biotechnol 98:629–639. https://doi.org/10.1007/s00253-013-5327-6
Dorel C, Vidal O, Prigent-Combaret C, Vallet I, Lejeune P (1999) Involvement of the Cpx signal transduction pathway of E. coli in biofilm formation. FEMS Microbiol Lett 178:169–175. https://doi.org/10.1111/j.1574-6968.1999.tb13774.x
Dutton RJ, Boyd D, Berkmen M, Beckwith J (2008) Bacterial species exhibit diversity in their mechanisms and capacity for protein disulfide bond formation. Proc Natl Acad Sci USA 105:11933–11938. https://doi.org/10.1073/pnas.0804621105
Fang FC, Libby SJ, Castor ME, Fung AM (2005) Isocitrate lyase (AceA) is required for Salmonella persistence but not for acute lethal infection in mice. Infect Immun 73:2547–2549. https://doi.org/10.1128/IAI.73.4.2547-2549.2005
Farr SB, Kogoma T (1991) Oxidative stress responses in Escherichia coli and Salmonella typhimurium. Microbiol Rev 55:561–585
Feng Y, Cronan JE (2012) Crosstalk of Escherichia coli FadR with global regulators in expression of fatty acid transport genes. PLoS ONE 7:e46275. https://doi.org/10.1371/journal.pone.0046275
Fujita Y, Matsuoka H, Hirooka K (2007) Regulation of fatty acid metabolism in bacteria. Mol Microbiol 66:829–839. https://doi.org/10.1111/j.1365-2958.2007.05947.x
Georgellis D, Kwon O, Lin EC (2001) Quinones as the redox signal for the Arc two-component system of bacteria. Science 292:2314–2316. https://doi.org/10.1126/science.1059361
Goldman BS, Gabbert KK, Kranz RG (1996) Use of heme reporters for studies of cytochrome biosynthesis and heme transport. J Bacteriol 178:6338–6347. https://doi.org/10.1128/jb.178.21.6338-6347.1996
Grabowicz M, Silhavy TJ (2017) Envelope stress responses: an interconnected safety net. Trends Biochem Sci 42:232–242. https://doi.org/10.1016/j.tibs.2016.10.002
Guest RL, Wang J, Wong JL, Raivio TL (2017) A bacterial stress response regulates respiratory protein complexes to control envelope stress adaptation. J Bacteriol 199:e00153-00117. https://doi.org/10.1128/JB.00153-17
Hernday AD, Braaten BA, Broitman-Maduro G, Engelberts P, Low DA (2004) Regulation of the pap epigenetic switch by CpxAR: phosphorylated CpxR inhibits transition to the phase ON state by competition with Lrp. Mol Cell 16:537–547. https://doi.org/10.1016/j.molcel.2004.10.020
Holyoake LV et al (2016) CydDC-mediated reductant export in Escherichia coli controls the transcriptional wiring of energy metabolism and combats nitrosative stress. Biochem J 473:693–701. https://doi.org/10.1042/BJ20150536
Humphreys S et al (2004) Role of the two-component regulator CpxAR in the virulence of Salmonella enterica serotype Typhimurium. Infect Immun 72:4654–4661. https://doi.org/10.1128/IAI.72.8.4654-4661.2004
Hung DL, Raivio TL, Jones CH, Silhavy TJ, Hultgren SJ (2001) Cpx signaling pathway monitors biogenesis and affects assembly and expression of P pili. EMBO J 20:1508–1518. https://doi.org/10.1093/emboj/20.7.1508
Imlay JA (2003) Pathways of oxidative damage. Annu Rev Microbiol 57:395–418. https://doi.org/10.1146/annurev.micro.57.030502.090938
Imlay JA (2013) The molecular mechanisms and physiological consequences of oxidative stress: lessons from a model bacterium. Nat Rev Microbiol 11:443–454. https://doi.org/10.1038/nrmicro3032
Isaac DD, Pinkner JS, Hultgren SJ, Silhavy TJ (2005) The extracytoplasmic adaptor protein CpxP is degraded with substrate by DegP. Proc Natl Acad Sci USA 102:17775–17779. https://doi.org/10.1073/pnas.0508936102
Iuchi S, Lin EC (1988) arcA (dye), a global regulatory gene in Escherichia coli mediating repression of enzymes in aerobic pathways. Proc Natl Acad Sci USA 85:1888–1892. https://doi.org/10.1073/pnas.85.6.1888
Jaswal K, Shrivastava M, Roy D, Agrawal S, Chaba R (2020) Metabolism of long-chain fatty acids affects disulfide bond formation in Escherichia coli and activates envelope stress response pathways as a combat strategy. PLoS Genet 16:e1009081. https://doi.org/10.1371/journal.pgen.1009081
Kadokura H, Tian H, Zander T, Bardwell JC, Beckwith J (2004) Snapshots of DsbA in action: detection of proteins in the process of oxidative folding. Science 303:534–537. https://doi.org/10.1126/science.1091724
Kang Y, Zarzycki-Siek J, Walton CB, Norris MH, Hoang TT (2010) Multiple FadD acyl-CoA synthetases contribute to differential fatty acid degradation and virulence in Pseudomonas aeruginosa. PLoS ONE 5:e13557. https://doi.org/10.1371/journal.pone.0013557
Kobayashi T, Kishigami S, Sone M, Inokuchi H, Mogi T, Ito K (1997) Respiratory chain is required to maintain oxidized states of the DsbA–DsbB disulfide bond formation system in aerobically growing Escherichia coli cells. Proc Natl Acad Sci USA 94:11857–11862. https://doi.org/10.1073/pnas.94.22.11857
Kumar P, Lee JH, Beyenal H, Lee J (2020) Fatty acids as antibiofilm and antivirulence agents. Trends Microbiol 28:753–768. https://doi.org/10.1016/j.tim.2020.03.014
Kwon O, Druce-Hoffman M, Meganathan R (2005) Regulation of the ubiquinone (coenzyme Q) biosynthetic genes ubiCA in Escherichia coli. Curr Microbiol 50:180–189. https://doi.org/10.1007/s00284-004-4417-1
Landeta C, Boyd D, Beckwith J (2018) Disulfide bond formation in prokaryotes. Nat Microbiol 3:270–280. https://doi.org/10.1038/s41564-017-0106-2
Lennen RM et al (2011) Membrane stresses induced by overproduction of free fatty acids in Escherichia coli. Appl Environ Microbiol 77:8114–8128. https://doi.org/10.1128/AEM.05421-11
Loiseau L et al (2017) The UbiK protein is an accessory factor necessary for bacterial ubiquinone (UQ) biosynthesis and forms a complex with the UQ biogenesis factor UbiJ. J Biol Chem 292:11937–11950. https://doi.org/10.1074/jbc.M117.789164
Mahan MJ, Tobias JW, Slauch JM, Hanna PC, Collier RJ, Mekalanos JJ (1995) Antibiotic-based selection for bacterial genes that are specifically induced during infection of a host. Proc Natl Acad Sci USA 92:669–673. https://doi.org/10.1073/pnas.92.3.669
Manta B, Boyd D, Berkmen M (2019) Disulfide bond formation in the periplasm of Escherichia coli. EcoSal Plus. https://doi.org/10.1128/ecosalplus.ESP-0012-2018
McKinney JD et al (2000) Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase. Nature 406:735–738. https://doi.org/10.1038/35021074
Messens J, Collet JF, Van Belle K, Brosens E, Loris R, Wyns L (2007) The oxidase DsbA folds a protein with a nonconsecutive disulfide. J Biol Chem 282:31302–31307. https://doi.org/10.1074/jbc.M705236200
Missiakas D, Georgopoulos C, Raina S (1993) Identification and characterization of the Escherichia coli gene dsbB, whose product is involved in the formation of disulfide bonds in vivo. Proc Natl Acad Sci USA 90:7084–7088. https://doi.org/10.1073/pnas.90.15.7084
Mitchell AM, Silhavy TJ (2019) Envelope stress responses: balancing damage repair and toxicity. Nat Rev Microbiol 17:417–428. https://doi.org/10.1038/s41579-019-0199-0
Miyadai H, Tanaka-Masuda K, Matsuyama S, Tokuda H (2004) Effects of lipoprotein overproduction on the induction of DegP (HtrA) involved in quality control in the Escherichia coli periplasm. J Biol Chem 279:39807–39813. https://doi.org/10.1074/jbc.M406390200
Munoz-Elias EJ, McKinney JD (2005) Mycobacterium tuberculosis isocitrate lyases 1 and 2 are jointly required for in vivo growth and virulence. Nat Med 11:638–644. https://doi.org/10.1038/nm1252
Nakayama S, Watanabe H (1995) Involvement of cpxA, a sensor of a two-component regulatory system, in the pH-dependent regulation of expression of Shigella sonnei virF gene. J Bacteriol 177:5062–5069. https://doi.org/10.1128/jb.177.17.5062-5069.1995
Nakayama SI, Kushiro A, Asahara T, Tanaka RI, Hu L, Kopecko DJ, Watanabe H (2003) Activation of hilA expression at low pH requires the signal sensor CpxA, but not the cognate response regulator CpxR, in Salmonella enterica serovar Typhimurium. Microbiology 149:2809–2817. https://doi.org/10.1099/mic.0.26229-0
Nevesinjac AZ, Raivio TL (2005) The Cpx envelope stress response affects expression of the type IV bundle-forming pili of enteropathogenic Escherichia coli. J Bacteriol 187:672–686. https://doi.org/10.1128/JB.187.2.672-686.2005
Oberg TS, Ward RE, Steele JL, Broadbent JR (2013) Genetic and physiological responses of Bifidobacterium animalis subsp. lactis to hydrogen peroxide stress. J Bacteriol 195:3743–3751. https://doi.org/10.1128/JB.00279-13
Pan X et al (2020) PvrA is a novel regulator that contributes to Pseudomonas aeruginosa pathogenesis by controlling bacterial utilization of long chain fatty acids. Nucleic Acids Res 48:5967–5985. https://doi.org/10.1093/nar/gkaa377
Papp-Szabo E, Firtel M, Josephy PD (1994) Comparison of the sensitivities of Salmonella typhimurium oxyR and katG mutants to killing by human neutrophils. Infect Immun 62:2662–2668. https://doi.org/10.1128/IAI.62.7.2662-2668.1994
Pradenas GA, Paillavil BA, Reyes-Cerpa S, Perez-Donoso JM, Vasquez CC (2012) Reduction of the monounsaturated fatty acid content of Escherichia coli results in increased resistance to oxidative damage. Microbiology (Reading) 158:1279–1283. https://doi.org/10.1099/mic.0.056903-0
Prigent-Combaret C, Brombacher E, Vidal O, Ambert A, Lejeune P, Landini P, Dorel C (2001) Complex regulatory network controls initial adhesion and biofilm formation in Escherichia coli via regulation of the csgD gene. J Bacteriol 183:7213–7223. https://doi.org/10.1128/JB.183.24.7213-7223.2001
Raivio TL (2014) Everything old is new again: an update on current research on the Cpx envelope stress response. Biochem Biophys Acta 1843:1529–1541. https://doi.org/10.1016/j.bbamcr.2013.10.018
Raivio TL, Silhavy TJ (1997) Transduction of envelope stress in Escherichia coli by the Cpx two-component system. J Bacteriol 179:7724–7733. https://doi.org/10.1128/jb.179.24.7724-7733.1997
Raivio TL, Leblanc SK, Price NL (2013) The Escherichia coli Cpx envelope stress response regulates genes of diverse function that impact antibiotic resistance and membrane integrity. J Bacteriol 195:2755–2767. https://doi.org/10.1128/JB.00105-13
Rhen M (2019) Salmonella and reactive oxygen species: a love-hate relationship. J Innate Immun 11:216–226. https://doi.org/10.1159/000496370
Rivera-Chavez F, Mekalanos JJ (2019) Cholera toxin promotes pathogen acquisition of host-derived nutrients. Nature 572:244–248. https://doi.org/10.1038/s41586-019-1453-3
Rodriguez JG et al (2014) Global adaptation to a lipid environment triggers the dormancy-related phenotype of Mycobacterium tuberculosis. mBio 5:e01125-01114. https://doi.org/10.1128/mBio.01125-14
Romeo T, Snoep JL (2005) Glycolysis and flux control. EcoSal Plus. https://doi.org/10.1128/ecosalplus.3.5.1
Schnappinger D et al (2003) Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: insights into the phagosomal environment. J Exp Med 198:693–704. https://doi.org/10.1084/jem.20030846
Shetty D, Abrahante JE, Chekabab SM, Wu X, Korber DR, Vidovic S (2019) Role of CpxR in biofilm development: expression of key fimbrial, O-antigen and virulence operons of Salmonella Enteritidis. Int J Mol Sci. https://doi.org/10.3390/ijms20205146
Skorko-Glonek J, Sobiecka-Szkatula A, Narkiewicz J, Lipinska B (2008) The proteolytic activity of the HtrA (DegP) protein from Escherichia coli at low temperatures. Microbiology 154:3649–3658. https://doi.org/10.1099/mic.0.2008/020487-0
Snyder WB, Davis LJ, Danese PN, Cosma CL, Silhavy TJ (1995) Overproduction of NlpE, a new outer membrane lipoprotein, suppresses the toxicity of periplasmic LacZ by activation of the Cpx signal transduction pathway. J Bacteriol 177:4216–4223. https://doi.org/10.1128/jb.177.15.4216-4223.1995
Søballe B, Poole RK (2000) Ubiquinone limits oxidative stress in Escherichia coli. Microbiology 146(Pt 4):787–796. https://doi.org/10.1099/00221287-146-4-787
Son MS, Matthews WJ Jr, Kang Y, Nguyen DT, Hoang TT (2007) In vivo evidence of Pseudomonas aeruginosa nutrient acquisition and pathogenesis in the lungs of cystic fibrosis patients. Infect Immun 75:5313–5324. https://doi.org/10.1128/IAI.01807-06
Spector MP, DiRusso CC, Pallen MJ, Del Portillo FG, Dougan G, Finlay BB (1999) The medium-/long-chain fatty acyl-CoA dehydrogenase (fadF) gene of Salmonella typhimurium is a phase 1 starvation-stress response (SSR) locus. Microbiology (Reading) 145(Pt 1):15–31. https://doi.org/10.1099/13500872-145-1-15
Unden G, Steinmetz PA, Degreif-Dunnwald P (2014) The aerobic and anaerobic respiratory chain of Escherichia coli and Salmonella enterica: enzymes and energetics. EcoSal Plus. https://doi.org/10.1128/ecosalplus.ESP-0005-2013
Zeng H, Snavely I, Zamorano P, Javor GT (1998) Low ubiquinone content in Escherichia coli causes thiol hypersensitivity. J Bacteriol 180:3681–3685. https://doi.org/10.1128/JB.180.14.3681-3685.1998
Zhang H, Javor GT (2003) Regulation of the isofunctional genes ubiD and ubiX of the ubiquinone biosynthetic pathway of Escherichia coli. FEMS Microbiol Lett 223:67–72. https://doi.org/10.1016/S0378-1097(03)00343-4
Acknowledgements
We thank Dr. Mahak Sharma and members of the RC lab for critical reading of the manuscript.
Funding
Research in RC laboratory is supported by the Department of Science and Technology-Science and Engineering Research Board (Grant number CRG/2018/000833), the Ministry of Human Resource Development-Scheme for Transformational and Advanced Research in Sciences (Grant number STARS/APR2019/BS/296/FS) and Indian Institute of Science Education and Research—Mohali.
Author information
Authors and Affiliations
Contributions
RC and KJ had the idea for the article; KJ, RC, and MS performed the literature search; and RC, KJ, and MS drafted and critically revised the work.
Corresponding author
Ethics declarations
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by Michael Polymenis.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Jaswal, K., Shrivastava, M. & Chaba, R. Revisiting long-chain fatty acid metabolism in Escherichia coli: integration with stress responses. Curr Genet 67, 573–582 (2021). https://doi.org/10.1007/s00294-021-01178-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-021-01178-z