
Abstract
The skin is the largest organ at the interface between the environment and the host. Consequently, the skin plays a central role in mounting effective host defense. In addition to pathogens, the microbiota and the host immune system are in permanent contact and communication via the skin. Consequences of this permanent interaction are a unique and partly symbiotic relationship, a tight interdependence between these partners, and also a functional “setting the clock,” in which, in the healthy steady state, an induction of protective responses to pathogens is guaranteed. At the same time, commensal microbes contribute to the alertness of the immune system and to the maintenance of immune tolerance. Atopic dermatitis (AD) is a chronic inflammatory skin disease based on a complex genetic trait with defects in cutaneous barrier, in stabilizing skin integrity. Most of AD patients develop deviated innate and adaptive immune responses. As a result, increased susceptibility to cutaneous infection is found in AD patients, and the interactions between these microbes and the skin participate in the development of chronic cutaneous inflammation. The role of the adaptive immune system was characterized in much detail, less though the contribution of innate immunity to AD pathogenesis. It is rather recent evidence that demonstrates a dominant role of components of the innate immune system not only for protecting from microbial invasion but also by orchestrating chronic skin inflammation. In this review we discuss the role of innate immune signaling and consecutive immune networks important for the pathogenesis and management of AD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Atopic dermatitis: a prototypic chronic inflammatory skin disease
Atopic dermatitis (AD) is a chronic inflammatory skin disease. It affects at least 15 % of children and is characterized by cutaneous hyper-reactivity to environmental triggers [1]. Various studies indicate that AD has a complex etiology, with activation of multiple immune and inflammatory pathways. Complex interactions between susceptibility genes, the host’s environment, defects in skin barrier function, and systemic and local immune responses contribute to the pathogenesis of AD [1, 2]. In recent years, genetic variants also of the innate immune system were detected by genome wide association studies (GWAS) as risk factors for AD and it has become clear that innate immune responses are part of the development and responsible for the severity of AD. Deviations of innate immune responses can be primary variants such as in the expression of antimicrobial peptides (AMPs) dermcidin and innate receptors and others are secondary to a deviation of the adaptive immune response and a consequence of a dominance of Th2 cytokines.
Cutaneous innate immunity
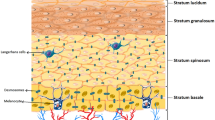
Skin functions have developed during evolution, preserving cutaneous integrity, and as the largest organ at the interface between the environment and the host, its dominant function is the orchestration of host defense mechanisms. The skin continuously encounters signals from the environment, which may act as triggers of defense. One major mechanism of immune defense is mounting tissue inflammation. Different functional compartments of the skin translate these signals into immune responses, both of the innate and the adaptive immune system. The skin displays not only a protective function as a physical barrier, but it is also a site of initial recognition of foreign substances, in which decisions about the induction or inhibition and the quality of immune responses take place. As in general, also the cutaneous immune system is divided into an innate and an adaptive part. It is now clear that the most effective antimicrobial response is based on a balance between the innate and adaptive immune system and that the skin is a site of greatest immune interactions. Proper immune function of the skin is crucial, as its dysfunction is implicated in the pathogenesis of a variety of inflammatory skin disorders, including AD, and even systemic disease as in food allergy. The skin’s innate immune system can be divided into three major components: anatomical/physical barrier (stratum corneum), secretory, and cellular elements (Fig. 1).

Cutaneous innate immune response. Structurally, the skin is divided into two main components: the epidermis on the surface and the underlying dermis. These components are separated by a basement membrane (lamina basale). The cellular compartment of the cutaneous innate immune system consists of resident cells, among them keratinocytes, which are capable to produce a vast repertoire of cytokines, chemokines and antimicrobial peptides (AMPs), different types of dendritic cells (DCs) of both, the epidermis and dermis, macrophages, and mast cells. In addition, many rapidly mobilized cells such as neutrophils, inflammatory DCs, T cells, and eosinophils can be immediately recruited to the skin and then can contribute to the composition and orchestration of cutaneous inflammation
Anatomical/physical barrier
Structurally, the skin is divided into two main components: the epidermis or epithelial component on the surface and dermis or connective component. These components are separated by a basement membrane (lamina basale), which provides a stabilizing and dynamic interface. Epidermis, a continually renewing epithelium, is subdivided in different layers with stratum corneum on the top (Fig. 1). Stratum corneum consists of corneocytes (final step of keratinocyte differentiation) which are surrounded by a protein envelope and touched together through corneodesmosomes. This layer has the main function as an anatomical barrier against pathogens, allergens, and shear force [3]. Keratinocytes, which comprise 90–95 % of the total epidermal cell population, play a pivotal role for the first defense. In addition to their function in the maintenance of the keratin barrier, they are themselves capable to produce a vast repertoire of cytokines, chemokines, and AMPs [4] and serve as initiators and amplifiers of immune responses. Following acute barrier disruption, an increase in the epidermal expression of tumor necrosis factor (TNF), IL-1, and IL-6 occurs, indicating original epidermal “inflammation” as defense mechanism [5]. Studies show that defects in epidermal barrier function contribute greatly to triggering and perpetuating skin inflammation in AD [6] and the extent of barrier dysfunction correlates with AD severity [7]. In vitro studies have shown that, in comparison to healthy cells, keratinocytes from patients with AD produce increased amounts of chemokines and cytokines, indicating, in addition to reactive, also intrinsic susceptibility to epidermal inflammation in AD [8]. The skin as complex organ in AD patients is characterized by increased transepidermal water loss (TEWL). Several underlying causes for increased TEWL can be identified. As one primary effect of terminal keratinocyte differentiation, ceramides are produced and placed into the stratum corneum. In the stratum corneum, ceramides act as dominant water-retaining molecules and major binders of structural proteins in the extracellular matrix. In AD patients, decreased levels of ceramides are found, which contribute to increased TEWL and malfunctioning of the stratum corneum [9]. One very important barrier protein in the skin is filaggrin. Filaggrin plays an integral role in maintaining the physical strength of the stratum corneum, and by minimizing entry of foreign antigens, it directly influences also the cutaneous immune response. In addition to lipids such as ceramides, filaggrin and other proteins participating in formation of the epidermal barrier determine the amount of the TEWL [4]. In addition, filaggrin helps to maintain an acidic pH in the skin by undergoing further processing in the outer layers of stratum corneum to release free amino acids [10]. It has also been demonstrated that the acidic filaggrin breakdown products urocanic acid and pyrrolidine carboxylic acid function as antimicrobials by exerting inhibitory effects on growth rate, cell density, and adhesion of Staphylococcus aureus [11]. The importance of skin barrier dysfunction as a causative factor for AD is highlighted in publications that identified loss-of-function mutations in the filaggrin gene (FLG) and found that they were associated with an increased risk of AD and correlate with its severity; however, only one third of AD patients carry FLG mutations, suggesting that other factors than FLG likely exist to be responsible for the barrier defects in AD patients [12, 13]. This could be other affected barrier proteins, metabolizing enzymes and proteases or factors that negatively regulate FLG expression independent of loss-of-function mutations. Indeed, it was demonstrated that filaggrin was downregulated in AD based on the overexpression of Th2 cytokines [14, 15]. Defects of the skin barrier as described can even be detected as dry and cracked skin, but more importantly on the molecular level, these defects result in uncovering of S. aureus-adhering extracellular matrix adhesins, such as fibrinogen and fibronectin [16] and decreased levels of sphinogosine, which exerts potent antimicrobial effect [17]. In addition, skin surface pH values change toward alkalinity when FLG is reduced allowing, e.g., bacteria to thrive [10, 18]. Indeed, in a murine model of an epicutaneous bacterial colonization, we showed that the level of skin barrier disruption correlates with persistence of S. aureus colonization and that the presence of S. aureus associated with profound cutaneous inflammation [19]. Thus, all these changes in AD skin compared to healthy skin favor S. aureus colonization and infection allowing a cascade of events to dominate that further orchestrates inflammation in the skin.
Secretory elements of the cutaneous innate immune system
Besides the physical barrier of the stratum corneum, to maintain the integrity of the skin compartment, the cutaneous immune system not only has an active and efficient defense system of responses to various infectious challenges, but also controls population density and composition of the cutaneous microbiome. For instance, keratinocytes are the main producer of AMPs in the skin. As antibiotic-like substances, AMPs play a decisive role in host innate immune defense, providing a rapid and direct first-line component to inhibit microbial growth [20]. Most AMPs carry an overall net positive charge. This ensures their interaction with the negatively charged phospholipids in the cell membranes of both Gram-positive and Gram-negative bacteria, as well as the anionic components of fungi and viruses. The peptides form a pore, which disrupt and destabilize the bacterial cell membrane, resulting in bacterial lysis. In addition, AMPs can also modify host immune responses, e.g., by acting as activating cytokines, by stimulating so called pattern recognition receptors (PRRs), and by promoting the recruitment of neutrophils, T cells, mast cells, and monocytes to the site of injury or infection [20].
There are many AMPs expressed in human and animal skin. Their main function in cutaneous host defense is to inhibit the growth of a wide spectrum of pathogens. The structures, expression, processing, induction, and antimicrobial and immunomodulatory properties vary between the peptides. AMPs can be subdivided into several families, among which β-defensins and cathelicidins are two major classes. In humans, β-defensin-1 (hBD-1) is constitutively expressed, while hBD-2 and hBD-3 and cathelicidin hCAP18/LL-37 are induced in response to inflammatory stimuli, e.g., via TLR signaling pathways [21]. HBD-2 kills predominately Gram-negative organisms such as Escherichia coli and Pseudomonas aeruginosa, and yeasts, but is relatively ineffective against Gram-positive bacteria such as S. aureus [22]. In contrast, HBD-3 and hCAP18/LL-37 are more potent, broad-spectrum AMPs that show microbicidal activity against both, Gram-positive and Gram-negative organisms and the yeast Candida albicans [23]. AMPs are induced in the skin upon injury, infection, and initiation of inflammation; however, comparing two inflammatory skin diseases, psoriasis and AD revealed that upregulation of AMPs in AD skin is much reduced compared to psoriasis. Further analyses demonstrated that AMP induction can be suppressed by Th2 cytokines, mainly IL-4, which is functional in AD skin [24, 25]. Dermcidin, another antibacterial and antimycotic AMP, is constitutively expressed in human eccrine sweat glands and secreted into sweat and has a broad spectrum of activity against a variety of pathogenic microorganisms. AD patients show a deficiency of dermcidin-derived antimicrobial peptides in sweat, which correlates with infection severity [26].
Thus, AMPs in AD may be reduced constitutively or their induction is suppressed by pleiotropic type 2 T helper (Th2) cell cytokine IL-4, thereby contributing to the pathogenesis of AD [27].
The role of Th2 cytokines for cutaneous innate immune responses in AD
Detailed characterization of AD inflammation reveals a biphasic cutaneous cytokine milieu with an initial recruitment of IL-4-producing Th2 cells, followed by a more mixed phenotype in the chronic phase [28]. Th2 cytokines mediate inflammatory reactions crucially involved in AD pathogenesis. Long established, Th2 cytokines IL-4 and IL-13 induce the isotype switching of B lymphocytes to produce IgE, which in the majority of AD patients can be found binding to ubiquitous environmental antigens. Th2 cytokine IL-5 and granulocyte–monocyte colony-stimulating factor (GM-CSF) are involved in the activation and the enhancement of cell survival of eosinophils and macrophages [29]. Human skin-derived Th2 cells were shown to be recruited to the skin by chemokines MDC and TARC (CCL22, CCL17) binding CCR4 on Th2 cells [30], chemokines that are induced by epithelial TSLP, and the Th2 cytokines IL-4 and IL-13, further amplifying the vicious circle of inflammation. These Th2 cytokines, above all IL-4, suppress AMPs [24, 25] and IL-4 not only downregulate the IL-17 response on the T cell level [31], but also deviate innate immune cells such as dendritic cells (DCs) to counter regulate Th17 responses in cutaneous inflammation as recently shown [32]. Since IL-17-producing cells are involved in protection against bacterial pathogens, IL-4 suppressing antibacterial immune responses on the innate (AMPs, DCs) and adaptive immune level [31, 33] demonstrates additional causes why patients with AD are cutaneously colonized with and infected by S. aureus. Even the binding of S. aureus to the skin was significantly increased in Th2-inflammatory skin lesions compared to Th1-inflammatory skin lesions [34]. There is also strong evidence that this Th2 bias also negatively affects the first line of resistance, the barrier, in the skin of AD patients. Th2 cytokines have been shown to downregulate FLG expression, and neutralization of IL-4 and IL-13 improves skin barrier integrity [15]. IL-4 has also been shown to inhibit ceramide synthesis in keratinocytes [35] and delay the recovery of the skin barrier in vivo [36]. AD patients with persistent S. aureus colonization show higher IgE levels despite therapy, suggesting that Th2 polarization adversely affects the immune response to this pathogen [37]. Indeed, IL-4 induces IgE production in B cells and suppresses anti-infectious immune responses by downregulating AMPs and inhibiting Th1 immunity [38]. A recent study of our group discovered an important further mechanism, how IL-4 and bacterial colonization promote AD: concerted activation of TLR2 through S. aureus components and IL4R causes an inhibition of anti-inflammatory IL-10 and consequently leads to exacerbation and persistence of AD [39]. In a model for AD, cutaneous inflammation following Th2 activation lasted for up to 48 h. Additional exposure to S. aureus-derived innate signals activating TLR2 amplified and expanded this dermatitis now lasting for 14 days [39]. As this switch to chronic dermatitis was completely dependent of IL-4, these investigations for the first time explain the biphasic cutaneous cytokine milieu as a spontaneous development of Th2 inflammation in the presence of innate TLR2 signals.
Dominant cells of the cutaneous innate immune system
Following a disruption of the physical barriers (filaggrin, tight junctions), a rapid, innate immune response needs to be initiated to prevent microbial invasion and replication. The cellular compartment of the cutaneous innate immune system consists of resident cells, among them keratinocytes, which contribute tremendously to immune functions, different types of DCs of both, the epidermis and dermis, macrophages, and mast cells. In addition, many rapidly mobilized cells such as neutrophils, inflammatory DCs, and eosinophils can be immediately recruited to the skin given the appropriate signals are present [40] (Figs. 1 and 2). All of these cells are able to recognize pathogens using different pathways. This recognition is managed by the binding of substances derived of pathogens to the so-called pattern recognition receptors on these immune cells [41]. This initiates a signaling cascade, leading to the production of pro-inflammatory cytokines, chemokines, AMPs, and inducible enzymes in the skin [42]. Activation of phagocytes leads to triggering of the respiratory burst and killing of engulfed organisms [43]. Stimulation of DCs results in the translation of innate signals to adaptive immune responses [44, 45].

Distinct functional consequences following cutaneous exposure to different TLR2 heterodimers. Cutaneous exposure to TLR2/TLR6 but not TLR2/TLR1 ligands induces systemic immune suppression following cutaneous bacterial exposure. Cutaneous exposure to Gram-positive bacteria activates TLR2/TLR6 on keratinocytes, which secrete IL-6 causing systemic induction of Gr1+CD11b+ myeloid-derived suppressor cells (MDSCs). MDSCs are recruited to the skin directly suppressing T cells. In AD, however, counter regulation of inflammation is not functional, rather suppression of anti-infectious cellular immunity by MDSCs leads to further amplification of dermatitis through bacterial or viral super-infection
Pathogen-associated molecular patterns and pattern-recognition receptors
Pattern-recognition receptors (PRRs) recognize highly conserved molecular patterns common to many classes of pathogens, known as pathogen-associated molecular patterns (PAMPs) [41]. PAMPs are nucleic acids, lipids, lipoproteins, carbohydrates, or peptidoglycans from bacteria, fungi, or protozoa. PRRs are expressed constitutively by the host, can be induced, and are germline-encoded. Both the epithelial barrier cells and resident innate immune cells in the skin express PRRs [46]. There are several classes of PRRs: Toll-like receptors (TLRs), NOD-like receptors (NLRs), RIG-I-like receptors (RLRs), and C-type lectin receptors (CLRs). All of these contribute to the innate sensing of microbes, the development of cutaneous inflammation as well as immune tolerance of the skin.
Toll-like receptors and their ligands
Among PRRs, Toll-like receptors are a well-characterized family with distinct recognition profiles [47]. TLR1–10 are the best characterized human PRRs. The recognition of PAMPs by TLRs occurs in different cell compartments, including the cell surface (TLR1, 2, 4–6, 10) and endosomes (TLR3, 7–9). The TLR family members are expressed on the cell membranes of innate immune cells (DCs, macrophages, natural killer cells) and of adaptive immunity cells (T and B cells) and of non-immune cells (epithelial and endothelial cells) [48]. When stimulated on innate immune sentinel cells such as DCs, most TLR ligands promote the development of Th1 or Th17 cells when these DCs activate and educate T helper cells. This regulation of the adaptive immune system by innate signals is very important because Th1 or Th17 cells are crucial for antibacterial, antifungal, and antiviral immunity. This emphasizes the role of TLRs’ function across the entire spectrum of innate and adaptive immunity.
TLRs are believed to function as homo- or hetero-dimers. Most TLRs transduce a signal through the intracellular adapter molecule called myeloid differentiation factor 88 (MyD88), activating transcription factors such as activator protein (AP)-1 and nuclear factor (NF)-κB, which results in the induction of pro-inflammatory cytokines, chemokines, AMPs and inducible enzymes in the skin [49].
TLR2 heterodimers and ligands
When compared to most other TLRs, TLR2 recognizes a remarkably broad range of PAMPs. These include bacterial lipopeptides (Lpp) from Gram-positive bacteria and lipoarabinomannan from mycobacteria and zymosan from yeast [50]. TLR2 has emerged as a principle receptor for Gram-positive bacteria, especially S. aureus [51] and it is now known that staphylococcal Lpp are the major ligands for TLR2 [52]. Purified native staphylococcal Lpp, including SitC, were shown to induce cytokines through the TLR2-MyD88 signaling pathway [53]. The use of S. aureus mutants deficient in maturation of lipoproteins (Δlgt) and improved Lpp purification methods show that TLR2 is activated by Lpp [52]. In vivo, different murine infection models showed that mice, deficient in TLR2, display increased susceptibility to staphylococcal infections with severe disease course, higher bacterial loads in tissue, and/or reduced inflammation [54]. This high diversity of ligand recognition by TLR2 comes possibly from its unique ability to homodimerize as well as heterodimerize with TLR1 and TLR6. Ozinsky et al. [55] were the first to show that TLR2, unlike other TLRs, has to form heterodimers with TLR1 or TLR6 to be able to initiate cell activation. Studies using knockout mice identified TLR1 as the coreceptor required for the recognition of bacterial triacylated lipoproteins such as Pam3Cys [56]. Diacylated components such as lipoprotein FSL-1 and Pam2Cys interact with TLR2/TLR6 heterodimers [57]. Using fluorescence resonance energy transfer (FRET) in human primary monocytes, Triantafilou et al. [58] have shown that a small pre-existing population of TLR2 heterodimers increases rapidly upon ligand treatment. Additionally, it was shown that TLR2/6 ligand binding reduced the percentage of preformed TLR2/1 heterodimers, but not vice versa. Employing lipid raft-disrupting agents [59], it was demonstrated that TLR2 heterodimers translocate to lipid rafts, depending on their interactions with specific ligands.
TLR2 heterodimers show functional differences
The unique ability of TLR2 to form heterodimers with TLR1 or TLR6 could be explained as evolutionary development either to expand a ligand spectrum or to induce different immune responses. Indeed, acylation patterns of Lpp are different among pathogens. Although the intracellular signaling seems to be identical following recognition of diacylated and triacylated Lpp [60], some co-receptor molecules such as CD14 and CD36 are known to increase the binding of Lpp and other PAMPs to TLRs and so to amplify the immune response. There is evidence that CD14 predominantly enhances the binding of TLR2/1 ligands [61], CD36 promotes the recognition of TLR2/6 ligands [62]. These studies suggest that the combination of different TLR2 ligands together with or without interaction with various co-receptors multiplies the ability of the immune system for adequate responses. There is evidence that TLR1 and TLR6 are not redundant as TLR1 mutation is associated with higher mortality in sepsis [63] and TLR6 has been shown to be protective for asthma [64]. Interestingly, a meta-analysis for TLR1, TLR2, and TLR6 polymorphisms toward pulmonary tuberculosis susceptibility shows that TLR1 was associated with increased risk, but TLR6 with decreased risk for tuberculosis [65]. The direct comparison of TLR2/6 and TLR2/1 ligands in vitro revealed that these substances show distinct activity in induction of gene expression and inflammatory mediators in lung tissue [66]. In the gut-associated lymphoid tissue (GALT), a TLR2/6 ligand FSL-1 was more effective than TLR2/1 ligand Pam3 at inducing Th1 and Th17 responses, while Pam3 was superior to FSL-1 at inducing Th1/Th17 in the spleen [67].
Much less research has been directed toward determining the functional differences of TLR2 heterodimers in vivo. Our group has demonstrated for the first time such distinct differences for ligands of the two TLR2 heterodimers in vivo. In this work, it is shown that even limited cutaneous exposure to TLR2/TLR6 but not TLR2/TLR1 ligands after cutaneous infection with S. aureus induces, following severe inflammation, systemic immune suppression [68, 69]. This immune suppression is due to systemic induction of Gr1+CD11b+ myeloid-derived suppressor cells (MDSCs) directly suppressing T cells (Fig. 2). Interestingly, signals through TLR2 on skin cells, but not on hematopoietic cells, as well as cutaneous IL-6 induction were necessary and sufficient for the expansion of MDSCs and for MDSCs to exert their immune suppression in this context (Fig. 2). These data from models are confirmed in human studies demonstrating MDSCs within peripheral blood mononuclear cells (PBMCs) and skin from AD patients, especially those with infectious complications such as eczema herpeticum. This increase of MDSCs especially in patients with severe AD indicates that the degree of inflammation determines an elevation in the frequency of immunosuppressive MDSCs as an attempt to stop severe inflammation. MDSCs are recruited to the skin, especially to sites of cutaneous exposure to TLR2/6 ligands from S. aureus. However, MDSCs fail to stop cutaneous inflammation in AD but allow, through temporary immune suppression, secondary infections to spread resulting in even enhanced inflammation. Thus, immune suppression in response to strong cutaneous inflammation predisposes AD patients to disseminated viral skin infections.

Preventing cutaneous inflammation in AD by non-pathogenic bacteria. Vitreoscilla filiformis components induce high levels of IL-10 in DCs via TLR2. These IL-10-DCs orchestrate the induction of IL-10high producing regulatory T (Tr1) cells, which in turn suppress dermatitis-mediating Th2 cells
This suggests that (i) the presence of certain TLR ligands, (ii) the ratio of different TLRs within a cell, or (iii) a possible interaction between TLR2 and TLR1 or TLR6 defines the nature of consecutive immune responses. Fine tuning of receptor specificity achieved by combination of different TLRs could be beneficial to the host cell, as the structure of bacterial Lpp is not constant in each bacterium. It was shown, recently, that the degree of Lpp-acylation depends on environmental factors and growth phase. Lipoprotein SitC was triacylated when S. aureus was in the exponential growth phase at neutral pH and diacylated in the post-exponential phase at low pH [70]. On the skin, where pH is low and chronic S. aureus colonization (which is almost always found in AD) is present, a post-exponential growth phase of S. aureus can be assumed. Consequently, Lpp from S. aureus on the skin are more diacylated. Based on our data, we hypothesize that diacylation of Lpp could have immune suppressive effects as a consequence. Further, one can also assume that pathogenic and non-pathogenic skin microflora may have different acylation properties and therefore different compositions of TLR2 ligands and thus overall differ in their immune consequences.
NOD-like receptors
Nucleotide-binding oligomerization domain-containing protein (NOD) 1 and NOD2 are intracellular receptors that respond to the bacterial cell degradation peptidoglycan (PGN) fragments. NOD1 responds selectively to Gram-negative bacteria, NOD2 senses muramyl dipeptide, a motif found in PGNs from all bacteria, including S. aureus [71]. Human keratinocytes were shown to express NOD1 and NOD2 [72]. Both NOD1 and NOD2 SNPs have been associated with increased IgE levels and AD [73]. Interestingly, NOD2 plays a critical role in clearance of S. aureus after subcutaneous or intraperitoneal infection [74]. We found that S. aureus-derived peptidoglycan fragments activating NOD2 are exclusively effective in the presence of TLR signals [45]. Those dual activated DCs displayed significantly enhanced IL-12p70 and IL-23 production compared to TLR agonist only stimulated cells and predominantly primed Th1 and Th17 cells while suppressing Th2 responses [45]. This points out that an activation of multiple PPRs is needed for initiation of inflammation, a situation which is a present in AD. In addition, this provides a first hint of how a Th2-dominated dermatitis switches to Th1/Th17 co-dominated inflammation as it is documented for AD.
Retinoic acid–inducible gene-like receptors
RLRs are intracellular innate receptors, containing a C-terminal helicase domain that recognizes viral genomic RNA and signals through an N-terminal CARD domain. The family of RLRs consists of retinoic acid–inducible gene (RIG)-I-helicase, MDA5, and LGP2. Activation of these receptors is crucial for eliciting antiviral responses, including induction of type I interferon gene expression [75]. Up to date, there is no evidence that RLRs play a role in AD pathogenesis.
C-type lectin receptors
CLRs contain one or more C-type lectin-like domains and recognize unique sugars present on both Gram-positive and Gram-negative bacteria, fungi, and viruses. Of the CLRs, mannan-binding lectin (MBL) is the best characterized. The transmembrane receptor dectin-1 is a receptor for β-glucans [76] found on fungal cell walls. Activation of dectin-1 leads to activation of NF-κB and secretion of pro-inflammatory cytokines [77]. The expression and function of dectin-1 in patients with AD have not been evaluated.
Polymorphisms in PPRs and associations with AD
The role of TLR2 for AD pathogenesis is discussed controversially. Genetic analyses revealed that one of the TLR2 polymorphisms (Arg753Gln) located within the intracellular part of the receptor correlated to infections with S. aureus [78], and importantly, this mutation was associated with a more severe phenotype of AD [79]. The subgroup of AD patients carrying this polymorphism had increased disease severity and was characterized by elevated IgE antibodies to S. aureus superantigens and HDM allergens. Interestingly, while lower TLR2 expression was observed in AD skin [68, 80] and on macrophages of AD patients and these produced less pro-inflammatory cytokines (IL-6, IL-8, IL-1β) after stimulation with PGN and lipoteichoic acid (LTA) [81], heterozygous carriers of TLR2 Arg753Gln with AD displayed an increased production of IL-6 and IL-12 by monocytes after TLR2 stimulation compared to wild-type AD patients and healthy controls [82]. This could be interpreted as follows: reduced responsiveness of TLR2 to S. aureus products may be beneficial in the special situation of AD pathogenesis. In contrast, increased responsiveness further amplifies inflammation in the skin. In other studies, the A allele in position 216934 was significantly associated with severe AD (scoring atopic dermatitis [SCORAD], >50) [83], and this polymorphism did not affect TLR2 mRNA expression; however, functional consequences were decreased TLR2-induced IL-6, but not TNF production [84]. This need for more individualized analyses is further indicated by a study of 275 German parent-offspring trios, which were analyzed for the four common TLR2 haplotypes and no association with AD was found [85]. Similarly, no associations between TLR1, TLR2, TLR4, and TLR6 polymorphisms and AD were found in other populations [86].
Two-fold risk for AD was found in children with Nod2/CARD15 polymorphism [87] and a study covering 11 SNPs of NOD1 found an association with AD [73]. One Nod1/CARD4 haplotype and three polymorphisms (rs2907748, rs2907749, rs2075822) were significantly associated with AD in a population-based cohort, case–control population, and/or family-based association analysis. Furthermore, AD has been associated with a polymorphism (Cys1237Thr) resulting in higher promoter activity in the gene encoding TLR9, which is crucial for the recognition of unmethylated CpG DNA sequences of bacteria, protozoa, and intracellular viral antigens [88]. It is obvious that receptors involved in the innate sensing of microbial substances play an important role in cutaneous regulation of inflammation. One important conclusion of these analyses regarding AD pathogenesis is that the complex genetic trait underlying AD phenotypes also involves PRRs, but that better stratification and more individualized phenotype/genotype analyses are needed to better disclose their role for cutaneous integrity and chronic inflammation of the skin.
The role of the microbiome for AD pathogenesis
Skin is a home to up to one billion bacteria per square centimeter [89]. The development of new technologies of microbial sequencing has brought new insights into the composition and distribution of cutaneous microbiota. It has revealed the presence of highly diverse skin microbiomes with specific location along distinct topographical sites of the skin [90]. Obviously, the exact composition of skin microbes varies between individuals, but intra-individuals comparison reveals that it appears to remain relatively stable over time [91]. Even between individuals (inter-individual comparison), the microbiome of similar/identical locations demonstrates some homology. At least 19 phyla are known to be part of the bacterial skin microbiome, with Actinobacteria, Firmicutes, Proteobacteria, and Bacteroidetes to predominate. The majority of the identified genera are Corynebacterium, Propionibacterium, and Staphylococcus [90]. The investigations of flora from distinct skin sites such hair follicles, sebaceous glands, and sweat glands have shown that skin location-specific physical metabolic and immunological factors are important for sustaining unique communities of microbes [92]. For instance, Propionibacterium and Staphylococcus species are present in skin sites enriched in sebaceous glands, Corynebacterium predominates moist sites such as the axilla, where Staphylococcus species are also present. In dry sites Proteobacteria and Flavobacteriales species dominate [93]. Thus, it is clear that there are pre-requisites of skin sites for specific skin colonization explaining why we find some inter-individual stability of microbiomes, even though the factors determining cutaneous microbiomes still need to be characterized in much more detail. In general, intrapersonal variation in microbial community and structure between symmetric skin sites is less than the interpersonal variation [92].
Recent evidence supports the idea that skin microbiota has a fundamental and complex role in the control of skin physiology, cutaneous immunity, and beyond. A large number of Gram-positive bacteria such as Lactococcus, Streptococcus, and Streptomyces species produce bactericidal factors that inhibit the growth of other bacterial strains [94]. Commensal Staphylococcus epidermidis also produces a variety of molecules that have antimicrobial activity. For example, peptides called phenosoluble modulins (PSMs) demonstrate selective activity against S. aureus, group A Streptococcus, and E. coli, but not to other S. epidermidis [95]. Interestingly, S. aureus strains also produce PSMs, but these have minimal antimicrobial activity and instead show chemotactic activity for neutrophils and induce lysis of these cells, whereas S. epidermidis PSMs show bacteria-killing activity, but no effect on neutrophils [96]. It has been also shown that S. epidermidis produces several AMPs and proteases that can limit biofilm formation of pathogenic species [97] and can induce AMP production by keratinocytes in TLR2-dependent manner [98]. Furthermore, S. epidermidis can also limit inflammatory responses and contribute to tissue repair. In a setting of skin injury, in which pathology is dependent on TLR3, a product of this bacterium, LTA, can suppress inflammation in a TLR2-dependent manner by inhibiting local production of various inflammatory mediators such as IL-6 and TNF [99]. These findings demonstrate that there is a well-balanced regulation in between the different components of the cutaneous microbiome. In addition to bacterial AMPs, cutaneous AMPs greatly contribute to this complex regulation and the stability of the compositions of the microbiomes living on healthy skin [100]. As a functional consequence of the persisting skin microbiota, shaping immune reactivity was identified. Thus, cutaneous microbiota provide substances locally that act like adjuvants orchestrating consecutive immune responses, starting from innate immune sensing, finally fine-tuning effector cells. However, in contrast to overt anti-infectious immune responses, microbiota “prepare for action” by conditioning the immune system, e.g., in the absence of a larger recruitment of immune cells. For example, S. epidermidis directly controls the activation of skin-resident T lymphocytes at steady state by production of IL-1α, which, in turn, facilitates the production of IFN-γ and IL-17 by dermal T cells. Some studies demonstrated that in the absence of skin commensals, the frequency of Foxp3+ Treg cells may be increased [101]. On the other hand, commensals are also important pathfinders for the induction and orchestration of immune tolerance. In the gut, commensals are critical and active inducer of regulatory responses, for example for the active suppression of inflammatory responses to food antigens or prevention of inflammatory diseases [102].
Thus, in general, the recognition of PAMPs of commensal bacteria through PRRs is not associated with pathogenic immune responses. It is still enigmatic how the same innate signaling results in even opposing immune consequences, in some circumstances shaping “defense,” in others initiating “tolerance,” but the general need for such differentiated regulation is obvious. Importantly, but often overlooked, complex innate immune sensing involves more than one single, e.g., TLR ligand, but it is rather a constellation of sensing via multiple PPRs, because ligands of multiple classes are usually present and differ in quantity or quality according to the circumstances. Their combination may be determining the outcome, i.e., when LPS on TLR4, RNA on TLR3, CpG DNA motifs on TLR9, etc. are active as double, triple, or in a multitude of activation pathways. It should also not be excluded that xenogeneic signals (delivered through a currently undefined mechanism) might synergize with microbial exposure for these effects. Another explanation could be that the strength of the innate immune signaling plays a role. A constant mild inflammation could be interpreted by the host immune system as a stimulus to counteract by inducing tolerance, a single pathway activation could result in receptor desensitization similar to the phenomenon of T cell anergy due to a low affinity antigen or missing second signals, and strong and chronic activation of the innate immune system induces will induce pathways of immune suppression or termination.
Overall, it is clear that the innate immune response is shaped by the skin microbiome and vice versa [92], and alterations of microbial communities that affect host-microbiome interactions have been associated with disease. AD is associated with changes in the composition of the skin microbiota, highlighting the importance of the microbiota in disease etiology [103]. Kong et al. [104] have recently performed 16S ribosomal RNA bacterial gene sequencing from serial skin sampling of children with AD to assess the relationship between skin microbiota and AD progression. In AD the proportion of S. aureus was greater during disease flares than at baseline or post-treatment and correlated with disease severity. Interestingly, increased AD severity and flares were associated with decreased bacterial diversity, whereas bacterial diversity (with increase of Streptococcus, Propionibacterium, and Corynebacterium) normalized during disease remissions and closely approximated to that observed in the skin of healthy individuals. Thus, bacterial diversity is associated with steady state and healthy skin, which indicates that diversity could prevent disease development and even promote improvement of disease.
AD shows increasing prevalence rates especially in western countries, and epidemiology indicated that absence on infections is associated with this increased incidence of AD. Consequently, the “hygiene hypothesis” was established. According to this hypothesis, increased hygiene standards with lack of exposure to infectious diseases early in life affect the development of the immune system and contribute to the development of AD and other atopic diseases. Today, this hypothesis is slightly changed as not necessarily infections, but just reduction in microbial diversity is believed to be underlying reduced immune training to resist atopic inflammation. Thus, exposure to an environment rich in microbes significantly reduces the risk to develop atopic diseases later in life indicating that exposure also to non-pathogenic microbes may prevent atopic and allergic diseases. Substitution of interactions with microbial substances or even microbes has since decades been investigated as preventive measure, but results are controversial. In most studies, oral administration of probiotics was used and an occurrence of atopic diseases was observed. Meta-analysis of randomized controlled studies shows that administration of lactobacilli or other probiotics during pregnancy prevents or decreased the risk for AD in children [105]. This protection from AD was provided for 4, 6 [106], or 7 [107] years of age. However, West et al. [108] did not find any long-term effects in regard to the prevention of AD in 8- and 9-year-olds in the cohort receiving Lactobacillus paracasei, despite a protective effect at 13 months. Daily intake of probiotic lactobacillus bacteria reduced the prevalence of AD at the age of 2 years by 50 % and this effect was stable until the age of 4 years [109]. It was subsequently demonstrated that Lactobacillus reuteri and L. casei prime monocyte-derived dendritic cells via DC-SIGN (DC-specific intercellular adhesion molecule 3-grabbing nonintegrin) to drive the development of IL-10-producing regulatory T cells [110]. A study, which investigated the oral treatment with combination of probiotic strains, significantly decreased skin inflammation in AD [111]. However, other strategies, reporting the oral use of probiotic bacteria, were questioned in regard to design of the study or failed to show significant effects in the prevention or treatment of AD [112]. Thus, the intestinal microbiome may also influence the skin. Studies investigating the effects of cutaneous administration of non-pathogens are rare. We recently demonstrated for the first time that signals derived from non-pathogenic bacteria are also functional as a treatment when applied to the skin [113]. In a prospective randomized placebo-controlled clinical trial application of a lysate of the Gram-negative non-pathogenic bacterium Vitreoscilla filiformis to inflamed AD skin leads to reduction of the AD disease score SCORAD and the patient-reported pruritus demonstrating that microbial signals can effectively alleviate T cell-mediated cutaneous inflammation in AD patients [44]. Investigating the underlying mechanism, Volz et al. found that the innate immune sensing of V. filiformis signals via TLR2-induced high levels of IL-10 in DCs functioning as the innate immune sentinel cells. These DCs orchestrated the induction of IL-10high, IFN-γlow producing regulatory T (Tr1) cells [44], which in turn suppressed dermatitis-mediating Th2 cell proliferation (Fig. 3). Thus, besides the inhibition of growth of pathogenic microbes, the other way, how commensal microorganisms contribute to host immunity, is induction of immune tolerance. Recently, it has been shown that the relative abundance of the Gram-negative gammaproteobacterium Acinetobacter correlates with IL-10 production in PBMCs from healthy individuals demonstrating that this co-existence is not based on immunological neglect but rather on active recognition resulting in tolerogenic cytokine production [114]. Strikingly, atopic individuals harbor significantly lower amounts of Acinetobacter on the skin and show diminished IL-10 production.
S. aureus as an initiation and exacerbation factor in AD
It has long been known that the skin of most patients with AD is colonized with S. aureus. S. aureus can be isolated from clinically affected and unaffected skin, and both acute and chronic AD lesions are colonized. Staphylococcal colonization density is significantly lower in healthy individuals than in patients with AD and bacterial counts on unaffected skin are lower than on affected skin [115]. S. aureus colonization is regarded as one of the most important initiating and exacerbating factors in AD [38, 103] and patients with more severe disease have been shown to have higher levels of S. aureus in their home environments [116]. S. aureus carries a wide repertoire of virulence factors, which are crucial for development of staphylococcal infections, which make them important targets for the host immune system in order to generate immune responses. For example, cell-surface proteins (including protein A) promote adhesion to damaged tissue and to the surface of host cells [117], which is a prerequisite for colonization and disease. S. aureus can exacerbate or contribute to persistent skin inflammation in patients with AD by secreting toxins with superantigenic properties, resulting in massive polyclonal activation of T cells and other immune cells. These superantigens and activated T cells correlate with the clinical severity of AD [118]. Interestingly, several types of superantigens exist and some disturb the normal humoral immune response, resulting also in anergy and immune suppression. Other products of S. aureus contribute largely to disease in patients with AD. Major cell wall components of S. aureus bind to TLR2, and in models, mice deficient in TLR2 were shown to be highly susceptible to S. aureus infection [54]. Lipoproteins and LTA were shown to be predominant staphylococcal TLR2 ligands [52, 119]. The work of S. Kaesler et al. for the first time described how the aggravation of cutaneous inflammation due to S. aureus occurs: the combination of the early AD cytokine IL-4 and activation of TLR2 on skin resident cells caused an inhibition of anti-inflammatory IL-10, which is normally induced via TLR2, and as a consequence inflammation of AD is amplified and massively prolonged, demonstrating chronification of AD [39]. These data show that TLR2 activation on skin resident cells aggravates cutaneous inflammation through the binding of ligands from pathogen bacteria. Children with lesions of AD were found to have increased levels of LTA that correlated with AD severity and S. aureus colony-forming units [119]. The amounts of LTA in the skin lesions were sufficient to exert biologic effects on various cell types in vitro, as 10 μg/ml LTA from S. aureus and more were shown to stimulate the production of multiple pro-inflammatory cytokines and chemokines in different leukocytes, especially in macrophages and monocytes [120]. Recently, we identified a new mechanism, how this S. aureus-derived cell wall component temporarily directly modulates the adaptive immune system. LTA activating the innate immune system through TLR2 signaling leads to amplification of inflammation; however, LTA when acting directly on T cells temporarily potently suppresses T cell activation. This temporary T cell paralysis functions independent of TLR signaling by means of transient cell cycle arrest [Chen et al., in revision]. This mechanism probably developed as means of S. aureus immune evasion and in the context of AD allows S. aureus to persist even when directly attacked by immune cells. These studies provide a further understanding by which S. aureus can exacerbate AD.
Therapeutic implications
Given the complex pathophysiology, the therapy of AD requires a multisided approach and includes several goals: protection of the skin barrier, control of microbial colonization, and suppression of inflammation. The current recommendations for AD therapy are dependent on the severity of the disease and the success of previous therapeutic regimen. The main axes of treatment are topical anti-inflammatory drugs followed by emollients to support skin barrier function; anti-inflammatory drugs already reduce cutaneous dominance of S. aureus by restoring skin functions. However, in many instances, disinfectants are added to the topical treatment in AD. In the case of overt cutaneous infection, combinations of systemic antibiotic treatment with topical anti-inflammatory drugs are recommended. However, antibiotic therapy non-specifically eradicates a variety of bacteria and thus decreases S. aureus predominance but, at the same time, affects bacterial diversity and may impact benefits derived from the non-pathogenic microbiota. Indeed, patients treated with antibiotics show a short-time improvement but become quickly recolonized, often with the same toxin-secreting organisms [121]. Increasing knowledge about changes in skin microbiome in AD [104] and the beneficial role of bacterial substances when applied to the skin [44, 113] provides new evidence to define preventive and therapeutic strategies to regulate rather than eradicate microbes in patients with AD. Consequently, re-establishing of a balanced microbiota or recolonization of the skin with commensals may be the promising novel therapies. Therefore, it is proposed to use antibiotics during flares, where S. aureus predominate, and use microbiota or substances derived thereof as therapeutic agents afterwards, in the post-flare stages of AD.
As some microbial AMPs specifically exert antimicrobial activity against some bacteria but not against others, these peptides may have also the potential to be used as a pathogen-specific antibiotic therapy for AD. Indeed, several recent clinical studies are currently testing the efficacy of AMPs (Table 1).
Numerous studies indicate that Th2 cells and their cytokines (with predominance of IL-4 and subsequent elevation of IgE levels) contribute to pathogenesis of AD by effects ranging from activation and enhancing of infiltration of immune cells to suppressing AMPs production, reducing the cutaneous barrier, and increasing of S. aureus binding to the skin. Our own studies further indicated that IL-4 also drives the chronic phase of AD inflammation [39]; therefore, clinical trials reporting significant improvement of AD inflammation by subcutaneous application of dupilumab, a human monoclonal antibody directed against IL-4Rα, the receptor for IL-4 and IL-13 could be promising [122] and is currently in clinical trials (Table 1). On the other hand, the latest study of Heil et al. with omalizumab, a humanized monoclonal mouse antibody against IgE, has not shown an improvement of AD [123]. A novel anti-IgE antibody called ligelizumab has been recently developed (Table 1). As the involvement of Th17 and Th22 cells in AD pathogeny has been recently published [124], ustekinumab (anti-IL12/IL-23) and an IL-22 inhibitor (ILV-094) are currently under investigation for AD (Table 1). IL-31 seems also to be involved in AD [125], and clinical studies with its inhibitors are already ongoing (Table 1) [126]. Likewise, the first trial investigating antibody that prevents interaction of thymic stromal lymphopoietin (TSLP) and its receptor has been finished recently (Table 1). Other innovative therapies aim to target signaling pathways for a variety of immune cells, cytokines and chemokines, for example, janus kinase (JAK)1, JAK2, JAK3, SHIP1, GATA-3, key enzymes for T cell activation such as phosphodiesterase 4 (PDE4) or a molecule responsible for Th2 activation prostaglandin D2 receptor (CRTH2) (Table 1). Another target of new therapies is NF-κB, a key molecule of TLR signaling (Table 1). Based on our study [68] and findings of others, IL-6 appears to be an important cytokine in AD pathogenesis. IL-6 was found to be increased in AD [127] and especially in AD skin lesion, in which the amount of IL-6 correlated with bacterial burden [119]. Genome wide association studies recently also identified an IL-6 receptor (IL-6R) variant as a new risk factor for AD [128], and we showed that IL-6 is the dominant cytokine-mediating MDSCs accumulation and immune suppression in severe AD [68]. Consequently, a small case series with three patients demonstrated therapeutic efficacy of an IL-6R blockade by tocilizumab, an IL-6R antibody [129]; however, blockade of IL-6 was associated with bacterial infections indicating possibly severe adverse events using such an approach. However, detecting MDSCs in the peripheral blood of patients with severe AD could also be further developed as biomarker for immune suppression and for stratified indication of IL-6 blockade or as new perspective of therapeutic options for depletion (apheresis) of MDSCs.
References
Novak N, Bieber T, Leung DY (2003) Immune mechanisms leading to atopic dermatitis. J Allergy Clin Immunol 112:S128–S139
Eyerich K, Eyerich S, Biedermann T (2015) The multi-modal immune pathogenesis of atopic eczema. Trends Immunol
Elias PM (2005) Stratum corneum defensive functions: an integrated view. J Invest Dermatol 125:183–200
Proksch E, Brandner JM, Jensen JM (2008) The skin: an indispensable barrier. Exp Dermatol 17:1063–1072
Wood LC, Jackson SM, Elias PM, Grunfeld C, Feingold KR (1992) Cutaneous barrier perturbation stimulates cytokine production in the epidermis of mice. J Clin Invest 90:482–487
Kuo IH, Yoshida T, De Benedetto A, Beck LA (2013) The cutaneous innate immune response in patients with atopic dermatitis. J Allergy Clin Immunol 131:266–278
Gupta J, Grube E, Ericksen MB, Stevenson MD, Lucky AW, Sheth AP, Assa’ad AH, Khurana Hershey GK (2008) Intrinsically defective skin barrier function in children with atopic dermatitis correlates with disease severity. J Allergy Clin Immunol 121:725–30.e2
Giustizieri ML, Mascia F, Frezzolini A, De Pita O, Chinni LM, Giannetti A, Girolomoni G, Pastore S (2001) Keratinocytes from patients with atopic dermatitis and psoriasis show a distinct chemokine production profile in response to T cell-derived cytokines. J Allergy Clin Immunol 107:871–877
Jungersted JM, Scheer H, Mempel M, Baurecht H, Cifuentes L, Hogh JK, Hellgren LI, Jemec GB, Agner T, Weidinger S (2010) Stratum corneum lipids, skin barrier function and filaggrin mutations in patients with atopic eczema. Allergy 65:911–918
Agrawal R, Woodfolk JA (2014) Skin barrier defects in atopic dermatitis. Curr Allergy Asthma Rep 14:433
Miajlovic H, Fallon PG, Irvine AD, Foster TJ (2010) Effect of filaggrin breakdown products on growth of and protein expression by Staphylococcus aureus. J Allergy Clin Immunol 126:1184–90.e3
Palmer CN, Irvine AD, Terron-Kwiatkowski A, Zhao Y, Liao H, Lee SP, Goudie DR, Sandilands A, Campbell LE, Smith FJ, O’Regan GM, Watson RM, Cecil JE, Bale SJ, Compton JG, DiGiovanna JJ, Fleckman P, Lewis-Jones S, Arseculeratne G, Sergeant A, Munro CS, El Houate B, McElreavey K, Halkjaer LB, Bisgaard H, Mukhopadhyay S, McLean WH (2006) Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet 38:441–446
Marenholz I, Nickel R, Ruschendorf F, Schulz F, Esparza-Gordillo J, Kerscher T, Gruber C, Lau S, Worm M, Keil T, Kurek M, Zaluga E, Wahn U, Lee YA (2006) Filaggrin loss-of-function mutations predispose to phenotypes involved in the atopic march. J Allergy Clin Immunol 118:866–871
Seguchi T, Cui CY, Kusuda S, Takahashi M, Aisu K, Tezuka T (1996) Decreased expression of filaggrin in atopic skin. Arch Dermatol Res 288:442–446
Howell MD, Kim BE, Gao P, Grant AV, Boguniewicz M, Debenedetto A, Schneider L, Beck LA, Barnes KC, Leung DY (2007) Cytokine modulation of atopic dermatitis filaggrin skin expression. J Allergy Clin Immunol 120:150–155
Cho SH, Strickland I, Boguniewicz M, Leung DY (2001) Fibronectin and fibrinogen contribute to the enhanced binding of Staphylococcus aureus to atopic skin. J Allergy Clin Immunol 108:269–274
Arikawa J, Ishibashi M, Kawashima M, Takagi Y, Ichikawa Y, Imokawa G (2002) Decreased levels of sphingosine, a natural antimicrobial agent, may be associated with vulnerability of the stratum corneum from patients with atopic dermatitis to colonization by Staphylococcus aureus. J Invest Dermatol 119:433–439
Hachem JP, Crumrine D, Fluhr J, Brown BE, Feingold KR, Elias PM (2003) pH directly regulates epidermal permeability barrier homeostasis, and stratum corneum integrity/cohesion. J Invest Dermatol 121:345–353
Wanke I, Skabytska Y, Kraft B, Peschel A, Biedermann T, Schittek B (2013) Staphylococcus aureus skin colonization is promoted by barrier disruption and leads to local inflammation. Exp Dermatol 22:153–155
Afshar M, Gallo RL (2013) Innate immune defense system of the skin. Vet Dermatol 24:32–8.e8-9
Schauber J, Gallo RL (2008) Antimicrobial peptides and the skin immune defense system. J Allergy Clin Immunol 122:261–266
Schroder JM, Harder J (1999) Human beta-defensin-2. Int J Biochem Cell Biol 31:645–651
Harder J, Bartels J, Christophers E, Schroder JM (2001) Isolation and characterization of human beta -defensin-3, a novel human inducible peptide antibiotic. J Biol Chem 276:5707–5713
Ong PY, Ohtake T, Brandt C, Strickland I, Boguniewicz M, Ganz T, Gallo RL, Leung DY (2002) Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N Engl J Med 347:1151–1160
Howell MD, Gallo RL, Boguniewicz M, Jones JF, Wong C, Streib JE, Leung DY (2006) Cytokine milieu of atopic dermatitis skin subverts the innate immune response to vaccinia virus. Immunity 24:341–348
Rieg S, Steffen H, Seeber S, Humeny A, Kalbacher H, Dietz K, Garbe C, Schittek B (2005) Deficiency of dermcidin-derived antimicrobial peptides in sweat of patients with atopic dermatitis correlates with an impaired innate defense of human skin in vivo. J Immunol 174:8003–8010
Homey B, Steinhoff M, Ruzicka T, Leung DY (2006) Cytokines and chemokines orchestrate atopic skin inflammation. J Allergy Clin Immunol 118:178–189
Hamid Q, Boguniewicz M, Leung DY (1994) Differential in situ cytokine gene expression in acute versus chronic atopic dermatitis. J Clin Invest 94:870–876
Spergel JM, Mizoguchi E, Oettgen H, Bhan AK, Geha RS (1999) Roles of TH1 and TH2 cytokines in a murine model of allergic dermatitis. J Clin Invest 103:1103–1111
Biedermann T, Schwarzler C, Lametschwandtner G, Thoma G, Carballido-Perrig N, Kund J, de Vries JE, Rot A, Carballido JM (2002) Targeting CLA/E-selectin interactions prevents CCR4-mediated recruitment of human Th2 memory cells to human skin in vivo. Eur J Immunol 32:3171–3180
Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C (2005) A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol 6:1133–1141
Guenova E, Skabytska Y, Hoetzenecker W, Weindl G, Sauer K, Tham M, Kim KW, Park JH, Seo JH, Ignatova D, Cozzio A, Levesque MP, Volz T, Koberle M, Kaesler S, Thomas P, Mailhammer R, Ghoreschi K, Schakel K, Amarov B, Eichner M, Schaller M, Clark RA, Rocken M, Biedermann T (2015) IL-4 abrogates TH17 cell-mediated inflammation by selective silencing of IL-23 in antigen-presenting cells. Proc Natl Acad Sci U S A 112:2163–2168
Biedermann T, Mailhammer R, Mai A, Sander C, Ogilvie A, Brombacher F, Maier K, Levine AD, Rocken M (2001) Reversal of established delayed type hypersensitivity reactions following therapy with IL-4 or antigen-specific Th2 cells. Eur J Immunol 31:1582–1591
Cho SH, Strickland I, Tomkinson A, Fehringer AP, Gelfand EW, Leung DY (2001) Preferential binding of Staphylococcus aureus to skin sites of Th2-mediated inflammation in a murine model. J Invest Dermatol 116:658–663
Hatano Y, Terashi H, Arakawa S, Katagiri K (2005) Interleukin-4 suppresses the enhancement of ceramide synthesis and cutaneous permeability barrier functions induced by tumor necrosis factor-alpha and interferon-gamma in human epidermis. J Invest Dermatol 124:786–792
Kurahashi R, Hatano Y, Katagiri K (2008) IL-4 suppresses the recovery of cutaneous permeability barrier functions in vivo. J Invest Dermatol 128:1329–1331
Guzik TJ, Bzowska M, Kasprowicz A, Czerniawska-Mysik G, Wojcik K, Szmyd D, Adamek-Guzik T, Pryjma J (2005) Persistent skin colonization with Staphylococcus aureus in atopic dermatitis: relationship to clinical and immunological parameters. Clin Exp Allergy 35:448–455
Biedermann T (2006) Dissecting the role of infections in atopic dermatitis. Acta Derm Venereol 86:99–109
Kaesler S, Volz T, Skabytska Y, Koberle M, Hein U, Chen KM, Guenova E, Wolbing F, Rocken M, Biedermann T (2014) Toll-like receptor 2 ligands promote chronic atopic dermatitis through IL-4-mediated suppression of IL-10. J Allergy Clin Immunol 134:92–99
Volz T, Kaesler S, Biedermann T (2011) Innate immune sensing 2.0—from linear activation pathways to fine tuned and regulated innate immune networks. Exp Dermatol 21:61–69
Medzhitov R, Janeway CA Jr (1997) Innate immunity: the virtues of a nonclonal system of recognition. Cell 91:295–298
Janeway CA Jr, Medzhitov R (2002) Innate immune recognition. Annu Rev Immunol 20:197–216
Pivarcsi A, Kemeny L, Dobozy A (2004) Innate immune functions of the keratinocytes. A review. Acta Microbiol Immunol Hung 51:303–310
Volz T, Skabytska Y, Guenova E, Chen KM, Frick JS, Kirschning CJ, Kaesler S, Rocken M, Biedermann T (2014) Nonpathogenic bacteria alleviating atopic dermatitis inflammation induce IL-10-producing dendritic cells and regulatory Tr1 cells. J Invest Dermatol 134:96–104
Volz T, Nega M, Buschmann J, Kaesler S, Guenova E, Peschel A, Rocken M, Gotz F, Biedermann T (2010) Natural Staphylococcus aureus-derived peptidoglycan fragments activate NOD2 and act as potent costimulators of the innate immune system exclusively in the presence of TLR signals. FASEB J 24:4089–4102
Kupper TS, Fuhlbrigge RC (2004) Immune surveillance in the skin: mechanisms and clinical consequences. Nat Rev Immunol 4:211–222
Kawai T, Akira S (2006) TLR signaling. Cell Death Differ 13:816–825
Akira S, Hemmi H (2003) Recognition of pathogen-associated molecular patterns by TLR family. Immunol Lett 85:85–95
Brikos C, O’Neill LA (2008) Signalling of toll-like receptors. Handb Exp Pharmacol 21–50
Zahringer U, Lindner B, Inamura S, Heine H, Alexander C (2008) TLR2—promiscuous or specific? A critical re-evaluation of a receptor expressing apparent broad specificity. Immunobiology 213:205–224
Stoll H, Dengjel J, Nerz C, Götz F (2005) Staphylococcus aureus deficient in lipidation of prelipoproteins is attenuated in growth and immune activation. Infect Immun 73:2411–2423
Hashimoto M, Tawaratsumida K, Kariya H, Aoyama K, Tamura T, Suda Y (2006) Lipoprotein is a predominant Toll-like receptor 2 ligand in Staphylococcus aureus cell wall components. Int Immunol 18:355–362
Kurokawa K, Lee H, Roh KB, Asanuma M, Kim YS, Nakayama H, Shiratsuchi A, Choi Y, Takeuchi O, Kang HJ, Dohmae N, Nakanishi Y, Akira S, Sekimizu K, Lee BL (2009) The triacylated ATP binding cluster transporter substrate-binding lipoprotein of Staphylococcus aureus functions as a native ligand for toll-like receptor 2. J Biol Chem 284:8406–8411
Takeuchi O, Hoshino K, Akira S (2000) Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. J Immunol 165:5392–5396
Ozinsky A, Underhill DM, Fontenot JD, Hajjar AM, Smith KD, Wilson CB, Schroeder L, Aderem A (2000) The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between toll-like receptors. Proc Natl Acad Sci U S A 97:13766–13771
Jin MS, Kim SE, Heo JY, Lee ME, Kim HM, Paik SG, Lee H, Lee JO (2007) Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell 130:1071–1082
Buwitt-Beckmann U, Heine H, Wiesmuller KH, Jung G, Brock R, Akira S, Ulmer AJ (2005) Toll-like receptor 6-independent signaling by diacylated lipopeptides. Eur J Immunol 35:282–289
Triantafilou M, Gamper FG, Haston RM, Mouratis MA, Morath S, Hartung T, Triantafilou K (2006) Membrane sorting of toll-like receptor (TLR)-2/6 and TLR2/1 heterodimers at the cell surface determines heterotypic associations with CD36 and intracellular targeting. J Biol Chem 281:31002–31011
Triantafilou M, Manukyan M, Mackie A, Morath S, Hartung T, Heine H, Triantafilou K (2004) Lipoteichoic acid and toll-like receptor 2 internalization and targeting to the Golgi are lipid raft-dependent. J Biol Chem 279:40882–40889
Farhat K, Riekenberg S, Heine H, Debarry J, Lang R, Mages J, Buwitt-Beckmann U, Roschmann K, Jung G, Wiesmuller KH, Ulmer AJ (2008) Heterodimerization of TLR2 with TLR1 or TLR6 expands the ligand spectrum but does not lead to differential signaling. J Leukoc Biol 83:692–701
Nakata T, Yasuda M, Fujita M, Kataoka H, Kiura K, Sano H, Shibata K (2006) CD14 directly binds to triacylated lipopeptides and facilitates recognition of the lipopeptides by the receptor complex of Toll-like receptors 2 and 1 without binding to the complex. Cell Microbiol 8:1899–1909
Hoebe K, Georgel P, Rutschmann S, Du X, Mudd S, Crozat K, Sovath S, Shamel L, Hartung T, Zahringer U, Beutler B (2005) CD36 is a sensor of diacylglycerides. Nature 433:523–527
Thompson CM, Holden TD, Rona G, Laxmanan B, Black RA, O’Keefe GE, Wurfel MM (2014) Toll-like receptor 1 polymorphisms and associated outcomes in sepsis after traumatic injury: a candidate gene association study. Ann Surg 259:179–185
Moreira AP, Cavassani KA, Ismailoglu UB, Hullinger R, Dunleavy MP, Knight DA, Kunkel SL, Uematsu S, Akira S, Hogaboam CM (2011) The protective role of TLR6 in a mouse model of asthma is mediated by IL-23 and IL-17A. J Clin Invest 121:4420–4432
Zhang Y, Jiang T, Yang X, Xue Y, Wang C, Liu J, Zhang X, Chen Z, Zhao M, Li JC (2013) Toll-like receptor −1, −2, and −6 polymorphisms and pulmonary tuberculosis susceptibility: a systematic review and meta-analysis. PLoS One 8, e63357
Barrenschee M, Lex D, Uhlig S (2010) Effects of the TLR2 agonists MALP-2 and Pam3Cys in isolated mouse lungs. PLoS One 5, e13889
Morgan ME, Koelink PJ, Zheng B, den Brok MH, van de Kant HJ, Verspaget HW, Folkerts G, Adema GJ, Kraneveld AD (2014) Toll-like receptor 6 stimulation promotes T-helper 1 and 17 responses in gastrointestinal-associated lymphoid tissue and modulates murine experimental colitis. Mucosal Immunol 7:1266–1277
Skabytska Y, Wolbing F, Gunther C, Koberle M, Kaesler S, Chen KM, Guenova E, Demircioglu D, Kempf WE, Volz T, Rammensee HG, Schaller M, Rocken M, Gotz F, Biedermann T (2014) Cutaneous innate immune sensing of Toll-like receptor 2–6 ligands suppresses T cell immunity by inducing myeloid-derived suppressor cells. Immunity 41:762–775
Skabytska Y, Biedermann T (2015) Cutaneous bacteria induce immunosuppression. Oncotarget 6:30441–30442
Kurokawa K, Kim MS, Ichikawa R, Ryu KH, Dohmae N, Nakayama H, Lee BL (2012) Environment-mediated accumulation of diacyl lipoproteins over their triacyl counterparts in Staphylococcus aureus. J Bacteriol 194:3299–3306
Girardin SE, Philpott DJ (2004) Mini-review: the role of peptidoglycan recognition in innate immunity. Eur J Immunol 34:1777–1782
Harder J, Nunez G (2009) Functional expression of the intracellular pattern recognition receptor NOD1 in human keratinocytes. J Invest Dermatol 129:1299–1302
Weidinger S, Klopp N, Rummler L, Wagenpfeil S, Novak N, Baurecht HJ, Groer W, Darsow U, Heinrich J, Gauger A, Schafer T, Jakob T, Behrendt H, Wichmann HE, Ring J, Illig T (2005) Association of NOD1 polymorphisms with atopic eczema and related phenotypes. J Allergy Clin Immunol 116:177–184
Hruz P, Zinkernagel AS, Jenikova G, Botwin GJ, Hugot JP, Karin M, Nizet V, Eckmann L (2009) NOD2 contributes to cutaneous defense against Staphylococcus aureus through alpha-toxin-dependent innate immune activation. Proc Natl Acad Sci U S A 106:12873–12878
Takeuchi O, Akira S (2010) Pattern recognition receptors and inflammation. Cell 140:805–820
Brown GD, Herre J, Williams DL, Willment JA, Marshall AS, Gordon S (2003) Dectin-1 mediates the biological effects of beta-glucans. J Exp Med 197:1119–1124
Reid DM, Gow NA, Brown GD (2009) Pattern recognition: recent insights from Dectin-1. Curr Opin Immunol 21:30–37
Lorenz E, Mira JP, Cornish KL, Arbour NC, Schwartz DA (2000) A novel polymorphism in the toll-like receptor 2 gene and its potential association with staphylococcal infection. Infect Immun 68:6398–6401
Ahmad-Nejad P, Mrabet-Dahbi S, Breuer K, Klotz M, Werfel T, Herz U, Heeg K, Neumaier M, Renz H (2004) The toll-like receptor 2 R753Q polymorphism defines a subgroup of patients with atopic dermatitis having severe phenotype. J Allergy Clin Immunol 113:565–567
Kuo IH, Carpenter-Mendini A, Yoshida T, McGirt LY, Ivanov AI, Barnes KC, Gallo RL, Borkowski AW, Yamasaki K, Leung DY, Georas SN, De Benedetto A, Beck LA (2013) Activation of epidermal toll-like receptor 2 enhances tight junction function: implications for atopic dermatitis and skin barrier repair. J Invest Dermatol 133:988–998
Niebuhr M, Lutat C, Sigel S, Werfel T (2009) Impaired TLR-2 expression and TLR-2-mediated cytokine secretion in macrophages from patients with atopic dermatitis. Allergy 64:1580–1587
Mrabet-Dahbi S, Dalpke AH, Niebuhr M, Frey M, Draing C, Brand S, Heeg K, Werfel T, Renz H (2008) The Toll-like receptor 2 R753Q mutation modifies cytokine production and Toll-like receptor expression in atopic dermatitis. J Allergy Clin Immunol 121:1013–1019
Potaczek DP, Nastalek M, Okumura K, Wojas-Pelc A, Undas A, Nishiyama C (2011) An association of TLR2-16934A >T polymorphism and severity/phenotype of atopic dermatitis. J Eur Acad Dermatol Venereol 25:715–721
Oh DY, Schumann RR, Hamann L, Neumann K, Worm M, Heine G (2009) Association of the toll-like receptor 2 A-16934T promoter polymorphism with severe atopic dermatitis. Allergy 64:1608–1615
Akdis CA, Akdis M, Bieber T, Bindslev-Jensen C, Boguniewicz M, Eigenmann P, Hamid Q, Kapp A, Leung DY, Lipozencic J, Luger TA, Muraro A, Novak N, Platts-Mills TA, Rosenwasser L, Scheynius A, Simons FE, Spergel J, Turjanmaa K, Wahn U, Weidinger S, Werfel T, Zuberbier T (2006) Diagnosis and treatment of atopic dermatitis in children and adults: European Academy of Allergology and Clinical Immunology/American Academy of Allergy, Asthma and Immunology/PRACTALL Consensus Report. J Allergy Clin Immunol 118:152–169
Weidinger S, Novak N, Klopp N, Baurecht H, Wagenpfeil S, Rummler L, Ring J, Behrendt H, Illig T (2006) Lack of association between Toll-like receptor 2 and Toll-like receptor 4 polymorphisms and atopic eczema. J Allergy Clin Immunol 118:277–279
Kabesch M, Peters W, Carr D, Leupold W, Weiland SK, von Mutius E (2003) Association between polymorphisms in caspase recruitment domain containing protein 15 and allergy in two German populations. J Allergy Clin Immunol 111:813–817
Novak N, Yu CF, Bussmann C, Maintz L, Peng WM, Hart J, Hagemann T, Diaz-Lacava A, Baurecht HJ, Klopp N, Wagenpfeil S, Behrendt H, Bieber T, Ring J, Illig T, Weidinger S (2007) Putative association of a TLR9 promoter polymorphism with atopic eczema. Allergy 62:766–772
Grice EA, Kong HH, Renaud G, Young AC, Bouffard GG, Blakesley RW, Wolfsberg TG, Turner ML, Segre JA (2008) A diversity profile of the human skin microbiota. Genome Res 18:1043–1050
Grice EA, Kong HH, Conlan S, Deming CB, Davis J, Young AC, Bouffard GG, Blakesley RW, Murray PR, Green ED, Turner ML, Segre JA (2009) Topographical and temporal diversity of the human skin microbiome. Science 324:1190–1192
Consortium HMP (2012) Structure, function and diversity of the healthy human microbiome. Nature 486:207–214
Grice EA, Segre JA (2011) The skin microbiome. Nat Rev Microbiol 9:244–253
Fitz-Gibbon S, Tomida S, Chiu BH, Nguyen L, Du C, Liu M, Elashoff D, Erfe MC, Loncaric A, Kim J, Modlin RL, Miller JF, Sodergren E, Craft N, Weinstock GM, Li H (2013) Propionibacterium acnes strain populations in the human skin microbiome associated with acne. J Invest Dermatol 133:2152–2160
Gallo RL, Hooper LV (2012) Epithelial antimicrobial defence of the skin and intestine. Nat Rev Immunol 12:503–516
Cogen AL, Yamasaki K, Sanchez KM, Dorschner RA, Lai Y, MacLeod DT, Torpey JW, Otto M, Nizet V, Kim JE, Gallo RL (2010) Selective antimicrobial action is provided by phenol-soluble modulins derived from Staphylococcus epidermidis, a normal resident of the skin. J Invest Dermatol 130:192–200
Wang R, Braughton KR, Kretschmer D, Bach TH, Queck SY, Li M, Kennedy AD, Dorward DW, Klebanoff SJ, Peschel A, DeLeo FR, Otto M (2007) Identification of novel cytolytic peptides as key virulence determinants for community-associated MRSA. Nat Med 13:1510–1514
Iwase T, Uehara Y, Shinji H, Tajima A, Seo H, Takada K, Agata T, Mizunoe Y (2010) Staphylococcus epidermidis Esp inhibits Staphylococcus aureus biofilm formation and nasal colonization. Nature 465:346–349
Lai Y, Cogen AL, Radek KA, Park HJ, Macleod DT, Leichtle A, Ryan AF, Di Nardo A, Gallo RL (2010) Activation of TLR2 by a small molecule produced by Staphylococcus epidermidis increases antimicrobial defense against bacterial skin infections. J Invest Dermatol 130:2211–2221
Lai Y, Di Nardo A, Nakatsuji T, Leichtle A, Yang Y, Cogen AL, Wu ZR, Hooper LV, Schmidt RR, von Aulock S, Radek KA, Huang CM, Ryan AF, Gallo RL (2009) Commensal bacteria regulate Toll-like receptor 3-dependent inflammation after skin injury. Nat Med 15:1377–1382
Wanke I, Steffen H, Christ C, Krismer B, Gotz F, Peschel A, Schaller M, Schittek B (2011) Skin commensals amplify the innate immune response to pathogens by activation of distinct signaling pathways. J Invest Dermatol 131:382–390
Naik S, Bouladoux N, Wilhelm C, Molloy MJ, Salcedo R, Kastenmuller W, Deming C, Quinones M, Koo L, Conlan S, Spencer S, Hall JA, Dzutsev A, Kong H, Campbell DJ, Trinchieri G, Segre JA, Belkaid Y (2012) Compartmentalized control of skin immunity by resident commensals. Science 337:1115–1119
Belkaid Y, Hand TW (2014) Role of the microbiota in immunity and inflammation. Cell 157:121–141
Biedermann T, Skabytska Y, Kaesler S, Volz T (2015) Regulation of T cell immunity in atopic dermatitis by microbes: the Yin and Yang of cutaneous inflammation. Front Immunol 6:353
Kong HH, Oh J, Deming C, Conlan S, Grice EA, Beatson MA, Nomicos E, Polley EC, Komarow HD, Murray PR, Turner ML, Segre JA (2012) Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res 22:850–859
Panduru M, Panduru NM, Salavastru CM, Tiplica GS (2015) Probiotics and primary prevention of atopic dermatitis: a meta-analysis of randomized controlled studies. J Eur Acad Dermatol Venereol 29:232–242
Wickens K, Stanley TV, Mitchell EA, Barthow C, Fitzharris P, Purdie G, Siebers R, Black PN, Crane J (2013) Early supplementation with Lactobacillus rhamnosus HN001 reduces eczema prevalence to 6 years: does it also reduce atopic sensitization? Clin Exp Allergy 43:1048–1057
Kalliomaki M, Salminen S, Poussa T, Isolauri E (2007) Probiotics during the first 7 years of life: a cumulative risk reduction of eczema in a randomized, placebo-controlled trial. J Allergy Clin Immunol 119:1019–1021
West CE, Hammarstrom ML, Hernell O (2013) Probiotics in primary prevention of allergic disease--follow-up at 8–9 years of age. Allergy 68:1015–1020
Kalliomaki M, Salminen S, Poussa T, Arvilommi H, Isolauri E (2003) Probiotics and prevention of atopic disease: 4-year follow-up of a randomised placebo-controlled trial. Lancet 361:1869–1871
Smits HH, Engering A, van der Kleij D, de Jong EC, Schipper K, van Capel TM, Zaat BA, Yazdanbakhsh M, Wierenga EA, van Kooyk Y, Kapsenberg ML (2005) Selective probiotic bacteria induce IL-10-producing regulatory T cells in vitro by modulating dendritic cell function through dendritic cell-specific intercellular adhesion molecule 3-grabbing nonintegrin. J Allergy Clin Immunol 115:1260–1267
Iemoli E, Trabattoni D, Parisotto S, Borgonovo L, Toscano M, Rizzardini G, Clerici M, Ricci E, Fusi A, De Vecchi E, Piconi S, Drago L (2012) Probiotics reduce gut microbial translocation and improve adult atopic dermatitis. J Clin Gastroenterol 46(Suppl):S33–S40
Lee J, Seto D, Bielory L (2008) Meta-analysis of clinical trials of probiotics for prevention and treatment of pediatric atopic dermatitis. J Allergy Clin Immunol 121(116–21), e11
Gueniche A, Knaudt B, Schuck E, Volz T, Bastien P, Martin R, Röcken M, Breton L, Biedermann T (2008) Effects of nonpathogenic gram-negative bacterium Vitreoscilla filiformis lysate on atopic dermatitis: a prospective, randomized, double-blind, placebo-controlled clinical study. Br J Dermatol 159:1357–1363
Hanski I, von Hertzen L, Fyhrquist N, Koskinen K, Torppa K, Laatikainen T, Karisola P, Auvinen P, Paulin L, Makela MJ, Vartiainen E, Kosunen TU, Alenius H, Haahtela T (2012) Environmental biodiversity, human microbiota, and allergy are interrelated. Proc Natl Acad Sci U S A 109:8334–8339
Monti G, Tonetto P, Mostert M, Oggero R (1996) Staphylococcus aureus skin colonization in infants with atopic dermatitis. Dermatology 193:83–87
Leung AD, Schiltz AM, Hall CF, Liu AH (2008) Severe atopic dermatitis is associated with a high burden of environmental Staphylococcus aureus. Clin Exp Allergy 38:789–793
Foster TJ, Höök M (1998) Surface protein adhesins of Staphylococcus aureus. Trends Microbiol 6:484–488
Bunikowski R, Mielke ME, Skarabis H, Worm M, Anagnostopoulos I, Kolde G, Wahn U, Renz H (2000) Evidence for a disease-promoting effect of Staphylococcus aureus-derived exotoxins in atopic dermatitis. J Allergy Clin Immunol 105:814–819
Travers JB, Kozman A, Mousdicas N, Saha C, Landis M, Al-Hassani M, Yao W, Yao Y, Hyatt AM, Sheehan MP, Haggstrom AN, Kaplan MH (2010) Infected atopic dermatitis lesions contain pharmacologic amounts of lipoteichoic acid. J Allergy Clin Immunol 125(146–52):e1–e2
Cleveland MG, Gorham JD, Murphy TL, Tuomanen E, Murphy KM (1996) Lipoteichoic acid preparations of gram-positive bacteria induce interleukin-12 through a CD14-dependent pathway. Infect Immun 64:1906–1912
Boguniewicz M, Sampson H, Leung SB, Harbeck R, Leung DY (2001) Effects of cefuroxime axetil on Staphylococcus aureus colonization and superantigen production in atopic dermatitis. J Allergy Clin Immunol 108:651–652
Beck LA, Thaci D, Hamilton JD, Graham NM, Bieber T, Rocklin R, Ming JE, Ren H, Kao R, Simpson E, Ardeleanu M, Weinstein SP, Pirozzi G, Guttman-Yassky E, Suarez-Farinas M, Hager MD, Stahl N, Yancopoulos GD, Radin AR (2014) Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N Engl J Med 371:130–139
Heil PM, Maurer D, Klein B, Hultsch T, Stingl G (2010) Omalizumab therapy in atopic dermatitis: depletion of IgE does not improve the clinical course - a randomized, placebo-controlled and double blind pilot study. J Dtsch Dermatol Ges 8:990–998
Gittler JK, Shemer A, Suarez-Farinas M, Fuentes-Duculan J, Gulewicz KJ, Wang CQ, Mitsui H, Cardinale I, de Guzman SC, Krueger JG, Guttman-Yassky E (2012) Progressive activation of T(H)2/T(H)22 cytokines and selective epidermal proteins characterizes acute and chronic atopic dermatitis. J Allergy Clin Immunol 130:1344–1354
Dillon SR, Sprecher C, Hammond A, Bilsborough J, Rosenfeld-Franklin M, Presnell SR, Haugen HS, Maurer M, Harder B, Johnston J, Bort S, Mudri S, Kuijper JL, Bukowski T, Shea P, Dong DL, Dasovich M, Grant FJ, Lockwood L, Levin SD, LeCiel C, Waggie K, Day H, Topouzis S, Kramer J, Kuestner R, Chen Z, Foster D, Parrish-Novak J, Gross JA (2004) Interleukin 31, a cytokine produced by activated T cells, induces dermatitis in mice. Nat Immunol 5:752–760
Werfel T, Biedermann T (2015) Current novel approaches in systemic therapy of atopic dermatitis: specific inhibition of cutaneous Th2 polarized inflammation and itch. Curr Opin Allergy Clin Immunol 15:446–452
Fedenko ES, Elisyutina OG, Filimonova TM, Boldyreva MN, Burmenskaya OV, Rebrova OY, Yarilin AA, Khaitov RM (2011) Cytokine gene expression in the skin and peripheral blood of atopic dermatitis patients and healthy individuals. Self Nonself 2:120–124
Esparza-Gordillo J, Schaarschmidt H, Liang L, Cookson W, Bauerfeind A, Lee-Kirsch MA, Nemat K, Henderson J, Paternoster L, Harper JI, Mangold E, Nothen MM, Ruschendorf F, Kerscher T, Marenholz I, Matanovic A, Lau S, Keil T, Bauer CP, Kurek M, Ciechanowicz A, Macek M, Franke A, Kabesch M, Hubner N, Abecasis G, Weidinger S, Moffatt M, Lee YA (2013) A functional IL-6 receptor (IL6R) variant is a risk factor for persistent atopic dermatitis. J Allergy Clin Immunol 132:371–377
Navarini AA, French LE, Hofbauer GF (2011) Interrupting IL-6-receptor signaling improves atopic dermatitis but associates with bacterial superinfection. J Allergy Clin Immunol 128:1128–1130
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is a contribution to the Special Issue on Immunodermatology – Guest Editors: Lars French and Alexander Navarini
Rights and permissions
About this article

Cite this article
Skabytska, Y., Kaesler, S., Volz, T. et al. The role of innate immune signaling in the pathogenesis of atopic dermatitis and consequences for treatments. Semin Immunopathol 38, 29–43 (2016). https://doi.org/10.1007/s00281-015-0544-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-015-0544-y