
Abstract
Purpose
This study aimed to develop a population pharmacokinetic (PPK) model to investigate the impact of GSTA1, GSTP1, and GSTM1 genotypes on busulfan pharmacokinetic (PK) variability in Chinese adult patients.
Methods
Forty-three and 19 adult patients who underwent hematopoietic stem cell transplantation (HSCT) were enrolled for modeling group and validation group, respectively. All patients received twice-daily intravenous busulfan as part of conditioning regimen before HSCT. The PPK model was developed by nonlinear mixed-effect modeling. Covariates investigated were age, sex, actual body weight, body surface area, diagnoses, hepatic function markers, GST genotypes and conditioning regimen.
Results
A total of 488 busulfan concentrations from 43 patients were obtained for the PPK model. The PK of intravenous busulfan was described by one-compartment model with first-order elimination with estimated clearance (CL) of 14.2 L/h and volume of distribution of 64.1 L. Inclusion of GSTA1 genotype as a covariate accounted for 1.1% of the inter-individual variability of busulfan CL (from 17.8% in the basic model to 16.7% in the final model). The accuracy and applicability of the final model were externally validated in the independent group. The difference of busulfan PK between Chinese patients and Caucasian patients existed because of the rarity of haplotype *B in Chinese population.
Conclusions
Although the GSTA1 genotype-based PPK model of intravenous busulfan was successfully developed and externally validated, the GSTA1 genotype was not considered to be clinically relevant to busulfan CL. We did not suggest the guidance of GSTA1 genotype on initial busulfan dose in Chinese adult patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
High-dose busulfan is commonly used as the cornerstone of chemotherapy regimen prior to hematopoietic stem cell transplantation (HSCT) [1]. Targeted busulfan is necessary because of the definite dosage–response relationship and narrow therapeutic index: subtherapeutic busulfan systemic exposure, expressed as area under the curve (AUC) < 900 μmol × min/L, results in relapse or graft failure, while supratherapeutic AUC > 1500 μmol × min/L, is associated with fatal toxicities, such as hepatic veno-occlusive disease (VOD) [2, 3]. Since very few busulfan AUCs achieve the therapeutic range after their initial standard doses [4, 5], an individualized busulfan initial dose is warranted for the targeted AUCs to improve clinical efficiency.
Noteworthily, studies [6,7,8] showed that glutathione S-transferase (GST) genotypes played a positive role in the individualization of busulfan dose for optimal clinical outcomes. GSTs, which are the enzymatic superfamily of detoxification, catalyze the conjugation of busulfan with glutathione (GSH) in liver [9]. Of all, GSTA1 is the predominant GST isoenzyme of busulfan metabolism, with minor contributions by GSTM1 and GSTP1 in vitro studies [10]. GST polymorphisms, gene deletion or single nucleotide polymorphism (SNP), cause a decreased metabolic activity of busulfan in patients with genetic variants [11]. In pediatric patients, Nava et al. [6] showed that GSTA1 diplotypes linked to poor busulfan metabolism and associated with AUCs within the toxic range. Consistently, GSTA1 diplotypes influenced the prediction errors of the weight- and age-based methods which are used to calculate the initial doses of busulfan. However, busulfan initial dosing guided by GST genotypes is not recommended in clinical practice due to inconsistent effect of the genetic polymorphism on the pharmacokinetic, efficacy, or toxicity of busulfan [12]. Studies about the influence of GST polymorphisms on busulfan pharmacokinetic (PK) are scant in Asian patients. The PK study of Yin et al. [8] demonstrated that GSTA1 and GSTP1 polymorphisms are significantly associated with busulfan exposure in Chinese adult patients. In addition, both Yin et al. [8] and our previous study [13] showed low busulfan exposure after the first dose in the Chinese population. We found that about half of the first AUCs were below the therapeutic range. These are a necessity to personalize the initial dose of busulfan to improve clinical outcome in Chinese patients.
This study aimed to quantify the impact of GSTA1, GSTP1, and GSTM1 genotypes on busulfan PK variability by population pharmacokinetic (PPK) analysis, and to clarify the role of genetic factors on the individualization of busulfan initial dose in Chinese adult patients undergoing HSCT.
Materials and methods
Patients
Forty-three adult patients who underwent HSCT for malignant diseases were enrolled in the modeling group at the department of Blood Marrow transplantation of Ruijin Hospital, Shanghai, China, from May 2011 to October 2014. Nineteen adult patients were enrolled in the validation group from March 2017 to August 2017. All procedures performed in this study were approved by the Ruijin Hospital Research Ethics Committee and in accordance with the 1964 Helsinki Declaration. Before the study, informed consent was obtained from the enrolled patients.
All patients received intravenous busulfan (Busulfex; Kyowa Hakko Kirin Co., Ltd, Tokyo, Japan) at 1.6 mg/kg twice daily. Busulfan was given as part of the BUCY, BUFLU, and CBV regimens. For BUCY regimen, busulfan was given over 2 h for 4 days on days − 8 to − 5 followed by intravenous cyclophosphamide (CY) 60 mg/kg once daily on days − 3 and − 2. For BUFLU regimen, intravenous fludarabine (FLU) 30 mg/m2 was given once daily with busulfan on day − 6 and − 3. The CBV regimen in validation group included 3 days of busulfan from day − 8 to − 6, VP16 at 400 mg/m2 daily on day − 5 and − 4 and CY 50 mg/kg daily on day − 3 and − 2. Then, gemcitabine was given at 75 mg/m2 on day − 9 and at 10 mg/m2 on day − 5, respectively.
Blood sampling and analysis
Blood samples were collected before busulfan infusion and at 0.5 h, 1 h, 1.5 h, 2 h, 2.5 h, 3 h, 4 h, 5 h, 6 h, 8 h, and 10 h after starting the first dosing. All the blood samples were drawn from a peripheral vein in the arm opposite to the central line where busulfan was infused. 3 mL of whole blood sample was collected in EDTA anticoagulant glass tubes. 1 ml of whole blood for DNA extraction was taken from the samples obtained before busulfan infusion. The residual whole blood was centrifuged at 3000 rpm for 10 min at 4 °C for plasma separation. Busulfan plasma concentrations were analyzed using a validated analytical method [13] by liquid chromatography–electrospray–tandem mass spectrometry. High-performance liquid chromatography separations were performed on an Agilent Eclipse XDB-C18 analytical column (100 mm × 2.1 mm, 3.5 μm) with a column temperature of 40 °C. The plasma was eluted using a mobile phase composed of 2 mmol/L ammonium formate solution (0.1% formic acid)-acetonitrile (60:40, V/V) at a flow rate of 0.2 mL/min. Electrospray ionization mass spectrometry was performed in multiple reaction monitoring mode with the target ions m/z 264.2 → 151.1 (busulfan) and m/z 278.3 → 69.2. The linearity range of busulfan standard curve was 25–2500 ng/mL (R = 0.999). The intraday and inter-day accuracies of busulfan quality control samples were from 96.4 to 109.2% and from 99.2 to 111.2%, respectively. The extraction recovery of busulfan was from 102.0 to 113.7%.
DNA extraction and glutathione S-transferases genotyping
The whole blood samples for DNA extraction were obtained before HSCT. DNA was extracted from peripheral blood lymphocytes by TIANamp Blood DNA Kit (Tiangen Biotech Co., Ltd, Beijing, China). The following genetic variants were determined: GSTA1 -69 C/T (rs3957357), GSTP1 313A/G (rs1695), GSTM1 (null allele). The GSTA1 haplotype (GSTA1*A and GSTA1*B) was determined by the -69 C/T variation in the prompter region of GSTA1. This SNP was previously shown in complete linkage disequilibrium with -631T/G, -567T/G, and -52G/A [14]. The SNPs in the GSTA1 and GSTP1 genes were genotyped by SNaPshot assay according to the manufacturer’s protocols (ABI SNaPshot Multiplex kit, CA, USA). The promoter region of the GSTA1 gene was amplified with the forward primer GSTA1-F (5′-GCTCGACAACTGAATTCCAGGTC-3′) and the reverse primer GSTA1-R (5′-CCCTAGTCTTTGCACCCAACTCAT-3′). The promoter region of the GSTP1 gene was amplified with the forward primer GSTP1-F (5′-CTCATCCTTCCACGCACATCCT-3′) and the reverse primer GSTP1-R (5′-TTTCTTTGTTCAGCCCCCAGTG-3′). Analysis was carried out using Genemapper software (version 4.1; Life Technologies). The GSTM1 gene was PCR amplified (215 bp) together with albumin gene (268 bp) as an internal control [15]. The primer pairs were as follows: GSTM1-F, 5′-GAA CTC CCT GAA AAG CTA AAG C-3′ and GSTM1-R, 5′-GTT GGG CTC AAA TAT ACG GTG G-3′, which yield a 215 bp fragment and β-actin-F, 5′- CAA CTTCAT CCA CGT TCA CC-3′ and β-actin-R, 5′- GAA GAG CCAAGG ACA GGT AC-3′, which yield a 268 bp fragment. PCR products were analyzed on a 2% agarose gel.
Development of a population pharmacokinetic model
A PPK model was developed by nonlinear mixed-effect modeling using Phoenix NLME software, Version 1.4 (Pharsight, A Certara Company, USA), according to the best practices and guidance [16]. The first-order conditional estimation method with extended least squares method (FOCE ELS) was adopted in the whole process to estimate PK parameters and their variability.
Structure model
Plasma concentration data were natural logarithm transformed before PPK modeling. Initially, a one-compartment model with first-order elimination was used as the base model, according to our previous study [13]. The model was parameterized in terms of total body clearance (CL) and volume of distribution (Vd). The inter-individual variability of the PK parameters was estimated using an exponential error model, where Pi represents the individual parameter estimate, P is the typical parameter estimate, and ηi is the unexplained inter-individual variability, which is assumed to follow a normal distribution with a mean of 0 and variance ω2:
A residual variability (additive, proportional, exponential and mixed random intra-individual residual error) model was explored to account for the intra-individual variability according to the log-likelihood (LL) difference between models [the improvement of objective function value (OFV)].
Covariate analysis
The effects of age, sex, actual body weight, body surface area (BSA), diagnoses, hepatic function markers [alanine transaminase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALP), total bilirubin (TBI)], GST genotypes, and conditioning regimen on pharmacokinetic parameters were investigated as potential covariates . The effects of continuous covariates were modeled in a linear or nonlinear relationship using the following equations:
where tv (P) is the typical value of P, Pi is the value of P for individual, and θ is the estimated effect of the covariate on P.
For categorical covariates, the effect on P was modeled using the following equation:
where indicator represents the binary covariate being assigned to 0 or 1.
The stepwise covariate modeling and likelihood ratio test were used to test the effect of each variable. The selection of covariates was initially selected by data visualization and biological plausibility. Further screening of potential covariates was performed by forward addition (P < 0.05) and backward elimination (P < 0.01) based on the changes in OFV: any decreases in OFV by more 3.84 and 6.63 (1 degree of freedom) denote an improved fit at P < 0.05 and P < 0.01, respectively. An additional criterion for covariate retention was reduction in the inter-individual variability and improvement in the parameter estimate precision. Clinical significance was also considered for retention of a potential covariate.
Model validation
Accuracy and stability of prediction of the final covariate model were validated both internally and externally.
Graphical inspection of the final model adopted goodness-of-fit plots, including observed concentrations (OBS) versus population predicted concentrations (PRED), OBS versus individual predicted concentrations (IPRED), conditional weighted residuals (CWRES) versus PRED, and CWRES versus time after the first dose. The CWRES results were summarized graphically using the quantile–quantile (Q–Q) plot and the histogram. Bootstrap analysis was performed for internal validation of model. 1000 resamples from the original data were performed. Mean values and 95% confidence interval (CI) of bootstrap parameters were compared with estimates of the final model. The final model was also evaluated graphically and statistically by visual predictive checks (VPC). The final model parameters were used to simulate a data set for 200 replicates and the 5th and 95th CIs of simulated values were processed for VPC.
To validate the developed model externally, the model was further evaluated in an independent group of adult patients undergoing HSCT. The individual PK parameters were predicted by Bayesian estimation (MAXEVAL = 0 in the estimation step, where MAXEVAL is the maximum number of model evaluations that can be used) with Phoenix NLME using the parameters of the final model. The predictive performance was evaluated by calculating the prediction error (PE) and absolute prediction error (APE) using the following equations:
Statistical analysis
The continuous variables were represented as mean with standard deviation or 95% CI. Statistical significance of the difference between groups was calculated by Chi square test. P values < 0.05 were considered significant.
Results
Patients’ characteristics and GST genetic frequencies
The mean age of patients was 31.5 years, and 74.4% of them were male. The mean weight was 64.3 kg in the modeling group (n = 43). There were 19 enrolled patients in the validation group. The age, sex, and weight were not significantly different (P > 0.05) between the validation group and modeling group. Patients’ characteristics are presented in Table 1. Most of the enrolled patients received allogenous HSCT from a human leukocyte antigen-matched donor using peripheral blood stem cells for their hematological malignancies. Five patients in the validation group received autologous HSCT.
The GST genetic frequencies are shown in Table 2. 10 of 43 (23.2%) patients were heterozygous genotype of GSTA1 (GSTA1*A*B), and only 1 patient (2.4%) was homozygous GSTA1 (GSTA1*B*B). Genetic frequency of GST A1*B haplotype was 14.0%. Both GSTA1 and GSTP1 genetic frequencies of patients in modeling group were in Hardy–Weinberg equilibrium (HWE). 2 of the 19 patients in validation group were heterozygous and homozygous GSTA1*B, respectively. The GSTA1 genetic frequency was not in HWE.
Structure model
A total of 488 busulfan concentrations from 43 patients were obtained for model development. A one-compartment model with first-order elimination best described the PK of intravenous busulfan. The intra-individual variability of the plasma concentration was estimated using the proportional error model finally. The estimated CL and Vd of basic model were 14.2L/h and 64.1L, respectively.
Covariate analysis
Age, actual body weight, body surface area, and hepatic function markers (ALT, AST, ALP, TBI) were tested as continuous covariates. Sex, diagnoses, GST genotypes, and conditioning regimen were tested as categorical covariates, while non-Hodgkin’s lymphoma, myelodysplastic syndromes, and hybrid acute leukemia patients were analyzed together with acute lymphoblastic leukemia patients relative to the patients with acute myelocytic leukemia and chronic myelocytic leukemia due to their small number. The patients with GSTA1*B*B or GSTP1*G*G were excluded to avoid statistical deviation.
GSTA1 genotype was a covariate on busulfan CL in a statistically significant manner. OFV decreased by 15.64 from 4942.75 (basic model) to 4927.11 (final model), adding the covariate of GSTA1 genotype. The individualized CL was estimated by the following equation:
where GGSTA1= 1 for GSTA1*A*B, 0 for GSTA1*A*A. Population-estimated CL of the final model was 15.0 L/h for a typical patient with GSTA1*A*A wild type. The inter-individual variability of CL declined from 17.8 to 16.8%, after inclusion of GSTA1 genotype as a covariate. The estimated PPK parameters and bootstrap results are listed in Table 3.
Model evaluation
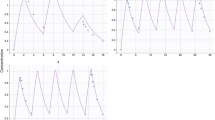
Goodness-of-fit plots of the final model in Fig. 1 showed that the PRED and IPRED were in reasonable agreement with OBS. Distribution of CWRES versus predicted concentration and time after dose were shown to be close to zero and uniformly distributed within the range (− 2 to 2) in the final model. Additionally, the CWRES distribution and histogram indicated that the assumption of normal distribution of the differences between PRED and OBS was acceptable, as shown in Figure S1. The robustness of the final model was internally validated by the bootstrap results. The population-estimated parameters were similar to the simulation values and within 95% CI from bootstrap analysis, as shown in Table 3. The VPC in Fig. 2 showed that the observed values were almost all positioned within the 5th and 95th CIs of simulated values. The results demonstrated the accuracy of the final model.

Goodness-of-fit plots of the final model. a Observed concentrations versus population predicted concentrations; b observed concentrations versus individual predicted concentrations; c conditional weighted residuals versus population predicted concentrations; d conditional weighted residuals versus time after the first dose. OBS means observed concentration; PRED means population predicted concentrations; IPRED means individual predicted (IPRED) concentrations; CWRES means conditional weighted residuals

The visual predictive check (VPC, n = 1000) of final model. The dots represent observed concentrations. The lines represent observed concentrations and the shadow represents the 5th and 95th percentiles of the simulated values
Busulfan concentrations from 19 patients in the external validation group were used to validate the final GSTA1 genotype-based model. The predictive performance of the final PPK model was evaluated by the comparison between the observed values (CLobs and AUCobs) and the simulated values (CLsim and AUCsim),as shown in Fig. 3. The Bayesian simulated PK parameters highly correlated with the observed data (r2 = 0.98 for the CLobs with CLsim and r2 = 0.84 for the AUCobs with AUCsim). The mean PE (MPE, %) and mean APE (MAPE, %) of the CLsim were 4.04% (95%CI: 4.02% to 4.06%) and 4.39% (95% CI: 4.57% to 4.51%). The MPE and MAPE of the AUCsim were − 2.55% (95%CI: − 2.58% to − 2.52%) and 4.93% (95%CI: 4.92% to 4.94%).

GSTA1 genotype-based simulations of CL and AUC in validation group (n = 19). a The box and whisker plot of observed CL and simulated CL. b The box and whisker plot of observed AUC and simulated AUC
Discussion
In this study, we developed the PPK model of intravenous busulfan, incorporating GST A1 genotype, and externally validated the accuracy and stability of the model in Chinese adult patients undergoing HSCT. The estimated CL of intravenous busulfan was 14.2 L/h (3.64 mL/min per kg) and consistent with the CL of 4.02 mL/min per kg in another study [8] of Chinese adult patients, higher than 1.9 mL/min per kg and 3.34 mL/min per kg in Caucasian patients [17, 18]. Inclusion of GSTA1 genotype as a covariate accounted for 1.1% of the inter-individual variability of busulfan CL in the final model.
The fast metabolism of busulfan might relate with the ethnic difference of GSTA1 genotype between Chinese patients and Caucasian patients. The patients with GST A1*B haplotype were the poorer metabolizers with lower CL of busulfan compared to those with wild type. The studies of Kusama et al. [19] and Kim et al. [20] showed significant impact of GST A1 genotype on busulfan CL. Genetic frequency of GST A1*B haplotype was 14.0% with 1 patient of homozygous excluded to avoid statistic deviation in our study. The distribution was consistent with the rarity of haplotype *B in the Asian population (42.9% versus 13.9% in the Caucasians and Asians) [21]. GSTA1-69C/T (rs3957357), with -631T/G, -567 T/G, and -52 G/A, is located in the promoter region of GSTA1 gene. GSTA1-69C/T can adequately identify GSTA1*B haplotype which showed significant decline of GSTA1 protein expression in vitro [22]. Ansari et al. [23] further distinguished *B 1b, *B 1a and *B 2 haplotypes by − 513 (rs11964968), − 1142 (rs58912740). Then, Nava et al. [6] classified *A*B and *B*B diplotypes into normal and poor metabolizers of busulfan. Based on this accurate GSTA1-genotype classification, the PPK model excellently predicted the initial busulfan doses and achieved the target AUC in 85.2% of the pediatric patients (95%CI 78.7–91.7%) [7]. However, this metabolic classification of busulfan was not appropriate for Chinese patients due to the low genetic frequencies of above GSTA1*B variants in the Asian population [21].
PPK analysis has shown an advantage of quantifying the effects of covariates on PK variability to assess the clinical relevance of those effects [24]. Our results showed that GSTA1 genotype explained a tiny part of inter-individual variability from 17.8% of the basic model to 16.7% of the final model. The low genetic frequency of GST A1*B may lead to minimal influence of GSTA1 genotype and limited variability of busulfan CL between Chinese adult patients. Additionally, that also might attribute to different GSTA1 enzymatic activities in patients at different age. Recent studies [6, 25, 27] reported that GSTA1 genotype played a pivotal role in prediction of initial busulfan doses in pediatric patients. Meanwhile, Abbasi et al. [28] showed that GSTA1 did not influence busulfan CL obviously in both oral and intravenous administration in adult patients. A higher impact of GSTA1 genotype on busulfan CL in pediatric patients could be explained by age-specific GSTA1 enzymatic activity [29, 30]. For GSTA1, one of drug-processing genes in liver, the highest fold increase in the mRNA expression was induced by the prototypical ligands of xenobiotic-sensing transcription factors during adolescent age. Li et al. [29] highlighted that the drug-processing genes regulation was critical to predict drug PK and to decrease drug toxicity in pediatric patients. This in vitro study demonstrated the importance of GSTA1 genotype to recommend the initial busulfan dose and to avoid toxicity in children.
We did not find that the other factors, such as GSTP1, GSTM1, conditioning regimen, body surface area (BSA), and weight, showed a significant impact on busulfan PK. Pharmacogenomics data about busulfan PK in different populations are summarized in Table 4 [6,7,8, 19, 20, 25,26,27,28, 31,32,33,34,35,36,37,38,39,40,41,42,43,44,45]. Of all, GSTM1 null genotype and GSTP1*G diplotype were commonly reported to have a significant association with busulfan CL [20, 34]. However, Ten Brink et al. [39] did not find an association between the two GST genotypes and busulfan CL. This may be related to their minor contribution to busulfan metabolism. For conditioning regimen, Yeh et al. [46] reported a greater interdose variability of busulfan CL in the targeted busulfan–FLU regimen compared to the targeted busulfan–CY regimen (P = 0.0016). The impact of FLU on busulfan CL was not found in the study of Perkins et al. [47]. PPK analysis of Wu et al. [48] found that BSA significantly influenced the CL and Vd of busulfan in Chinese patients (P < 0.001, n = 53). The PPK or PK studies of busulfan in Asian patients were scanty and limited by small sample size and single center. A large-scale and multicenter clinical trial was needed to develop the busulfan PPK model to validate the influencing factors on busulfan PK and to be applicable for more Asian patients.
The GSTA1 genotype-based PPK model of intravenous busulfan was successfully developed and externally validated in Chinese adult patients. However, the influence of GSTA1 genotype on busulfan CL is tiny and unlikely to be clinically relevant. We did not suggest the guidance of GSTA1 genotype on initial busulfan dose in Chinese adult patients undergoing HSCT.
References
Ciurea SO, Andersson BS (2009) Busulfan in hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 15(5):523–536
Copelan EA, Bechtel TP, Avalos BR et al (2001) Busulfan levels are influenced by prior treatment and are associated with hepatic veno-occlusive disease and early mortality but not with delayed complications following marrow transplantation. Bone Marrow Transplant 27:1121–1124
Bolinger AM, Zangwill AB, Slattery JT et al (2000) An evaluation of engraftment, toxicity and busulfan concentration in children receiving bone marrow transplantation for leukemia or genetic disease. Bone Marrow Transplant 25:925–930
Lee JW, Kang HJ, Lee SH et al (2012) Highly variable pharmacokinetics of once-daily intravenous busulfan when combined with fludarabine in pediatric patients: phase I clinical study for determination of optimal once-daily busulfan dose using pharmacokinetic modeling. Biol Blood Marrow Transplant. 18(6):944–950
Paci A, Vassal G, Moshous D et al (2012) Pharmacokinetic behavior and appraisal of intravenous busulfan dosing in infants and older children. Ther Drug Monit 34(2):198–208
Nava T, Rezgui MA, Uppugunduri CRS et al (2017) GSTA1 genetic variants and conditioning regimen: missing key factors in dosing guidelines of busulfan in pediatric hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 23(11):1918–1924
Nava T, Kassir N, Rezgui MA et al (2018) Incorporation of GSTA1 genetic variations into a population pharmacokinetic model for IV busulfan in paediatric hematopoietic stem cell transplantation. Br J Clin Pharmacol 84(7):1494–1504
Yin J, Xiao Y, Zheng H, Zhang YC (2015) Once-daily i.v. BU-based conditioning regimen before allogeneic hematopoietic SCT: a study of influence of GST gene polymorphisms on BU pharmacokinetics and clinical outcomes in Chinese patients. Bone Marrow Transplant. 50(5):696–705
Gibbs JP, Czerwinski M, Slattery JT (1996) Busulfan-glutathione conjugation catalyzed by human liver cytosolic glutathione S-transferases. Cancer Res 56(16):3678–3681
Czerwinski M, Gibbs JP, Slattery JT (1996) Busulfan conjugation by glutathione S-transferases alpha, mu, and pi. Drug Metab Dispos 24:1015–1019
Hayes JD, Strange RC (2000) Glutathione S-transferase polymorphisms and their biological consequences. Pharmacology 61(3):154–166
Palmer J, Mccune JS, Perales MA et al (2016) Personalizing busulfan-based conditioning: considerations from the American Society for Blood and Marrow Transplantation Practice Guidelines Committee. Biol Blood Marrow Transplant 22(11):1915–1925
Huang J, Li Z, Liang W et al (2019) Accurate prediction of initial busulfan exposure using a test dose with 2- and 6-hour blood sampling in adult patients receiving a twice-daily intravenous busulfan-based conditioning Regimen. J Clin Pharmacol 59(5):638–645
Matsuno K, Kubota T, Matsukura Y, Ishikawa H, Iga T (2004) Genetic analysis of glutathione S-transferase A1 and T1 polymorphisms in a Japanese population. Clin Chem Lab Med 42:560–562
Arand M, Muhlbauer R, Hengstler J et al (1996) A multiplex polymerase chain reaction protocol for the simultaneous analysis of the glutathione S-transferase GSTM1 and GSTT1 polymorphisms. Anal Biochem 236:184–186
Byon W, Smith MK, Chan P et al (2013) Establishing best practices and guidance in population modeling: an experience with an internal population pharmacokinetic analysis guidance. CPT Pharmacometr Syst Pharmacol 3(2):e51
Fernandez HF, Tran HT, Albrecht F et al (2002) Evaluation of safety and pharmacokinetics of administering intravenous busulfan in a twice-daily or daily schedule to patients with advanced hematologic malignant disease undergoing stem cell transplantation. Biol Blood Marrow Transplant 8(9):486–492
Mamlouk K, Saracino G, Berryman RB, Fay JW, Pineiro LA, Vance EA et al (2005) Modification of the Bu/Cy myeloablative regimen using daily parenteral busulfan: reduced toxicity without the need for pharmacokinetic monitoring. Bone Marrow Transplant 35:747–754
Kusama M, Kubota T, Matsukura Y et al (2006) Influence of glutathione S-transferase A1 polymorphism on the pharmacokinetics of busulfan. Clin Chim Acta 368(1–2):93–98
Kim SD, Lee JH, Hur EH et al (2011) Influence of GST gene polymorphisms on the clearance of intravenous busulfan in adult patients undergoing hematopoietic cell transplantation. Biol Blood Marrow Transplant 17(8):1222–1230
Coles BF, Morel F, Rauch C et al (2001) Effect of polymorphism in the human glutathione s-Transferase A1 promoter on hepatic GSTA1 and GSTA2 expression. Pharmacogenet Genom. 11(8):663–669
Ansari M, Lauzon-Joset JF, Vachon MF et al (2010) Influence of GST gene polymorphisms on busulfan pharmacokinetics in children. Bone Marrow Transplant. 45:261–267
Sun H, Fadiran EO, Jones CD et al (1999) Population pharmacokinetics. A regulatory perspective. Clin Pharmacokinet. 37(1):41–58
Ansari M, Curtis PH, Uppugunduri CRS et al (2017) GSTA1 diplotypes affect busulfan clearance and toxicity in children undergoing allogeneic hematopoietic stem cell transplantation: a multicenter study. Oncotarget. 8(53):90852–90867
Ansari M, Huezo-Diaz P, Rezgui MA et al (2016) Influence of glutathione S-transferase gene polymorphisms on busulfan pharmacokinetics and outcome of hematopoietic stem-cell transplantation in thalassemia pediatric patients. Bone Marrow Transplant 51(3):377–383
Johnson L, Orchard PJ, Baker KS et al (2008) Glutathione S-transferase A1 genetic variants reduce busulfan clearance in children undergoing hematopoietic cell transplantation. J Clin Pharmacol 48(9):1052–1062
Abbasi N, Vadnais B, Knutson JA et al (2011) Pharmacogenetics of intravenous and oral busulfan in hematopoietic cell transplant recipients. J Clin Pharmacol 51(10):1429–1438
Li CY, Renaud HJ, Klaassen CD et al (2016) Age-specific regulation of drug-processing genes in mouse liver by ligands of xenobiotic-sensing transcription factors. Drug Metab Dispos Biol Fate Chem 44(7):1038–1049
Lu H, Gunewardena S, Cui JY, Yoo B, Zhong XB, Klaassen CD (2013) RNA-sequencing quantification of hepatic ontogeny and tissue distribution of mRNAs of phase II enzymes in mice. Drug Metab Dispos 41:844–857
Terakura S, Onizuka M, Fukumoto M et al (2019) Analysis of glutathione S-transferase and cytochrome P450 gene polymorphism in recipients of dose-adjusted busulfan-cyclophosphamide conditioning. Int J Hematol. https://doi.org/10.1007/s12185-019-02741-8
Michaud V, Tran M, Pronovost B et al (2019) Impact of GSTA1 polymorphisms on busulfan oral clearance in adult patients undergoing hematopoietic stem cell transplantation. Pharmaceutics. https://doi.org/10.3390/pharmaceutics11090440
Nishikawa T, Yamaguchi H, Ikawa K et al (2019) Influence of GST polymorphisms on busulfan pharmacokinetics in Japanese children. Pediatr Int 61(6):558–565
Choi B, Kim MG, Han N et al (2015) Population pharmacokinetics and pharmacodynamics of busulfan with GSTA1 polymorphisms in patients undergoing allogeneic hematopoietic stem cell transplantation. Pharmacogenomics. 16(14):1585–1594
Ten Brink MH, Swen JJ, Böhringer S et al (2013) Exploratory analysis of 1936 SNPs in ADME genes for association with busulfan clearance in adult hematopoietic stem cell recipients. Pharmacogenet Genom 23(12):675–683
Ten Brink MH, Van BT, Swen JJ et al (2013) Effect of genetic variants GSTA1 and CYP39A1 and age on busulfan clearance in pediatric patients undergoing hematopoietic stem cell transplantation. Pharmacogenomics. 14(14):1683–1690
Uppugunduri CRS, Rezgui MA, Diaz PH et al (2014) The association of cytochrome P450 genetic polymorphisms with sulfolane formation and the efficacy of a busulfan-based conditioning regimen in pediatric patients undergoing hematopoietic stem cell transplantation. Pharmacogenomics J. 14(3):263–271
Bonifazi F, Storci G, Bandini G et al (2014) Glutathione transferase-A2 S112T polymorphism predicts survival, transplant-related mortality, busulfan and bilirubin blood levels after allogeneic stem cell transplantation. Haematologica 99(1):172–179
Ten Brink MH, Wessels JA, Hartigh JD et al (2012) Effect of genetic polymorphisms in genes encoding GST isoenzymes on BU pharmacokinetics in adult patients undergoing hematopoietic SCT. Bone Marrow Transplant 47(2):190–195
Elhasid R, Krivoy N, Rowe JM et al (2010) Influence of glutathione S-transferase A1, P1, M1, T1 polymorphisms on oral busulfan pharmacokinetics in children with congenital hemoglobinopathies undergoing hematopoietic stem cell transplantation. Pediatr Blood Cancer 55(6):1172–1179
Kim MG, Kwak A, Choi B et al (2019) Effect of glutathione S-transferase genetic polymorphisms on busulfan pharmacokinetics and veno-occlusive disease in hematopoietic stem cell transplantation: a meta-analysis. Basic Clin Pharmacol Toxicol 124(6):691–703
Srivastava A, Poonkuzhali B, Shaji RV et al (2004) Glutathione S-transferase M1 polymorphism: a risk factor for hepatic venoocclusive disease in bone marrow transplantation. Blood 104(5):1574–1577
Bremer S, Fløisand Y, Brinch L et al (2015) Glutathione transferase gene variants influence busulfan pharmacokinetics and outcome after myeloablative conditioning. Ther Drug Monit 37(4):493–500
Ansari M, Rezgui MA, Théoret Y et al (2013) Glutathione S-transferase gene variations influence BU pharmacokinetics and outcome of hematopoietic SCT in pediatric patients. Bone Marrow Transplant 48(7):939–946
Zwaveling J, Press RR, Bredius RG et al (2008) Glutathione S-transferase polymorphisms are not associated with population pharmacokinetic parameters of busulfan in pediatric patients. Ther Drug Monit 30(4):504–510
Yeh RF, Pawlikowski MA, Blough DK et al (2012) Accurate targeting of daily intravenous busulfan with 8-hour blood sampling in hospitalized adult hematopoietic cell transplant recipients. Biol Blood Marrow Transplant 18(2):265–272
Perkins JB, Kim J, Anasetti C et al (2012) Maximally tolerated busulfan systemic exposure in combination with fludarabine as conditioning before allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant 18(7):1099–1107
Wu X, Xie H, Lin W et al (2017) Population pharmacokinetics analysis of intravenous busulfan in Chinese patients undergoing hematopoietic stem cell transplantation. Clin Exp Pharmacol Physiol 44(5):529–538
Acknowledgments
The authors thank the staff of the Department of Bone Marrow Transplantation, Ruijin Hospital, Shanghai Jiaotong University School of Medicine, for their collaboration and the use of their facilities.
Funding
This work was funded by the National Natural Science Foundation in China (Grant No. 81503137).
Author information
Authors and Affiliations
Contributions
Jiong Hu and Wanhua Yang were in charge of the entire project and reviewed the manuscript. Yidan Sun designed the study and wrote the manuscript. Jingjing Huang performed the study and wrote the manuscript. Chenxia Hao, Ziwei Li, and Wu Liang analyzed the data and interpreted the results. Weixia Zhang and Bing Chen provided the reagents and materials.
Corresponding author
Ethics declarations
Conflict of interest
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical approval
The study protocol was approved by the Ruijin Hospital Research Ethics Committee.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
280_2019_4001_MOESM1_ESM.tif
Supplementary material 1 Figure S1 The histogram and the quantile–quantile (Q–Q) plot of conditional weighted residuals (CWRES) (TIFF 341 kb)
Rights and permissions
About this article

Cite this article
Sun, Y., Huang, J., Hao, C. et al. Population pharmacokinetic analysis of intravenous busulfan: GSTA1 genotype is not a predictive factor of initial dose in Chinese adult patients undergoing hematopoietic stem cell transplantation. Cancer Chemother Pharmacol 85, 293–308 (2020). https://doi.org/10.1007/s00280-019-04001-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-019-04001-2