
Abstract
Here, we reviewed clinical-morphological data and investigated mutational profiles by NGS in a single-center series of 58 consecutive MPN-SVT patients admitted to our hospital between January 1979 and November 2021. We identified 15.5% of PV, 13.8% of ET, 34.5% of PMF, 8.6% of SMF and 27.6% of MPN-U. Most cases (84.5%) carried JAK2V617F mutation, while seven patients were characterized by other molecular markers, namely MPL in four and CALR mutations in three cases. NGS was performed in 54 (93.1%) cases: the most frequent additional mutations were found in TET2 (27.8%) and DNMT3A (16.7%) genes, whereas 25 (46.3%) patients had no additional mutation. Cases with JAK2V617F homozygosity had a higher median number of additional mutations than those with low allele burden. More importantly, all cases of leukemic evolution were characterized by a higher median number of co-mutations, and a co-mutational pattern of high-risk lesions, such as truncating mutations of ASXL1, bi-allelic TP53 loss, and CSMD1 mutations. Nevertheless, no difference was found between cases with and without additional somatic mutations regarding fibrotic progression, SVT recurrence, other thrombo-hemorrhagic complications, or death. After a median follow-up of 7.1 years, ten deaths were recorded; fibrotic progression/leukemic evolution was ascertained in one (1.7%) and six (10.3%) patients, respectively, while 22 (37.9%) patients suffered from recurrent thrombosis. In conclusion, our data underline the importance of using NGS analysis in the management of MPN-related SVT as it can support the MPN diagnosis, particularly in “triple-negative” cases, and provide additional information with potential consequences on prognosis and therapeutic strategies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
BCR::ABL1-negative myeloproliferative neoplasms (MPN) are characterized by a defined risk of disease progression to secondary myelofibrosis (SMF) or acute myeloid leukemia (also known as blast-phase MPN—MPN-BP) and show an increased thrombotic risk [1, 2]. Arterial and venous thrombotic events at initial presentation are reported in approximately 11%-25% of patients with essential thrombocythemia (ET) [3,4,5], 12%-39% of those with polycythemia vera (PV) [6], and in approximately 9.5% of those with primary myelofibrosis (PMF) [1].
Venous thrombosis can also involve unusual sites, including cerebral veins [7, 8] and splanchnic vessels (SVT). The latter includes Budd-Chiari syndrome (BCS), portal vein thrombosis (PVT) and other thrombotic events that can occur in isolated or multiple vessels of the portal venous axis, most commonly in the splenic (ST) and mesenteric ones (MT), with a prevalence between 1 and 23% [9,10,11].
Accordingly, BCS and PVT in the absence of underlying liver disease share many risk factors, with MPNs being the most common [12]: more specifically, as confirmed in a recent meta-analysis [13], BCR::ABL1-negative MPNs are the most frequent underlying prothrombotic factor in BCS and PVT, with a reported prevalence of 30%-50% and 15%-30%, respectively [14,15,16,17]. Therefore, given the frequency of MPNs in SVT patients, referral to a hematologist to discuss molecular screening and bone marrow (BM) biopsy should be systematically considered since targeting the underlying malignant condition, primarily with generally long-term anticoagulation, positively influences patients’ outcomes [18].
However, recognition of an underlying MPN can be difficult [19]: indeed, hypersplenism, hemodilution, iron deficiency, hepatic ischemia and/or occult bleeding can affect hematological parameters or inappropriately increase serum erythropoietin levels [20]. Furthermore, MPNs associated with SVT appear to have atypical intrinsic clinical-laboratory features compared to “classic” MPNs, including onset at a younger age, a female predominance, and the presence of normal peripheral blood (PB) cell count [21].
All these patients should be evaluated with a careful investigation, including a JAK2V617F mutation test. In this context, in a meta-analysis of 831 patients with unprovoked SVT, the aforementioned mutation was detected in 33% of overall and in 49% of idiopathic cases [21]. Similarly, a previous study involving 93 consecutive SVT cases diagnosed from 1992 to 2005 identified the JAK2V617F mutation in 35.6% of 73 patients with PVT and in 40% of 20 with BCS [19]. Furthermore, even though JAK2 exon 12 mutations have previously been described only anecdotally in SVT patients [13, 22,23,24], Magaz et al. [25] have recently reported that in non-cirrhotic, non-malignant SVT next-generation sequencing (NGS) can identify these mutations, including those in the hotspot exon 12 region of JAK2, not previously detected by conventional techniques, for example by direct Sanger sequencing.
As for the MPL gene, its mutations have also been documented, although in significantly fewer SVT patients [13, 26]. Similarly, after the description of CALR deletions and/or insertions [27, 28], Turon et al. [29] and Haslam et al. [30] first assessed the incidence of these mutations in two large cohorts of patients with SVT of different etiologies but found them only in a minority of cases. Consequently, although CALR and MPL mutations should be investigated in all JAK2V617F-negative SVT patients, this approach has lower sensitivity [26]. Conversely, BM biopsy could represent a key component of the diagnostic algorithm of SVT-related MPNs [19, 23]. However, a diagnosis of MPN on histological grounds alone and the distinction between PV, ET and PMF is quite complex and difficult [31]. Recently, NGS has contributed to a thorough description of the mutational spectrum of MPNs and a classification on the basis of the causal biological mechanisms has been proposed [32]. Also, the order of acquisition of somatic mutations influences the thrombotic risk [33], but how this is relevant in the context of MPNs associated with SVT is still unclear.
Clinical features and outcome of MPN-SVT patients have already been reported in several studies [34,35,36,37,38,39]. However, due to the rarity of the disease and the several unmet clinical needs related to its management [40], a more in-depth analysis is required to accurately estimate the risk of SVT recurrence and evolution (fibrotic or leukemic) of the underlying MPN, and to test the effectiveness of current therapeutic strategies in reducing the associated vascular risk.
Here, we conducted a retrospective, cohort study with the aim of reviewing clinical-morphological features and correlating them with mutational profiles by NGS in MPN patients who suffered from SVT.
Methods
Patients
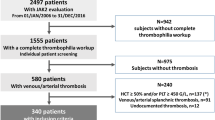
A single-center consecutive series of 58 patients admitted to our hospital between January 1979 and November 2021 with a diagnosis of SVT associated with BCR::ABL1-negative MPN was evaluated. Inclusion criteria were as follows: available demographic, clinical, hematological, and histological data at diagnosis; stored BM biopsy; and at least one granulocyte DNA sample (collected at the time of JAK2V617F mutational screening, after a median time from MPN diagnosis of 6.1 years) to perform NGS analysis. The protocol was approved by the local IRB (439_2022 BIS). Baseline clinical characteristics and outcome measures [thrombosis, hemorrhages, fibrotic progression, leukemic evolution, death, and overall survival (OS)] were assessed. Major thromboses and bleeding events were recorded when objectively documented and according to standard definitions as detailed by the CYTO-PV Collaborative Group [41]. Diagnosis of SMF was made according to the International Working Group for Myelofibrosis Research and Treatment (IWG-MRT) criteria [42]. Diagnosis of MPN-BP was made with a 20% threshold of BM or PB blasts [43]. Patients were treated with antiplatelet/anticoagulant and/or cytoreductive agents according to institutional guidelines which were based on current recommendations.
Follow-up information was updated in April 2022.
Methods
Molecular analyses
The JAK2V617F mutation was detected by allele-specific PCR according to the protocol of Baxter et al. [44] and confirmed by direct Sanger sequencing. Quantitative analysis of the allele burden of the JAK2V617F mutation was performed by RQ-PCR using JAK2 MutaQuant (Ipsogen Inc, New Haven, CT, USA). The cut-off used for defining a case as negative for JAK2V617F mutation was 0.5%.
MPL mutations were tested by direct sequencing of exon 10. Primer used were: MPL10F 5’ TAGCCTGGATCTCCTTGGTG 3’; MPL10R 5’ CCTGTTTACAGGCCTTCGGC 3’.
Mutations in exon 9 of CALR gene were also assessed by bidirectional sequencing approach as previously described [28]. All sequencing analyses were performed on ABI PRISM 310 Genetic Analyzer (Applied Biosystems, Warrington, UK) using the Big Dye Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems, Warrington, UK).
Sequencing, mutation calling and filtering
NGS analysis was performed using DNA extracted from PB granulocytes. 100 ng of tumor DNA was used to prepare libraries using the HyperPlus kit (KAPA Biosystems). The targeted gene panel included coding exons and splice sites of 73 genes involved in myeloid malignancies based on literature data. Libraries were sequenced using 150 bp paired end reads, to a mean total depth of 1006x.
Mutect 2 GATK 4.1.5.0, LoFreq (v. 2.1.5) and VarDict (2019.06.04) were used for variant calling, and only variants called by at least 2/3 of these variant callers were further considered. To filter out artifacts and germline polymorphisms [45, 46], we first excluded variants with at least one of the following characteristics:—variant allele frequency (VAF) lower than 0.02 and less than 15 supporting reads;—coverage < 300x;—occurrence in > 5% of a panel of normal controls (PoN, analyzed for the same gene panel and in the same sequencing conditions) at a VAF < 5%;—expected minor germline allele frequency > 0.001 based on the information retrieved from the public database gnomAD (http://gnomad.broadinstitute.org; gnomAD r3.1.2);—or occurrence in at least one sample of our PoN at a VAF > 45% (both features being suggestive of polymorphic variants).
The shortlist of variants was therefore considered as containing bona fide somatic mutations, whose significance was evaluated at the amino acid level in order to differentiate known/putative pathogenic mutations from variants of unknown significance (VUS) on the basis of data available in literature and reported in the publicly accessible Catalogue Of Somatic Mutations In Cancer (COSMIC, version 69) (http://cancer.sanger.ac.uk/cancergenome/projects/cosmic).
Variants deemed as VUS were excluded from downstream analyses. VAF was used to estimate the mutation clonality and mutation precedence by analyzing the confidence intervals (CI) of VAF values, inferred by assuming a binomial distribution of VAF and calculated against the hypothesis of a random VAF of 0.5. Variants with non-overlapping CIs were estimated to be acquired in different time windows, under the assumption that mutations with higher VAF are present in more cancer cells and thus were acquired earlier [47,48,49].
Cytogenetic analyses
G-banding with trypsin performed on fresh BM aspirates was the standard technique for chromosome analysis with at least 20 metaphases described [50]. Normal karyotype was defined as the absence of any chromosomal abnormality in a minimum of 20 metaphases examined. Chromosomal abnormalities were considered clonal if the same structural or extra-chromosome abnormality appears in at least two and monosomy in at least three metaphases.
Bone marrow biopsy
Histological confirmation of the clinical diagnosis of MPN as defined by the 2016 WHO classification was performed by an experienced pathologist (U.G.). Formalin-fixed, paraffin-embedded BM biopsy specimens were available for all patients. Sections were stained with hematoxylin–eosin, Giemsa, and Gomori’s silver impregnation for the evaluation of morphology and BM fibrosis grade.
Statistical approach
Descriptive statistics were generated for all clinical characteristics and outcome measures as appropriate: for continuous variables, median and range; for categorical variables, frequency and percentage.
We used Fisher’s exact test and non-parametric methods (Wilcoxon and Kruskal–Wallis) tests to analyze categorical and quantitative variables, respectively. We evaluated death risk (with follow-up time truncated at 25 years) according to selected variables by fitting univariate Cox models to calculate hazard ratios (HR) and 95% CIs. Statistical analysis was performed with Stata 17 (StataCorp. 2021).
Results
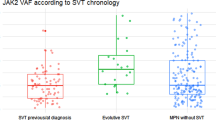
Overall, 58 patients with MPN-SVT were analyzed (Table 1). In 24 (41.3%) cases the diagnosis of MPN and SVT were coincident, in 19 (32.8%) an SVT index event preceded the diagnosis of MPN by a median of 2.8 years (range, 0.3–27.9), while in the remaining 15 (25.9%) patients it occurred after a median of 13.3 years from MPN diagnosis (range, 3.0–25.4).
Female patients (32/58, 55.2%) were prevalent, with a median age at MPN diagnosis of 47.1 years (range, 18.5–78.7). Notably, only a minority of patients (20.7%) were over 60 at the time of the index event, while 32.8% were under 40.
Portal vein thrombosis occurred in 19 (32.8%) cases, ST in four (6.9%), MT in one (1.7%) and BCS in eight (13.8%). An association of PVT and/or ST and/or MT was found in 26 (44.8%) patients.
Hematological parameters at MPN and SVT diagnosis are summarized in Supplementary Table S1: at MPN diagnosis significantly higher median hemoglobin (Hb) levels were detected (13.7 vs. 13.2 g/dL; p = 0.01) as compared to SVT diagnosis, with a trend towards a statistically significant difference in median platelet (PLT) counts (405 vs. 392 × 109/L; p = 0.06).
Pre-PMF along with myeloproliferative neoplasm, unclassifiable (MPN-U) were the most frequent MPN subtypes (27.6%, n = 16 each), followed by PV (15.5%, n = 9), ET (13.8%, n = 8), SMF (8.6%, n = 5), and overt PMF (6.9%, n = 4).
As reported in Supplementary Table S2, hematological parameters were consistent with those already reported for each MPN subtype: in detail, Hb levels were significantly higher in PV (p = 0.01), while PLT counts in ET patients (p = 0.0006). PMF cases showed significantly higher lactate dehydrogenase (LDH) levels (p = 0.0008), with a trend towards a statistically significant difference in white blood cells (WBC) count (p = 0.07). As expected, PMF patients also exhibited significantly larger spleen size (p = 0.003), albeit with no robust differences in the number of cases with splenomegaly.
No difference was found also for gender, type of SVT, constitutional symptoms, abnormal karyotype, and therapies (either anticoagulant or cytoreductive agents).
Regarding driver mutations, JAK2V617F was found in most patients (49/58, 84.5%). Three (5.2%) had a CALR mutation (including two patients with CALR type 1 mutation and the remaining case with a CALR type 1-like mutation) and four (6.9%) an MPLW515L/K mutation. The remaining two (3.4%) patients were “triple-negative”. As reported in Supplementary Table S2, a higher median JAK2V617F allele burden was detected among patients with SMF, followed by PMF cases (p = 0.003).
As detailed in Fig. 1, NGS analysis—performed on 54/58 patients—showed no additional mutations in 25 (46.3%) cases, including the two “triple-negative” subjects, with the remaining 29 (53.7%) patients harboring at least one additional pathogenic variant. In detail, most of the subjects (28/29, 96.5%) had one or two mutations, whereas only one (3.5%) patient showed three concomitant variants. The most frequent sequence variants involved TET2 gene (15/29, 51.7%), followed by DNMT3A (9/29, 31.0%) and ASXL1 (6/29, 20.7%). Other variants detected in at least one case also include U2AF1, TP53, EZH2, SF3B1, SETBP1, RUNX1, and CUX1 (Fig. 1).

Oncoplot depicting the mutational profile in the study cohort. Each column corresponds to a unique sample. The left-side bar plots indicate VAF gene-wise, the side bar plots the number of samples affected by mutations gene-wise, and the top side bar plots the mutation number per sample. Diagnosis annotation is included at the bottom of the oncoplot
As reported in Supplementary Table S3, when considering the number of additional mutations, no specific association was found with each MPN subtype; similarly, there was no difference in their distribution according to hematological parameters, hepato-splenomegaly, constitutional symptoms, gender, or age at MPN diagnosis. Interestingly, when driver and additional mutations are considered, a different co-mutational spectrum emerged (Fig. 2A): specifically, JAK2V617F mutation occurred concurrently with DNMT3A mutations, while MPL mutations were associated with ASXL1 mutations (2/4 cases). Our cohort was also enriched in cases with high VAF (≥ 50%) of JAK2V617F (13/49 cases), suggesting that acquired uniparental disomy (aUPD) was more frequent than observed in non-SVT MPN cases. Furthermore, high VAF JAK2V617F mutations had a higher median number of additional mutations than cases with low allele burden (2 vs. 1, p = 0.0007, two-tailed). More importantly, as shown in Fig. 2B-C, the co-mutational spectrum was also different between patients with low and high VAF of JAK2V617F, with DNMT3A being the most frequent partner of JAK2V617F low-burden cases, while TET2 and ASXL1 mutations were more frequently associated with JAK2V617F high-burden cases. Overall, high molecular risk (HMR) mutations were more frequently detected among patients with at least two non-driver mutations. Indeed, this correlated with clinical outcomes, as the six cases of leukemic evolution were all characterized by a higher JAK2V617F allele burden, a higher median number of co-mutations, and a co-mutational pattern of high-risk lesions showing themselves a high VAF, such as truncating mutations of ASXL1 (3/5), bi-allelic loss of TP53 (1/6), and CSMD1 mutations (1/6), confirming that genomics can help predict clinical outcomes (Fig. 3). Nevertheless, no difference was found between cases with and without additional somatic mutations regarding fibrotic progression, SVT recurrence, other thrombo-hemorrhagic complications, or death.

(A) Interaction of somatic variants from a selected pool of genes. Patterns of co-occurrence are highlighted in blue while mutual exclusion in brown. Statistically significant pairs are highlighted with either dots (p < 0.05) or stars (p < 0.01). (B) and (C) depict chord diagrams gauging the different mutational patterns seen in samples with respectively high and low JAK2V617F allelic ratio

Kaplan Meier curve with leukemic progression cumulative risk over a 40 years follow-up (95%CI, gray ribbons). On the right, an oncoplot showing the mutational distribution among samples from patients who evolved into acute myeloid leukemia (AML). Mutations in non-driver MPN genes are annotated for the amino acid change
The study of mutations VAF also allowed to estimate the order of acquisition of the mutations by analyzing the overlap between VAF CIs, under the assumption that mutations with a higher VAF would be present in a larger number of cells and therefore be acquired earlier in the clonal phylogeny of the tumor. This was particularly useful for JAK2V617F and TET2, the two most frequently mutated genes: indeed, we could estimate that JAK2V617F mutation preceded those of TET2 in 13/14 cases (Fig. 4 and Supplementary Fig. S4). This has relevant implications, since JAK2-first cases are associated with a more pro-thrombotic phenotype and more aUPD, features that are enriched in our MPN-SVT cohort.

Stacked barplot showing driver gene clonal composition. Per-sample clones were defined as Driver-First in the setting of at least one additional mutation where driver genes were mutated at higher VAF with no CI overlap; Driver-Only, where no additional mutations were found; Driver-Second, where additional mutations were deemed to be antecedent to the driver ones; Uncertain, where precedence could not be determined with confidence; NA, where VAF could not be reliably assessed. Figure 4 detail provides two examples of our cohort samples clonal architecture
Cytogenetic data were available at diagnosis in all patients (Table 1), with a normal karyotype reported in the majority of cases (53/58, 91.4%). In particular, as regards chromosomal abnormalities, they were represented by 20q- in two cases and 5q- or + 1 in one case each.
Information on concomitant hypercoagulable disorders was available in 49 (84.5%) patients: thrombophilia abnormalities were detected in 20 (40.8%) cases, the most frequent including heterozygous prothrombin G20210A mutation and hyperhomocysteinemia in five (10.2%) cases each, heterozygous factor V Leiden mutation in two (4.1%) and protein S deficiency in nine (18.4%).
Most SVT events (82.8%) developed in the absence of any antithrombotic prophylaxis. In contrast, most patients (53/58, 91.4%) received anticoagulant and/or antiplatelet agents for secondary prophylaxis. In detail, anticoagulants alone were administered to 43 (81.1%) patients [vitamin K antagonists (VKAs) in 69.8% and direct oral anticoagulants (DOACs) in 30.2% of cases], antiplatelets alone (aspirin in 75% and clopidogrel in 25% of cases) in eight (15.1%) and a combination of anticoagulants and antiplatelets in two (3.8%).
Globally, cytoreductive agents were used in 45 (77.6%) cases (including nine patients already on treatment at the time of SVT), mostly hydroxyurea (75.9% of cases), with only three (5.2%) patients treated with ruxolitinib.
Median follow-up for the whole cohort was 7.1 years (range, 0.3–29.3) after MPN diagnosis and 8.8 years (range, 0.6–35.8) after the diagnosis of SVT.
A total of ten (17.2%) patients experienced SVT recurrence, involving more than one site in three cases, while isolated recurrences were PVT in one case, ST, MT and BCS in two cases each; at the time of SVT recurrence, six patients were on anticoagulation with VKAs, while one patient was on antiplatelet agents alone. Excluding SVT recurrences, 12 new episodes of thrombosis occurred in 11 patients, more frequently involving the arterial district (8/12, 66.7%), in course of anticoagulant therapy with VKAs in most cases (9/11, 81.8%) (Table 1). Focusing specifically on patients with recurrent arterial thrombosis, they were on antiplatelet therapy only in a minority of cases (3/8, 37.5%) while additional cardiovascular risk factors (i.e., diabetes, hypertension, dyslipidemia or smoking habit) were reported in most patients (6/8, 75%).
Overall, 48.3% (28/58) of patients developed esophageal varices, with 35.7% of them experiencing one or more variceal bleeding episodes (range, 1–5) during anticoagulant therapy with VKAs in most cases (8/10, 80%).
At the last follow up, 10/58 (17.2%) patients with MPN-SVT had died; causes of death are summarized in Table 1. Progression to SMF and MPN-BP was ascertained in one (1.7%) and six (10.3%) patients, respectively.
As reported in Supplementary Table S4, clinical features and the type and number of driver or non-driver mutations, as well as cytoreductive or anticoagulant therapies did not have a significant impact on OS of these patients.
Discussion
In this real-life, single-center cohort study, we investigated mutational profiles using NGS and reviewed clinical-morphological data of 58 consecutive MPN-SVT patients with a median follow-up of 7.1 years after MPN diagnosis.
First, we confirmed a consistent association of SVT with a distinct phenotype characterized by female predominance and younger age, across all MPN subtypes, and a striking association with the JAK2V617F mutation (84.5% in our cohort).
Second, pre-PMF and MPN-U were the most frequent underlying diagnoses. In this regard, we recognize that an accurate differentiation among MPN subtypes in SVT patients can represent a diagnostic challenge: as already reported in a previous study by our group [31], peculiar clinical-laboratory features were found in the three morphology-defined MPN subtypes when associated with SVT. More precisely, there was an almost complete correspondence between the clinical and morphological features of PMF patients, whereas discrepancies were frequent in patients with an ET- or, above all, a PV-like morphology, since only a minority of cases showed a clinical phenotype typical of PV. This finding is also in line with previous studies indicating the lack of specificity of current diagnostic criteria for PV in SVT patients [40, 51,52,53]. Consistent with this notion, up to 27.6% of MPN-SVT cases included in our study received a primary diagnosis of MPN-U based on BM findings. Overall, past and current evidence suggests that a histology-based classification has limitations and does not reflect a genomic classification that would better fit the actual biology of these diseases.
It should also be emphasized that the distribution by gender and age differs from that observed in the general population and in MPN patients without SVT, suggesting that traditional risk factors for thrombosis are of limited value in the context of MPN-SVT [39, 54], with consequent growing interest in other possible biomarkers of vascular injury and thrombotic risk, with circulating endothelial cells among the most promising [55]. Indeed, we found that MPN-SVT cases had a not negligible risk of subsequent vascular events, even though most of our cases were treated with both anticoagulant/antiplatelet and/or cytoreductive drugs, mainly hydroxyurea. Although used only in a minority of our cases (5.2%), ruxolitinib, the first JAK1/2 inhibitor to become commercially available, could represent an effective therapeutic option also in this subgroup of patients, reducing disease-related symptoms and spleen size, as well as the resistivity indices of the hepatic and splenic intraparenchymal arteries, with a substantial stabilization of esophageal varices grade [56].
According to our data, the onset of SVT should not be considered a feature that suggests an underlying aggressive MPN. In the past, MPN-SVT have in fact been reported as atypical myeloproliferative disorders with high thrombotic risk and slow disease progression [57]. In particular, our cohort of 16 MPN-U patients appears to represent a distinct clinical group, since SVT was the initial manifestation in all of these cases: moreover, they showed a very indolent clinical course, without evidence of fibrotic progression or leukemic evolution, and requiring the introduction of cytoreductive agents in fewer cases. Importantly, none of these patients developed clinical features during the follow-up that allowed physicians to reclassify them as one of the classical BCR::ABL1-negative MPNs. Similar results have also been recently reported in previous studies [34, 35, 39] supporting the notion that MPN-U with SVT may represent a very early stage of hematological neoplasm without overt histopathological and hematological features indicative of a specific disease subtype according to WHO criteria.
Consequently, survival of MPN-SVT patients is primarily influenced by the recurrent thrombotic event and/or the consequences on splanchnic circulation and liver function, rather than the natural history of the underlying malignancy. In support of this statement, our cohort was also enriched in cases with JAK2V617F homozygosity; furthermore, as we could estimate that JAK2V617F mutation preceded those of TET2 in most cases (Fig. 4 and Supplementary Fig. S4), we confirm that mutations order has relevant implications also in MPN-SVT patients, since JAK2-first cases have already been demonstrated to be typically associated with an increased risk of thrombosis [33]. Consistently, the frequency of JAK2-first cases was much lower in unselected, non-SVT MPN cases [32]. In addition, no obvious genetic thrombophilia conditions were found despite thorough investigation in our patients. Indeed, prothrombin G20210A and factor V Leiden mutations were present with a frequency not different from the general population, and no high-risk mutations were found, once more stressing the role of genomics of the underlying MPN as the main driver of the clinical outcome.
Furthermore, in agreement with previous studies [25, 32, 58] the most frequently mutated gene was TET2 (15/29 patients, 51.7%), followed by DNMT3A (9/29, 31.0%). Some of these genes have essential functions independent of their enzymatic activity, with TET2 appearing to be an important regulator of hematopoietic stem cell biology [59]. TET2 mutations have been identified in a high proportion of patients with JAK2V617F-positive and JAK2V617F-negative MPNs, suggesting that the occurrence of these mutations may represent an early event in disease pathogenesis [60]. Among others, Colaizzo et al. identified previously unreported TET2 mutations in 3/23 non-cirrhotic JAK2V617F-positive MPN-SVT patients [61]. Interestingly, only patient no. 3 showed overt MPN, while the others developed an MPN-specific phenotype only years after SVT diagnosis, thus further supporting the notion of TET2 mutations as an early event along with JAK2V617F in the natural history of MPNs. Classically, MPNs diagnosed at the time of an SVT are considered to be early in their disease course, and our clinical and histological data support this hypothesis. However, our genomic findings show a rather complex genetic architecture, also supporting in our cohort the recent view that the preclinical evolution of these malignancies dates back well before the time of diagnosis [62].
Furthermore, TET2 deficiency results in an increased pro-inflammatory phenotype in murine macrophages which could favor the development of atherosclerosis and thrombosis [63]. More specifically, even in the context of clonal hematopoiesis of indeterminate potential (CHIP) [64,65,66,67], DNMT3A/TET2/ASXL1 gene mutations have already been shown to cause a 1.9-fold higher risk of developing coronary disease and a 4-fold higher risk of myocardial infarction [68].
Interestingly, it has recently been reported that TET2 and DNMT3A genes are both associated with an increased thrombotic risk in PV [58]. Indeed, our cases show a high prevalence of TET2 mutations in our patients’ population (28.5%) compared to other previous studies (roughly 14%) [61, 69, 70]. This evidence together with the “JAK2-first” genotype may confer a genetically-determined prothrombotic phenotype in MPN patients affected by SVT.
However, when considering HMR mutations, which are already well-known to be associated with poor prognosis in BCR::ABL1-negative MPNs [71], particularly PMF, they were more frequently detected among patients with at least two non-driver mutations, and correlate with clinical outcomes, as all the cases of leukemic evolution were characterized by a higher JAK2V617F allele burden, a higher median number of co-mutations, and a co-mutational pattern of high-risk lesions (Fig. 3). A previous study has already demonstrated that a JAK2V617F allele burden ≥ 50% and presence of chromatin/spliceosome/TP53 mutations are associated with a higher incidence of fibrotic progression/leukemic evolution or death in MPN-SVT patients [72]. Nevertheless, while identifying at least one additional mutation in 53.7% of our patients, the type and number of non-driver mutations were not shown to have any significant impact on fibrotic progression, SVT recurrence, other thrombo-hemorrhagic complications, or death. A possible explanation for the observed differences between our cohorts could be due to a higher frequency of HMR mutations in the Debureaux series (14 cases vs. 8 in our cohort) [72]. Another point to consider is the different representation of specific MPN subtypes: in particular, while Debureaux et al. [72] did not include any patient with MPN-U among their case series, this diagnosis was made in nearly 30% of our subjects, thus supporting the notion of the potential impact of morphological features on the prognostic role of molecular profile also in the MPN-SVT setting.
All this considered, while acknowledging the limited availability of the method and its high cost, our data underline the importance of using NGS analysis in the management of MPN-related SVT as it can support the MPN diagnosis, particularly in “triple-negative” cases, i.e., without JAK2V617F or exon 12, CALR, or MPL mutations. Although no additional mutations were found in our two “triple-negative” cases, targeted NGS myeloid analysis has proven useful in highlighting alternative drivers in these subgroups of patients in the past [73, 74] that may be helpful for diagnosis. Furthermore, in the future whole genome approaches are likely to highlight different genomic lesions, possibly in the form of copy-number changes or rearrangements even in the absence of recurrent gene mutations.
We are aware that our considerations are partially limited by the relatively small number of patients with MPN and SVT. Additionally, the presence of the JAK2V617F mutation in most of our cases may represent a selection bias; however, since the discovery of JAK2 mutation in 2005, all patients who were admitted to our hospital due to idiopathic SVT have so far routinely undergone BM biopsy, along with molecular screening for CALR and MPL mutations as well. Finally, NGS analysis was performed using DNA extracted from PB granulocytes collected at the time of JAK2V617F mutational screening, i.e., after a median time from MPN diagnosis of 6.1 years. This was primarily due to the inclusion in our series of 21 (36.2%) patients diagnosed with MPN prior to the discovery of JAK2 mutation in 2005 (mainly between 1990 and 2004), a period in which collection of onset samples for molecular testing was not included in routine clinical practice. While admitting that this latter aspect may represent a limitation of our study, however, as reported for the first time in the pivotal study by Grinfeld et al. [32], the acquisition of additional molecular abnormalities is typically associated with disease progression (fibrotic progression or leukemic evolution), which is a rare event in patients with MPN and SVT and was also rare in our cohort.
In conclusion, our results suggest that a comprehensive approach is always required to make a diagnosis of MPN in patients presenting or suffering from SVT during follow-up, considering BM morphology, clinical and molecular data (either on driver or non-driver mutations), with potential consequences also on prognosis and therapeutic management.
Data Availability
The data that support the findings of this study are available from the senior authors upon reasonable request.
References
Barbui T, Carobbio A, Cervantes F et al (2010) Thrombosis in primary myelofibrosis: incidence and risk factors. Blood 115:778–782
Falanga A, Marchetti M (2012) Thrombotic disease in the myeloproliferative neoplasms. Hematology (Am Soc Hematol Educ Program) 2012:571–581
Harrison CN, Campbell PJ, Buck G et al (2005) Hydroxyurea compared with anagrelide in high-risk essential thrombocythemia. N Engl J Med 353:33–45
Gisslinger H, Gotic M, Holowiecki J et al (2013) Anagrelide compared to hydroxyurea in WHO-classified essential thrombocythemia: the ANAHYDRET Study, a randomized controlled trial. Blood 121:1720–1728
Carobbio A, Thiele J, Passamonti F et al (2011) Risk factors for arterial and venous thrombosis in WHO-defined essential thrombocythemia: an international study of 891 patients. Blood 117:5857–5859
Marchioli R, Finazzi G, Landolfi R et al (2005) Vascular and neoplastic risk in a large cohort of patients with polycythemia vera. J Clin Oncol 23:2224–2232
Dentali F, Ageno W, Rumi E et al (2014) Cerebral venous thrombosis and myeloproliferative neoplasms: results from two large databases. Thromb Res 134:41–43
Martinelli I, De Stefano V, Carobbio A et al (2014) Cerebral vein thrombosis in patients with Philadelphia-negative myeloproliferative neoplasms. An European Leukemia Net study. Am J Hematol 89:E200–E205
Thatipelli MR, McBane RD, Hodge DO, Wysokinski WE (2010) Survival and recurrence in patients with splanchnic vein thrombosis. Clin Gastroenterol Hepatol 8:200–205
Finazzi G, De Stefano V, Barbui T (2013) Are MPNs Vascular Diseases? Curr Hematol Malig Rep 8:307–316
Cortelezzi A, Moia M, Falanga A et al (2005) Incidence of thrombotic complications in patients with haematological malignancies with central venous catheters: a prospective multicentre study. Br J Haematol 129:811–817
Elkrief L, Payancé A, Plessier A et al (2023) Management of splanchnic vein thrombosis. JHEP Rep. https://doi.org/10.1016/j.jhepr.2022.100667
Smalberg JH, Arends LR, Valla DC, Kiladjian JJ, Janssen HL, Leebeek FW (2012) Myeloproliferative neoplasms in Budd-Chiari syndrome and portal vein thrombosis: a meta-analysis. Blood 120:4921–4928
Darwish Murad S, Plessier A, Hernandez-Guerra M et al (2009) Etiology, management, and outcome of the Budd-Chiari syndrome. Ann Intern Med 151:167–175
Bayraktar Y, Harmanci O, Buyukasik Y et al (2008) JAK2V617F mutation in patients with portal vein thrombosis. Dig Dis Sci 53:2778–2783
Valla D, Casadevall N, Huisse MG et al (1988) Etiology of portal vein thrombosis in adults: a prospective evaluation of primary myeloproliferative disorders. Gastroenterology 94:1063–1069
Donadini MP, Dentali F, Ageno W (2012) Splanchnic vein thrombosis: new risk factors and management. Thromb Res 129:S93–S96
Chagneau-Derrode C (2013) Impact of cytoreductive therapy on the outcome of patients with myeloproliferative neoplasms and hepato-splanchnic vein thrombosis. Hepatology 58:847A-898A
Primignani M, Barosi G, Bergamaschi G et al (2006) Role of the JAK2 mutation in the diagnosis of chronic myeloproliferative disorders in splanchnic vein thrombosis. Hepatology 44:1528–1534
Jones C, Levy Y, Tong AW (2014) Elevated serum erythropoietin in a patient with polycythaemia vera presenting with Budd-Chiari syndrome. BMJ Case Rep 2014:1–4
Sekhar M, McVinnie K, Burroughs AK (2013) Splanchnic vein thrombosis in myeloproliferative neoplasms. Br J Haematol 162:730–747
Bergamaschi GM, Primignani M, Barosi G et al (2008) MPL and JAK2 exon 12 mutations in patients with the Budd-Chiari syndrome or extrahepatic portal vein obstruction. Blood 111:4418
Kiladjian JJ, Cervantes F, Leebeek FW et al (2008) The impact of JAK2 and MPL mutations on diagnosis and prognosis of splanchnic vein thrombosis: a report on 241 cases. Blood 111:4922–4929
Fiorini A, Chiusolo P, Rossi E et al (2009) Absence of the JAK2 exon 12 mutations in patients with splanchnic venous thrombosis and without overt myeloproliferative neoplasms. Am J Hematol 84:126–127
Magaz M, Alvarez-Larrán A, Colomer D et al (2021) Next-generation sequencing in the diagnosis of non-cirrhotic splanchnic vein thrombosis. J Hepatol 74:89–95
Iurlo A, Cattaneo D, Gianelli U, Fermo E, Augello C, Cortelezzi A (2015) Molecular analyses in the diagnosis of myeloproliferative neoplasm-related splanchnic vein thrombosis. Ann Hematol 94:881–882
Klampfl T, Gisslinger H, Harutyunyan AS et al (2013) Somatic mutations of calreticulin in myeloproliferative neoplasms. N Eng J Med 369:2379–2390
Nangalia J, Massie CE, Baxter EJ et al (2013) Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Eng J Med 369:2391–2405
Turon F, Cervantes F, Colomer D, Baiges A, Hernández-Gea V, Garcia-Pagán JC (2015) Role of calreticulin mutations in the etiological diagnosis of splanchnic vein thrombosis. J Hepatol 62:72–74
Haslam K, Langabeer SE (2015) Incidence of CALR mutations in patients with splanchnic vein thrombosis. Br J Haematol 168:459–460
Gianelli U, Iurlo A, Cattaneo D et al (2015) Discrepancies between bone marrow histopathology and clinical phenotype in BCR-ABL1-negative myeloproliferative neoplasms associated with splanchnic vein thrombosis. Leuk Res 39:525–529
Grinfeld J, Nangalia J, Baxter EJ et al (2018) Classification and personalized prognosis in myeloproliferative neoplasms. N Engl J Med 379:1416–1430
Ortmann CA, Kent DG, Nangalia J et al (2015) Effect of mutation order on myeloproliferative neoplasms. N Engl J Med 372:601–612
Lavu S, Szuber N, Mudireddy M et al (2018) Splanchnic vein thrombosis in patients with myeloproliferative neoplasms: the Mayo clinic experience with 84 consecutive cases. Am J Hematol 93:E61–E64
Cattaneo D, Gianelli U, Bianchi P, Cortelezzi A, Iurlo A (2018) Heterogeneity among splanchnic vein thrombosis associated with myeloproliferative neoplasms. Eur J Intern Med 52:e25–e26
Hoekstra J, Bresser EL, Smalberg JH, Spaander MC, Leebeek FW, Janssen HL (2011) Long-term follow-up of patients with portal vein thrombosis and myeloproliferative neoplasms. J Thromb Haemost 9:2208–2214
Gangat N, Wolanskyj AP, Tefferi A (2006) Abdominal vein thrombosis in essential thrombocythemia: prevalence, clinical correlates, and prognostic implications. Eur J Haematol 77:327–333
De Stefano V, Vannucchi AM, Ruggeri M et al (2016) Splanchnic vein thrombosis in myeloproliferative neoplasms: risk factors for recurrences in a cohort of 181 patients. Blood Cancer J 6:e493
Sant’Antonio E, Guglielmelli P, Pieri L et al (2020) Splanchnic vein thromboses associated with myeloproliferative neoplasms: An international, retrospective study on 518 cases. Am J Hematol 95:156–166
Barosi G, Vannucchi AM, De Stefano V et al (2014) Identifying and addressing unmet clinical needs in Ph-neg classical myeloproliferative neoplasms: a consensus-based SIE, SIES, GITMO position paper. Leuk Res 38:155–160
Marchioli R, Finazzi G, Specchia G et al (2013) Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med 368:22–33
Barosi G, Mesa RA, Thiele J et al (2008) Proposed criteria for the diagnosis of post-polycythemia vera and post-essential thrombocythemia myelofibrosis: a consensus statement from the International Working Group for Myelofibrosis Research and Treatment. Leukemia 22:437–438
Swerdlow SH, Campo E, Harris L et al. (2008) WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed. IARC, Lyon, 127–129
Baxter EJ, Scott LM, Campbell PJ et al (2005) Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 365:1054–1061
Bolli N, Manes N, McKerrell T et al (2015) Characterization of gene mutations and copy number changes in acute myeloid leukemia using a rapid target enrichment protocol. Haematologica 100:214–222
McKerrell T, Moreno T, Ponstingl H et al (2016) Development and validation of a comprehensive genomic diagnostic tool for myeloid malignancies. Blood 128:e1–e9
Nik-Zainal S, Van Loo P, Wedge DC et al (2012) The life history of 21 breast cancers. Cell 149:994–1007
Bolli N, Biancon G, Moarii M et al (2018) Analysis of the genomic landscape of multiple myeloma highlights novel prognostic markers and disease subgroups. Leukemia 32:2604–2616
Ziccheddu B, Biancon G, Bagnoli F et al (2020) Integrative analysis of the genomic and transcriptomic landscape of double-refractory multiple myeloma. Blood Adv 4:830–844
Rack KA, van den Berg E, Haferlach C et al (2019) European recommendations and quality assurance for cytogenomic analysis of haematological neoplasms. Leukemia 33:1851–1867
Gianelli U, Iurlo A, Vener C et al (2008) The significance of bone marrow biopsy and JAK2V617F mutation in the differential diagnosis between the early pre-polycythemic phase of polycythemia vera and essential thrombocythemia. Am J Clin Pathol 130:336–342
Spivak JL, Silver RT (2011) The treatment of essential thrombocytosis revisited. Blood 28:1179–1180
Silver RT, Chow W, Orazi A, Arles SP, Goldsmith SJ (2013) Evaluation of WHO criteria for diagnosis of polycythemia vera: a prospective analysis. Blood 122:1881–1886
Rosti V, Villani L, Riboni R et al (2013) Spleen endothelial cells from patients with myelofibrosis harbor the JAK2V617F mutation. Blood 121:360–368
Giordano G, Napolitano M, Cellurale M et al (2022) Circulating Endothelial Cell Levels Correlate with Treatment Outcomes of Splanchnic Vein Thrombosis in Patients with Chronic Myeloproliferative Neoplasms. J Pers Med 12:364
Pieri L, Paoli C, Arena U et al (2017) Safety and efficacy of ruxolitinib in splanchnic vein thrombosis associated with myeloproliferative neoplasms. Am J Hematol 92:187–195
Barosi G, Buratti A, Costa A et al (1991) An atypical myeloproliferative disorder with high thrombotic risk and slow disease progression. Cancer 68:2310–2318
Segura-Díaz A, Stuckey R, Florido Y et al (2020) Thrombotic risk detection in patients with polycythemia vera: the predictive role of DNMT3A/TET2/ASXL1 mutations. Cancers (Basel) 12:934
Solary E, Bernard OA, Tefferi A, Fuks F, Vainchenker W (2014) The Ten-Eleven Translocation-2 (TET2) gene in hematopoiesis and hematopoietic diseases. Leukemia 28:485–496
Delhommeau F, Dupont S, Della Valle V et al (2009) Mutation in TET2 in myeloid cancers. N Engl J Med 360:2289–2301
Colaizzo D, Tiscia GL, Pisanelli D et al (2010) New TET2 gene mutations in patients with myeloproliferative neoplasms and splanchnic vein thrombosis. J Thromb Haemost 8:1142–1144
Sousos N, Ní Leathlobhair M, Simoglou Karali C et al (2022) In utero origin of myelofibrosis presenting in adult monozygotic twins. Nat Med 28:1207–1211
Fuster JJ, MacLauchlan S, Zuriaga MA et al (2017) Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 355:842–847
Laurie CC, Laurie CA, Rice K et al (2012) Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat Genet 44:642–650
Jaiswal S, Fontanillas P, Flannick J et al (2014) Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 371:2488–2498
Genovese G, Kähler AK, Handsaker RE et al (2014) Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 371:2477–2487
Steensma DP, Bejar R, Jaiswal S et al (2015) Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 126:9–16
Jaiswal S, Natarajan P, Silver AJ et al (2017) Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med 377:111–121
Westbrook R, Lea N, Mohamedali A et al (2012) Prevalence and clinical outcomes of the 46/1 haplotype, Janus kinase 2 mutations, and ten-eleven translocation 2 mutations in Budd-Chiari syndrome and their impact on thrombotic complications post liver transplantation. Liver Transpl 18:819–827
How J, Zhou A, Oh ST (2017) Splanchnic vein thrombosis in myeloproliferative neoplasms: pathophysiology and molecular mechanisms of disease. Ther Adv Hematol 8:107–118
Iurlo A, Cattaneo D, Gianelli U (2019) Blast Transformation in Myeloproliferative Neoplasms: Risk Factors, Biological Findings, and Targeted Therapeutic Options. Int J Mol Sci 20:1839
Debureaux PE, Cassinat B, Soret-Dulphy J et al (2020) Molecular profiling and risk classification of patients with myeloproliferative neoplasms and splanchnic vein thromboses. Blood Adv 4:3708–3715
Cattaneo D, Croci GA, Bucelli C et al (2021) Triple-Negative Essential Thrombocythemia: Clinical-Pathological and Molecular Features. A Single-Center Cohort Study. Front Oncol 11:637116
Maddali M, Venkatraman A, Kulkarni UP et al (2022) Molecular characterization of triple-negative myeloproliferative neoplasms by next-generation sequencing. Ann Hematol 101:1987–2000
Acknowledgements
This study was partially funded by Italian Ministry of Health—Current research IRCCS to NB. NB is recipient of an Investigator grant from the Associazione Italiana Ricerca sul Cancro (AIRC IG n. 25739).
Author information
Authors and Affiliations
Contributions
D. Cattaneo, N. Bolli, and A. Iurlo were responsible for study concept and design and wrote the paper. D. Consonni was responsible for statistical analyses. A. Marchetti, M. Lionetti, E. Fermo, A. Maeda, A. Marella, A. Neri, and N. Bolli made and interpreted mutational analyses. U. Gianelli reviewed bone marrow biopsies. D. Cattaneo, C. Bucelli, V. Bellani, C. De Magistris, M. Primignani, L. Baldini, and A. Iurlo followed the patients and collected data. All authors critically reviewed and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest disclosures
All the authors declare they have no potential conflicts of interest.
Ethical statement
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Niccolò Bolli and Alessandra Iurlo co-contributing senior authors.
Supplementary Information
Below is the link to the electronic supplementary material.

277_2023_5217_Fig5_ESM.png
Supplementary Fig. S4 VAF mutation study with flanking CIs in our patient cohort. VAF was modeled according to a binomial distribution. (PNG 234 kb)
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

{kind=link}
Cite this article
Cattaneo, D., Bucelli, C., Marchetti, A. et al. Pathological and genomic features of myeloproliferative neoplasms associated with splanchnic vein thrombosis in a single-center cohort. Ann Hematol 102, 1409–1420 (2023). https://doi.org/10.1007/s00277-023-05217-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-023-05217-2