
Abstract
Membrane-bound, pyrroloquinoline quinone (PQQ)-dependent glycerol dehydrogenase (GLDH, or polyol dehydrogenase) of Gluconobacter sp. oxidizes various secondary alcohols to produce the corresponding ketones, such as oxidation of D-sorbitol to L-sorbose in vitamin C production. Substrate specificity of GLDH is considered limited to secondary alcohols in the D-erythro configuration at the next to the last carbon. Here, we suggest that L-ribose, D- and L-lyxoses, and L-tagatose are also substrates of GLDH, but these sugars do not meet the substrate specificity rule of GLDH. The oxygen consumption activity of wild-type Gluconobacter frateurii cell membranes depends on several kinds of sugars as compared with that of the membranes of a GLDH-negative variant. Biotransformation of those sugars with the membranes was examined to determine the reaction products. A time course measuring the pH in the reaction mixture and the increase or decrease in substrates and products on TLC suggested that oxidation products of L-lyxose and L-tagatose were ketones with unknown structures, but those of L-ribose and D-lyxose were acids. The oxidation product of L-ribose was purified and revealed to be L-ribonate by HRMS and NMR analysis. Biotransformation of L-ribose with the membranes and also with the whole cells produced L-ribonate in nearly stoichiometric amounts, indicating that the specific oxidation site in L-ribose is recognized by GLDH. Since purified GLDH produced L-ribonate without any intermediate-like compounds, we propose here a reaction model where the first carbon in the pyranose form of L-ribose is oxidized by GLDH to L-ribonolactone, which is further hydrolyzed spontaneously to produce L-ribonate.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Acetic acid bacteria oxidize various sugars and alcohols on the periplasmic surface of the cytoplasmic membrane and accumulate the oxidation products in the medium in a nearly stoichiometric manner (Matsushita et al. 1994). This ability results in incomplete oxidation unique to acetic acid bacteria and is accomplished by a strong oxidation ability, tolerance against oxidized products, and weak metabolic activity on the oxidized product (Nakano and Ebisuya 2016). The metabolism on the product may have a negative effect on productivity. Rather than growing or resting cells, cell membranes can be used to improve productivity (Adachi et al. 2006). The oxidation reaction system consists of the membrane-bound, ubiquinone-reducing dehydrogenase, and terminal ubiquinol oxidase(s) as well as ubiquinone. Membrane-bound, PQQ-dependent polyol dehydrogenase (encoded in the sldBA gene) is known to oxidize various secondary alcohols such as glycerol, D-arabitol, D-mannitol, and D-sorbitol to produce the corresponding ketones, dihydroxyacetone, D-xylulose, D-fructose, and L-sorbose, respectively (Matsushita et al. 2003). For such a wide array of substrates, this enzyme has multiple names: glycerol dehydrogenase (GLDH) (Ameyama et al. 1985), arabitol dehydrogenase (Adachi et al. 2001), sorbitol dehydrogenase (Miyazaki et al. 2002), gluconate 5-dehydrogenase (Salusjärvi et al. 2004), and polyol dehydrogenase (Shinjoh et al. 2002). Because this enzyme oxidizes glycerol at a significant rate, which is the simplest of these structures and found in nature, hereafter, we abbreviate the polyol dehydrogenase as GLDH. The common structure of the substrates of GLDH is “secondary alcohols” in the D-erythro configuration at the penultimate carbon, which is known as the Bertrand-Hudson rule (Hann et al. 1938; Kulhánek 1989; Matsushita et al. 2003). Therefore, products of the oxidation reactions with the enzyme are ketones of the second carbon.
GLDH oxidizes D-lyxose and L-fucose, which do not meet the Bertrand-Hudson rule (Peters et al. 2013). Peters et al. constructed a series of multiple deletion mutant strains and compared the enzyme activity of the constructed bacterial strains. They used whole-cell suspensions as an enzyme source, and evaluated enzyme activity with phenazine methosulfate (PMS) and 2,6-dichlorophenolindophenol (DCPIP), which are an artificial electron mediator and final electron acceptor, respectively. We were inspired by their work on revisiting the substrate specificity of GLDH. As the first step of substrate specificity evaluation, the substrate specificity of GLDH of Gluconobacter thailandicus strains NBRC3258 and NBRC3255 was recently reported to include the oxidations of D-pentonates, D-fructose, and D-psicose producing 4-keto-D-pentonates, 5-keto-D-fructose, and 5-keto-D-psicose, respectively (Ano et al. 2017). In the present study, we focused on the oxidation of sugars by GLDH in our thermotolerant Gluconobacter frateurii strain CHM43 (Moonmangmee et al. 2000) as the second stage of the evaluation. To identify the reaction products, cell membranes instead of the intact cell suspensions were used for the enzyme assays in this study. The sugar oxidation ability of the GLDH-linked respiratory chain, i.e., sugar-dependent oxygen consumption activity, was adopted to evaluate the substrate specificity, to avoid any issues of membrane transport of artificial electron mediators and acceptors.
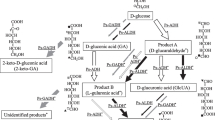
Here, we compared the sugar oxidase activities on the membranes of the wild type Gluconobacter sp. and its ∆sldBA variant. L-lyxose, L-ribose, and L-tagatose were identified as new substrates of GLDH. The reaction product of L-ribose was revealed to be L-ribonate. As shown in Fig. 1, we suggest that GLDH oxidizes the anomeric carbon of L-ribonopyranose to produce L-ribonolactone.

Aldopentoses and ketohexose as new substrates for GLDH of Gluconobacter frateurii proposed in this study. The structures of D-arabitol, D- and L-lyxose, D- and L-ribose, and D- and L-tagatose are shown. D-Arabitol and the open chain form of D-ribose follow the Bertrand-Hudson rule (pale blue) and are oxidized by GLDH to produce ketones at the second carbon, D-xylulose and likely 4-keto-D-ribose, respectively. This study suggests three new substrates for GLDH: L-lyxose, L-ribose, and L-tagatose. D-Lyxose was reported as a substrate for GLDH by Peters et al. This study also shows that D-tagatose is not a substrate for GLDH. L-Lyxose and L-tagatose assume a pyranose form in solution, and one of the secondary alcohols in L-lyxopyranose and L-tagatopyranose have a similar structure (highlighted in pink) and are dehydrogenated by GLDH to produce 2-, 3-, or 4-keto-L-lyxopyranose and 3-, 4-, or 5-keto-L-tagatopyranose, respectively. D-lyxose and L-ribose have a similar structure (highlighted in lime green) in the pyranose form. The anomeric carbon of L-ribopyranose is dehydrogenated to produce L-ribono-1,5-lactone, followed by spontaneous hydrolysis into L-ribonic acid. Oxidation of D-lyxose likely proceeds in a similar manner to that of L-ribose
Materials and methods
Chemicals
D, L-Lyxoses, D, L-riboses, and D, L-tagatoses were purchased from Wako Pure Chemical Industries (Osaka, Japan) at greater than 98% purity. D-Ribonolactone was purchased from Sigma-Aldrich (St. Louis, MO, USA) at greater than 97% purity and used as the calibration standard for L-ribonate quantification. Yeast extract was kindly supplied by Oriental Yeast (Osaka, Japan). MYDOL 10 (40% solution of C9-11-alkyl glucosides) is a kind gift from Kao (Tokyo, Japan). Endonucleases and genetic engineering kits were kind gifts from Toyobo (Osaka, Japan). All other materials used were analytical grade from commercial sources.
Microorganisms and cultivation
G. frateurii strain CHM43 (Moonmangmee et al. 2000), which has been deposited in NBRC (http://www.nite.go.jp/en/nbrc/index.html) as NBRC101659, and its ∆adhBA derivative SEI46, ∆sldBA derivative TORI3, and ∆adhBA ∆sldBA derivative TORI4 were used in this study. YPS medium (3 g of yeast extract, 3 g polypeptone, and 50 g D-sorbitol per liter), G-GA medium (3 g of yeast extract, 3 g polypeptone, 10 g D-glucose, and 10 g Na-D-gluconate per liter), and GY medium (2.5 g of yeast extract and 10 g glycerol per liter) were used. For membrane preparation, cells were cultivated in 500 mL culture medium in a 3-L baffle flask at 30 °C with shaking at 200 rpm. The Escherichia coli strain DH5α was used for plasmid construction (Hanahan 1983). E. coli strains were grown on modified Luria-Bertani medium (Sambrook and Russel 2001), which consists of 10 g of polypeptone, 5 g of yeast extract, and 5 g of NaCl, filled to 1 L with distilled water, and pH adjusted to 7.0 with NaOH. Kanamycin was used at final concentrations of 50 μg mL−1 for both E. coli and G. frateurii. Ampicillin was used at final concentrations of 50 and 500 μg mL−1 for E. coli and G. frateurii, respectively.
Construction of deletion variants of G. frateurii strain CHM43
A 5′ region of the adhAB genes (GLF_0091-0092), the structural gene of membrane-bound alcohol dehydrogenase, from G. frateurii strain CHM43 was amplified by two specific primers, CHM-HinXho-∆adhAB-5(+) and CHM-∆adhAB-5-RV(−) by using Herculase II fusion DNA polymerase (Agilent Technologies, CA, USA). Oligonucleotides used in this study are listed in supplementary Table S1. The amplified 0.8-kb PCR products were digested with HindIII and EcoRV. The whole adhAB genes from the G. frateurii strain CHM43 was amplified by two specific primers, CHM-HinXho-∆adhAB-5(+) and CHM-∆adhAB-3-Xba(−), by using Herculase II fusion DNA polymerase. The amplified 5.0-kb PCR products were digested with EcoRV and XbaI to obtain 0.8-kb DNA fragments that contain the 3′ region of the adhB gene. These two DNA fragments, the 5′ and 3′ regions of the adhAB genes were inserted into the HindIII and XbaI site of pKOS6b (Kostner et al. 2013), a suicide vector that cannot replicate in the G. frateurii CHM43 cells. The resulting plasmid pJ738 carrying the ∆adhAB allele was used for gene disruption.
A 5′ region of the sldBA gene (GLF_2777-2776), the structural gene of GLDH, from the G. frateurii strain CHM43 was amplified by two specific primers, CHM-∆sldB-5-Sal(+) and CHM-∆sldB-5-Sac(−), by using Herculase II fusion DNA polymerase. The amplified 0.9-kb PCR products were digested with SalI and SacI. A 3′ region of the sldA gene from G. frateurii strain CHM43 was amplified by two specific primers, CHM-∆sldA-3-Sac(+) and CHM-∆sldA-3(−), by using Herculase II fusion DNA polymerase. The amplified 0.9-kb PCR products were digested with SacI and EcoRI. These two DNA fragments, 5′ and 3′ regions were inserted into the SalI and EcoRI site of pKOS6b (Kostner et al. 2013) to construct pTT2 carrying the ∆sldBA allele for the gene disruption. The nucleotide sequences of the inserted DNA constructed in this study were confirmed by sequencing.
Fresh G. frateurii CHM43 cells grown on YPS medium were washed with sterile 10% (w/v) glycerol to prepare competent cells. The competent cells were transformed with pTT2 via electroporation at 1.8 kV cm−1, 200 Ω, and 25 μF by using a 1-mm gap cuvette on GenePluser (BIO-RAD Laboratories, Hercules, CA, USA). After incubation of cell suspension in the G-GA medium for 5 h at 30 °C with vigorous shaking, the cell suspension was inoculated on G-GA agar containing 50 μg mL−1 kanamycin and incubated for 2 days at 30 °C. The first recombinant strains were obtained as kanamycin-resistant colonies and were individually inoculated in G-GA medium containing 50 μg mL−1 kanamycin. The ∆sldBA alleles in the first recombinant strain candidates were confirmed by PCR. The first recombinant strain was grown on a G-GA medium free of kanamycin and spread on G-GA agar containing 120 μg mL−1 fluorocytosine (Fluorochem, Glossop, UK). After a 2-day incubation, fluorocytosine-resistant colonies appeared and were examined for kanamycin susceptibility. Fluorocytosine-resistant, kanamycin-sensitive second recombinant strain candidates were examined to determine which sldBA allele exists in the genome by PCR, wild type, or ∆sldBA. The second recombinant strain having the ∆sldBA allele was named TORI3 and used in this study as the derivative of the CHM43 strain defective in the sldBA genes.
Construction of a GLDH-overproducing variant of G. frateurii
The whole sldBA genes from G. frateurii strain CHM43 was amplified using two specific primers, CHM-∆sldB-5-Sal(+) and CHM-∆sldA-3(−), and the Herculase II fusion DNA polymerase. The amplified 4.2-kb PCR products were digested with SalI and EcoRI for insertion behind the lac promoter in pBBR1MCS-4 (Kovach et al. 1995), which had been treated with SalI and EcoRI, to construct pTT1. The G. frateurii strain TORI4, the ∆adhAB ∆sldBA variant of CHM43, was transformed with pTT1 by electroporation as described above. The resulting transformant was used as a GLDH-overproducer.
Preparation of membranes
G. frateurii CHM43 and TORI3 (ΔsldBA) strains were cultivated in either G-GA or GY medium. The cultivation period was 12 h for G-GA medium or 18 h for GY medium, respectively, which corresponds to the late exponential growth phase. The cells were harvested by centrifugation at 9000×g and 4 °C for 10 min and re-suspended in 10 mM 2-(N-morpholino)ethanesulfonate (MES) (K+, pH 6.0) containing 2 mM CaCl2. The re-suspended cells were collected by centrifugation. Cell paste was re-suspended in four volumes (4 ml for 1-g wet weight cell) of 10 mM K+-MES (pH 6.0) containing 2 mM CaCl2 and 0.5 mM phenylmethylsulfonyl fluoride. The cell suspension was passed through a French pressure cell press (1000 kg cm−2). After centrifugation at 10,000×g and 4 °C for 10 min to remove intact cells; the supernatants were centrifuged at 100,000×g and 4 °C for 1 h. The precipitate was re-suspended in the same buffer and used as membranes.
Oxidase assay
Oxidase activity was measured by a Clark-type oxygen electrode (YSI model 5300, Yellow Spring Instrument, Yellow Springs, OH, USA) at 25 °C. The electrode was calibrated by using air-saturated 50 mM Na+-acetate (pH 5.0), assuming the concentration of molecular oxygen to be 249 μM (Mitchell et al. 1979). Sodium dithionite was used for calibration to reduce molecular oxygen completely. The reaction mixture (total volume of 1.5 mL) contained membranes, 50 mM Na+-acetate (pH 5.0), and 100 mM substrate. One unit was defined as a micromolar of half a molecular oxygen (equivalent to oxygen atom) consumed per minute.
Analytical biotransformation
Five milliliters of the reaction mixture consisted of 1.0 mg protein mL−1 of the membranes and 100 mM substrate, D-ribose, L-ribose, D-lyxose, L-lyxose, D-tagatose, or L-tagatose, in 50 mM Na+-acetate (pH 5.0), in a disposable 50-mL plastic tube with a cap that has eight holes with 2-mm diameter, which was shaken at 150 rpm and 30 °C for 24 h. An aliquot (500 μL) of the reaction mixture was taken periodically, and the membranes were removed after centrifugation at 100,000×g and 4 °C for 1 h.
Determination of evaporation factor
Significant amounts of water evaporate, and the reaction mixture is concentrated during the analytical biotransformation described above. Thus, we determined evaporation factors to estimate accurate concentrations of L-ribose and the oxidation product. Five milliliters of 100 mM D-ribose was shaken, and the reaction mixture was withdrawn periodically in the same manner as described above. The amounts of D-ribose were determined as described below to calculate evaporation factors as follows: 0 h, 1.00; 1.5 h, 1.01; 3 h, 1.04; 6 h, 1.05; 12 h, 1.13; 24 h, 1.38.
Preparative biotransformation and purification of the oxidation product from L-ribose
Fifteen milliliters of the reaction mixture consisted of 2.5 mg mL−1 of the CHM43 membranes, and 100 mM of L-ribose were stirred in 50-mL glass beaker at room temperature for 4.5 h. During the stirring, the pH of the reaction mixture was maintained between 4.5 and 6.1 with 1 N NaOH. The reaction mixture was then transferred to a 300-mL flask and shaken at 200 rpm and 30 °C for 19 h. The total reaction period was 24.5 h, and pH of the reaction mixture was adjusted to 5.6 with 1 N NaOH. The membranes were removed by centrifugation at 100,000×g and 4 °C for 1 h.
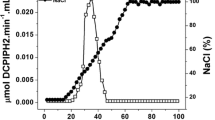
All of the biotransformation mixture (14 mL) was applied to a 2-mL Dowex 1 × 4 (100–200 Mesh) column, which had been activated with 0.1 N NaOH. After washing the column with 10 mL of distilled water, a linear concentration gradient consisting of 20 mL each of distilled water and 50 mM NaCl, and the eluate was fractionated by 2 mL.
NMR analysis
The purified reaction products were freeze-dried and dissolved in D2O (Sigma-Aldrich, St. Louis, MO, USA). 1H NMR spectra were recorded on a Bruker AVANCE 400 (400 Hz).
Analytical procedures
Protein concentrations were determined by the modified Lowry method with bovine serum albumin as the standard (Dulley and Grieve 1975). The amount of L-ribose was determined using a high-performance liquid chromatography (HPLC) system equipped with a Pb2+ cation-exchange column (SUGAR SP0810, 8.0 mm I.D. × 300 mm L; Shodex, Showa Denko KK, Kawasaki, Japan) and a refractive index (RI) detector. The chromatography was run by using distilled water as mobile phase at a flow rate of 0.5 mL min−1 at 80 °C. The amount of the oxidation product produced from L-ribose was determined using an HPLC system equipped with an ion-exclusion column (RSpak KC-811, 8.0 mm I.D. × 300 mm L; Shodex, Showa Denko KK, Kawasaki, Japan) and a diode array detector at 210 nm. The chromatography was run by using 0.1% (w/v) H3PO4 as the mobile phase at flow rate of 0.7 mL min−1 at 60 °C.
For thin layer chromatography (TLC) analysis, samples (typically 2.0 μL) were spotted on the analytical TLC silica gel sheet (aluminum sheet 20 × 20 cm, Merck, Darmstadt, Germany), which had been dried at 121 °C for 10 min prior to use. The chromatography was developed at room temperature with a solvent system consisting of ethyl acetate: acetic acid: methanol: distilled water ratio of 6:1.5:1.5:1. After the silica gel sheet was dried at 121 °C for 10 min, molecules possessing ketone or aldehyde groups were stained by spraying a color-developing reagent consisting of 0.2 g diphenylamine, 200 μL aniline, 10 mL acetone, and 1.5 mL phosphoric acid over the TLC sheet, followed by drying at 121 °C for 2 min.
Purification of GLDH
The membrane fraction containing 279 mg of proteins from the TORI4 strain (CHM43 ΔadhAB ΔsldBA) harboring pTT1 (sldBA+) was suspended with 10 mM MES-KOH (pH 6.0) to a protein concentration of 10 mg ml−1. To avoid releasing PQQ from GLDH, 20 mM CaCl2 and 2 μM PQQ were added and incubated for 10 min on ice before solubilization. MYDOL 10 was added to a final concentration of 0.4%, and incubated for 1 h on ice. After centrifuging at 120,000×g for 1 h, the supernatant including the solubilized enzyme was dialyzed in 10 mM Na+-acetate (pH 5.0) containing 2 mM CaCl2 and 0.1% MYDOL 10. The dialysate was applied to a 5-ml DEAE-Toyopearl column equilibrated with 10 mM Na+-acetate (pH 5.0), containing 2 mM CaCl2 and 0.1% MYDOL 10. After washing with 5 ml of the same buffer, bound proteins were eluted with a linear-gradient system each consisting of 15 ml of 10 mM Na+-acetate (pH 5.0) containing 2 mM CaCl2 and 0.1% MYDOL 10, and 50 mM Na+-acetate (pH 5.0) containing 30 mM CaCl2 and 0.1% MYDOL 10. GLDH activities were detected in the flow-though and gradient fractions, presumably due to over saturation. The active fractions in the flow-through were pooled and dialyzed in 10 mM Na+-acetate (pH 4.5), containing 5 mM CaCl2 and 0.1% MYDOL 10.
The dialysate was applied to a 3-ml CM-Toyopearl column equilibrated with 10 mM Na+-acetate (pH 4.5), containing 5 mM CaCl2 and 0.1% MYDOL 10. After washing with 3 ml of the same buffer, bound proteins were eluted with a linear-gradient system each containing 9 ml of 10 mM Na+-acetate (pH 4.5) containing 5 or 30 mM CaCl2 and 0.1% MYDOL 10. GLDH activities were detected in the flow-through and gradient fractions, presumably due to over saturation. The active fractions in the flow-through were pooled, concentrated with ultrafiltration Amicon Ultra Ultracel-100 K (Millipore, Billerica, MA, USA), and used as purified GLDH.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed on a 12.5% (w/v) acrylamide slab gel by the method described by Laemmli (Laemmli 1970). The proteins were stained with 0.1% (w/v) of Coomassie Brilliant Blue R-250. The following proteins were used as reference for estimation of molecular mass: phosphorylase b (94 kDa), bovine serum albumin (68 kDa), ovalbumin (43 kDa), carbonic anhydrase (31 kDa), trypsin inhibitor (21 kDa), and lysozyme (14 kDa).
Biotransformation of L-ribose with purified GLDH
The reaction mixture (1 mL) consisted of 0.62 mg/ml purified GLDH, corresponding to 0.51 oxygen consumption U/ml, 100 mM L-ribose, 50 mM Na+-acetate (pH 5.0), and 0.2 mM phenazine methosulfate (PMS) with or without 650 U/ml bovine catalase (Wako Pure Chemical Industries, Osaka, Japan) was shaken at 150 rpm and 30 °C for 18 h. The reaction mixtures were withdrawn periodically at 200 μL. In order to remove proteins, the samples were mixed with 30 μL of 20% 5-sulfosalicylic acid and filtered with Millex-LH 0.45 μm (Millipore, Billerica, MA, USA). L-Ribose and the reaction products were quantified by HPLC as described above.
Biotransformation of L-ribose with the whole-cell suspension
G. frateurii CHM43 and TORI3 (ΔsldBA) strains were cultivated in G-GA medium for 12 h, which corresponds to the late exponential growth phase. The cells were harvested by centrifugation at 9000×g and 4 °C for 10 min and re-suspended in 10 mM K+-MES (pH 6.0) containing 2 mM CaCl2. The re-suspended cells were collected by centrifugation, and again re-suspended in one volume (1 ml for 1 g of wet cell) of 10 mM K+-MES (pH 6.0) containing 2 mM CaCl2. A 5-mL suspension, consisting of whole cells, with OD600 of 0.37 units (corresponding to 0.54 oxygen consumption U/mL on the wild-type cells), 100 mM L-ribose, and 50 mM Na+-acetate (pH 5.0) was shaken at 150 rpm and 30 °C for 24 h, as described under the Analytical biotransformation section. Aliquots (500 μL) of the reaction mixtures were withdrawn periodically; the cells were pelleted by centrifugation at 9000×g and 4 °C for 10 min, and the supernatant was filtered with Millex-LH 0.45 μm. L-Ribose and L-ribonate, in the filtrate, were quantified by HPLC, as described above.
Results
Oxygen consumption ability of G. frateurii membranes on various substrates
Peters et al. (Peters et al. 2013) found that D-lyxose and L-fucose are substrates for GLDH but do not follow the Bertrand-Hudson rule. This finding inspired us to search for new reaction products to understand the position of the oxidation site(s) and reaction mechanism underlying oxidations of new substrates. First, we examined GLDH activity on D- and L-fucoses, D- and L-tagatoses, and aldopentoses, but not on xylose and arabinose because D-xylose and L-arabinose are well known as the substrates of the PQQ-dependent, membrane-bound glucose dehydrogenase (Buchert 1991; Meyer et al. 2013). In order to examine substrate specificity of the G. frateurii GLDH using the cell membranes of the wild-type CHM43 strain and its ∆sldBA derivative constructed in this study, oxygen consumption activity was measured with several substrates, D-arabitol (as a positive control substrate), D- and L-lyxoses, D- and L-riboses, D- and L-tagatoses, and D- and L-fucoses. As shown in Fig. 2, oxidase activity with the wild-type membranes on either D-tagatose or L-fucose was negligible, thus indicating that these sugars are not substrates for any dehydrogenases in the cell membranes of the wild type. Other substances were oxidized with the wild-type membranes at different rates. Of these substrates, the oxidation ability on D-lyxose, L-ribose, L-tagatose, and D-arabitol decreased to negligible levels with the deletion of the sldBA genes. Moreover, oxidation activities of L-lyxose and D-ribose were also reduced with the ∆sldBA membranes. Loss of, or reduction in, oxidation activity with the ∆sldBA membranes for D-arabitol, D- and L-lyxoses, D- and L-riboses, and L-tagatose suggests that these substrates are oxidized by GLDH. It is noteworthy that the structure of D- and L-lyxoses, L-riboses, and L-tagatose do not follow the Bertrand-Hudson rule (see Fig. 1).

Substrate specificity of GLDH. Oxidase activity of the membranes from the wild-type (gray bar) and ∆sldBA (white bar) strains grown on G-GA medium were determined by using an oxygen electrode with 100-mM substrate in 50 mM Na+-acetate (pH 5.0) at 25 °C. Mean values with standard deviation from triplicate oxidase assay are shown
Biotransformation of the various substrates by the membranes
Oxidation of D-ribose can be explained by the substrate specificity of GLDH, because the chain form of D-ribose meets the Bertrand-Hudson rule (Fig. 1, highlighted in pale blue), i.e., presumably producing 4-keto-D-ribose. However, because other substrates do not meet the Bertrand-Hudson rule, we anticipated a mechanism where the closed-ring structures of the other aldopentoses and L-tagatose would be involved in the oxidation reactions. L-lyxose and L-tagatose share a common structural element (Fig. 1, highlighted in pink); and D-lyxose and L-ribose have a structural element in common as well (Fig. 1, highlighted in lime green). We supposed that GLDH oxidizes them at other site(s) than hydroxyl group on the penultimate carbon under different rule(s) than the Bertrand-Hudson rule. In order to validate this hypothesis, biotransformation experiments of these five substrates were conducted with wild-type and ∆sldBA membranes, where the pH in the reaction mixtures was recorded, and the substrates and products in the reaction mixtures were analyzed by TLC (Fig. 3).

Time course of the pH change and TLC assay for biotransformation of L-tagatose (a, b), D-ribose (c, d), L-lyxose (e, f), L-ribose (g, h), and D-lyxose (i, j) with the G. frateurii membranes. Membranes (1 mg protein/ml) of the wild-type (closed circle) G. frateurii and ∆sldBA (open circle) strains grown on G-GA medium were incubated with 100 mM each of L-tagatose, D-ribose, L-lyxose, L-ribose, or D-lyxose in 50 mM Na+-acetate (pH 5.0). The mixtures were shaken at 150 rpm and 30 °C for 24 h. The reaction mixtures were withdrawn at the indicated times, and the supernatants were obtained after ultracentrifugation. The pH change of reaction mixtures is shown in a, c, e, g, and i, and TLC analysis for 2 μL of the reaction mixture are shown in b, d, f, h, and j; 50 mM each of L-tagatose, D-ribose, L-lyxose, L-ribose, or D-lyxose as a standard compound was also loaded on the TLC. As for L-ribose (g), mean values with standard deviation from triplicate biotransformation experiments are shown. Aniline-staining solution was sprayed on the plate to visualize ketones and aldehydes. New spots appeared during the biotransformations are indicated by arrowheads
Principally two types of results were obtained: from L-tagatose, D-ribose, and L-lyxose and the other from L-ribose and D-lyxose. Changes in the pH values of reaction mixtures as a function of time in the wild-type and ∆sldBA membranes were similar to each other in the case of L-tagatose until 6 h (Fig. 3a), D-ribose (Fig. 3c), and L-lyxose (Fig. 3e). Conversely, the pH values decreased exclusively with the wild-type membranes but not with the ∆sldBA membranes in the case of L-ribose (Fig. 3g) and D-lyxose (Fig. 3i). On evaluation of the TLC assay, intensities of the substrates would increase gradually as a function of time, if no reaction occurr. It may be because our biotransformation experiments were accompanied by significant amounts of evaporation (see the Materials and Methods section). As the amount of substrate decreased, some faint spots appeared on the TLC plate with the wild-type membranes but not with the ∆sldBA membranes in L-tagatose (Fig. 3b), D-ribose (Fig. 3d), and L-lyxose (Fig. 3f) oxidations. However, no new spots were observed from the oxidation of L-ribose (Fig. 3h) and D-lyxose (Fig. 3j). These results suggest that the reaction products of L-tagatose, D-ribose, and L-lyxose are likely ketone or aldehyde compounds, while those of L-ribose and D-lyxose are likely carbonic acids (see the Discussion section). Since we unexpectedly found that GLDH of G. frateurii CHM43 strain produces an acidic compound by oxidization of L-ribose, the oxidation product was subjected to structural analysis.
Accumulation of the oxidation product in a reciprocal manner to consumption of L-ribose upon biotransformation
For quantitative measurements, L-ribose and the oxidation product in the biotransformation mixture with the wild-type membranes were analyzed by HPLC as described in the Materials and Methods section. Elution profiles of the reaction mixture of L-ribose for 0- and 24-h reactions are shown in Supplementary Fig. S1. L-Ribose eluted at 44.1 min in the sugar column, but it was not detected in the organic acid column. After the 24-h reaction, two peaks were detected that derived from the oxidation product (12.2-min retention time) and acetate (15.4-min retention time) of which the latter was added as the buffer in the biotransformation mixture. The peak for the oxidation product increased by approximately 20 times in a 24-h reaction period. A time-lapse experiment of biotransformation of L-ribose was conducted with both the wild-type and ∆sldBA membranes (Fig. 4). Significant amounts of water seemed to evaporate in this experiment. Thus, we determined the “evaporation factor” in our biotransformation experiments to adjust the quantification of L-ribose and the oxidation products as described in the Materials and Methods section. With the wild-type membranes, the consumption of L-ribose ceased around 10 mM L-ribose left after a 6-h incubation, and the oxidation product also reached a plateau at that time. Thus, a decrease in L-ribose levels was reciprocal with the accumulation of the oxidation product.

Time course of biotransformation of L-ribose and the oxidation product. Membranes of wild-type (closed circle) and ∆sldBA (open circle) G. frateurii strains grown on G-GA medium were incubated with 100-mM L-ribose in 50 mM Na+-acetate (pH 5.0) with shaking at 150 rpm and 30 °C for 0 h (a) and 24 h (b). Reaction products were prepared by ultracentrifugation, and HPLC was performed. Elution profiles of the organic acid column are shown in a and b. The amount of L-ribose (c) and L-ribonate (d) were measured by HPLC. Mean values with standard deviation from triplicate biotransformation experiments are shown
Unlike the wild type, consumption of L-ribose by the ∆sldBA membranes could hardly be observed (Fig. 4a). However, a small accumulation of the oxidation product with the ΔsldBA membranes was observed, even though consumption of L-ribose was limited. A compound with a retention time at 12.2 min in the reaction with the ∆sldBA membranes had a high absorption peak around 260 nm (Supplementary Fig. S2), suggesting that this is different from the compound with the same retention time at 12.2 min in the wild-type reaction mixture showing a relatively low absorption peak around 260 nm (Supplementary Fig. S2). This compound might be produced from acetate, because the absorption peak around 260 nm of the compound with the same retention time at 12.2 min was eliminated, if acetate was omitted from the reaction mixture (Supplementary Fig. S2).
Purification of the oxidation product
In order to purify the reaction product, we omitted the Na+-acetate buffer from the biotransformation mixture, because acetate was one of the major impurities in this experimental setup. Instead, to keep the pH value where the GLDH functions, we titrated the pH in the reaction mixture with NaOH as the biotransformation proceeded. When the reaction mixture was analyzed by HPLC, instead of acetate, two additional peaks were detected at retention times of 17.5 and 25.5 min (Supplemental Fig. S3a). The reaction product was purified with Dowex 1 × 4 anion exchange column chromatography as described in the Materials and Methods section. The purified materials showed the peak of the oxidation product (12.2-min retention time) as the major constituent (Supplementary Fig. S3b).
Identification of the oxidation product of L-ribose
The structure of the purified oxidation product was determined by HRMS and NMR analysis (Supplementary Fig. S4). Analytical data with the oxidation product are summarized below, HRMS: calcd. C5H9O6 for (M-H)−, 165.0399; found, 165.0412. 1H NMR δH (400 MHz, D2O): 3.63–3.68 (m, 1H, H5), 3.80–3.86 (m, 2H, H4, and H5), 3.91 (dd, 1H, J = 7.0, 3.4 Hz, H3), and 4.18 (d, 1H, J = 3.4 Hz, H2); 13C NMR δC (100 MHz, D2O): 62.91 (C5), 71.58 (C4), 73.32 (C3), 73.55 (C2), and 178.26 (C1).
In addition, the presence of -CH(OH)-CH(OH)-CH(OH)-CH2OH is confirmed by the H-H COSY spectrum which revealed the cross peaks of the four signals at δ3.63–3.68, 3.80–3.86, 3.91, and 4.18. The HMBC spectrum showed correlations of H-2 to C-1, C-3, and C-4; H-3 to C-1, C-4, and C-5; H-4 to C-3 and C-5; and H-5 to C-4. From the data above, the oxidation product was identified to be L-ribonate.
Transformation of L-ribose with whole-cell biocatalysis
In order to avoid cytoplasmic catabolism of L-ribose and its oxidation product L-ribonate, we had, so far, examined oxidative biotransformation with cell membranes. However, it is practically preferable that the whole-cell suspensions, rather than cell membranes, catalyze this biotransformation, without any side-reactions, with a reasonably high yield. Thus, we tried to transform L-ribose with a whole-Gluconobacter cell biocatalysis. To facilitate a comparison between the two catalyses, with membranes and whole cells, the wild-type cell suspension with L-ribose oxidase activity (0.54 U/mL) similar to that used in the biotransformation with membranes, was employed in the current experiment. The ∆sldBA cells, having the same cell density unit (as per OD600), were employed for the control experiment. A decrease in pH was observed in the reaction with the wild-type cells, similar to that with the membranes (compare Figs. 3g and 5a), which likely corresponds to a decrease in L-ribose and an increase in L-ribonate (Fig. 5). Chromatograms of the sugar and organic acid columns suggested that the whole-cell biotransformation did not produce high amounts of by-products (data not shown). The yield of biotransformation was 82% (mol/mol), and 4.5% of input L-ribose was retained after the 24-h reaction. Thus, the whole-cell biocatalysis resulted into a fair-yield transformation of L-ribose to L-ribonate, which is lower than, but comparable to, that with the cell membranes (Fig. 4).

Time course of L-ribose and L-ribonate in the whole-cell biotranformation. The whole-cell suspensions of wild-type (closed circle) and ∆sldBA (open circle) G. frateurii strains, grown on G-GA medium, were incubated with 100-mM L-ribose in 50 mM Na+-acetate (pH 5.0), being shaken at 150 rpm and 30 °C for 24 h. Reaction products were prepared by centrifugation, following which HPLC was performed. The pH change of the reaction mixtures is shown in a. The amount of L-ribose (b) and L-ribonate (c) was measured by HPLC. Mean values with standard deviation from triplicate biotransformation experiments are shown
In the biotransformation using ∆sldBA cells, a significant decrease in pH was observed, contrary to that with the membranes (compare Figs. 3g and 5a), suggesting the formation of some acidic products. The ∆sldBA cells produced the compound with retention time of 12 min, similar to that with the membranes. However, this compound did not have an absorption peak around 260 nm, indicating that it is different from that produced in the biotransformation with the membranes (Supplementary Fig. S5). Moreover, similar to L-ribonate, this compound showed an absorption shoulder around 220 nm, but the absorption ratio between the peaks at 190 and at 220 nm was much less than that of the corresponding compound produced with the wild-type cell suspension (Supplementary Fig. S5a).
Biotransformation of L-ribose with purified GLDH
When GLDH oxidizes L-ribose, L-ribonolactone is expected to be produced as the reaction product (see Fig. 1). However, the major reaction product (12.2-min retention time) in the biotransformation of L-ribose with the membrane was not L-ribonolactone but L-ribonate, although it is reasonable to postulate that hydrolysis of L-ribonolactone is involved in this biotransformation. In order to know whether the hydrolysis occurs spontaneously or in an enzyme-dependent manner, we tried biotransformation of L-ribose by using purified GLDH supplemented with phenazine methosulfate (PMS) as an electron mediator between GLDH and molecular oxygen.
We constructed a GLDH-overproducing G. frateurii strain (TORI4 harboring pTT1) as described in the Materials and Methods section. GLDH was solubilized from the membranes of the overproducing strain with alkylglucosides and purified by using two kinds of column chromatography, DEAE-Toyopearl and CM-Toyopearl, as described previously (Adachi et al. 2001). The procedures were modified as described in the Materials and Methods section. The purities at each purification step were evaluated by SDS-PAGE (Fig. 6). The purified enzyme solution had a specific GLDH activity of 16.54 U/mg and was used for biotransformation of L-ribose.

Time course of biotransformation of L-ribose with purified GLDH. a A model for the reaction of the transformation of L-ribose with purified GLDH assisted by catalase. PMSOx, oxidized form of PMS; PMSRed, reduced form of PMS; Cat, catalase. b The reaction mixture consisting of purified GLDH and catalase (closed circles) and purified GLDH only (open circles) were incubated with 100-mM L-ribose and 0.2 mM PMS in 50 mM Na+-acetate (pH 5.0). Mixtures were shaken at 150 rpm and 30 °C for 18 h. Aliquots of the reaction mixture (200 μl) were withdrawn periodically and treated with 5-sulfosalicylic acid to remove proteins. The amount of L-ribose and L-ribonate were measured by HPLC
It was reported that the reduced form of PMS reacts with molecular oxygen and produces a one-electron reduced form of molecular oxygen, the superoxide radical (Nishikimi et al. 1972). If that is the case in our experimental setup, superoxide dismutase (SOD) would decrease oxygen consumption according to the catalysis: 2O2• + 2H+ ➔ O2 + H2O2. However, an excess amount of SOD did not affect the PMS-dependent oxygen consumption of GLDH, instead, an excess amount of catalase decreased the oxygen consumption velocity to about half of that without it (data not shown). These data suggest that PMS-dependent oxidation by GLDH produces hydrogen peroxide. Another report suggests that hydrogen peroxide production via reduction of PMS by the action of succinate dehydrogenase (Kearney and Singer 1956). If that is the case in our experiment, catalase “regenerates” molecular oxygen as follows: 2H2O2 ➔ O2 + H2O. Because hydrogen peroxide is a reactive molecular species and may inactivate GLDH activity during the biotransformation reaction, we added a reasonable amount of catalase to the reaction as described in the Materials and Methods section to eliminate hydrogen peroxide. Overall, the reaction in our biotransformation experiment with the catalase can be assumed to be as seen in Fig. 6a.
Because a significant amount of water evaporated, we estimated the evaporation factor by using the concentration of acetate to adjust the quantification of L-ribose and the oxidation products. In this biotransformation experiment, acetate was not involved in the production of the unknown compound that was described in the biotransformation of L-ribose with the membranes; therefore, acetate can be considered a stable solute. L-ribose was oxidized at a slower rate than expected, but L-ribonate was produced reciprocally (Fig. 6b). No significant signals in HPLC analysis were observed other than L-ribose, acetate, and L-ribonate in the sugar and organic acid columns. These results suggest that L-ribonate can be produced without any lactone-hydrolyzing enzymes.
Discussion
By comparing the oxygen-consumption activities of the wild-type and ∆sldBA membranes, we examined aldopentoses and ketohexoses as the substrates for GLDH. L-lyxose, L-ribose, and L-tagatose were found to be new substrates of GLDH. A similar but more comprehensive experimental setup was reported by Peters et al. to identify substrates for characterized and uncharacterized membrane-bound dehydrogenases of G. oxydans strain 621H (Peters et al. 2013). Peters et al. constructed a series of gene-deletion mutants of the dehydrogenases and concluded that L-fucose, D-lyxose, and D-ribose are substrates of GLDH by comparison of dehydrogenase activities of the ∆sldBA derivative and its parental strain by using an artificial electron acceptor with whole-cell preparations. In order to avoid a cytoplasmic catabolism of various substrates examined in this study, we prepared cell membranes and assayed them for oxygen consumption activity. Our Gluconobacter strain showed D-lyxose and D-ribose oxidase activity by GLDH, but did not show L-fucose oxidase activity in the membranes. Oxidation of L-lyxose will be discussed later, but oxidation of D-ribose follows the Bertrand-Hudson rule, producing an open-chain form of 4-keto-D-ribose as suggested in Fig. 1.
As for D-fucose oxidation, it was highly oxidized by the membranes of the ∆sldBA strain. Membrane-bound glucose dehydrogenase (GDH) oxidizes D-fucose (Meyer et al. 2013). GDH activity was elevated in the cell membranes of our ∆sldBA or ∆adhAB strain (data not shown) presumably due to limited space on the cytoplasmic membrane of the wild-type strain, so that it is assumed that elevated D-fucose oxidase activity is attributed to elevated levels of GDH.
We suggest that L-lyxose, L-ribose, and L-tagatose are new substrates for GLDH. Open-chain forms of L-lyxose and L-tagatose have a common structural feature, the threo configuration in the third and fourth carbon in L-lyxose and fourth and fifth carbon in L-tagatose. Thus, they do not follow the Bertrand-Hudson rule in the open-chain forms. However, the sugars undergo a structural change into the closed-ring structure; aldopentoses and ketohexoses often take the pyranose from within the solution (Angyal 1969; Que and Gray 1974). GLDH (polyol dehydrogenase, the sldBA gene products) of strain 621H oxidizes cyclic alcohols 1,2-cyclopentanediol, 1,3-cyclopentanediol, and 1,2-cyclohexanediol in the whole-cell dehydrogenase assays (Peters et al. 2013). Moonmangmee et al. (Moonmangmee et al. 2001) also reported that the PQQ-dependent “cyclic alcohol dehydrogenase” that oxidizes a wide variety of polyols including cyclic alcohols was purified from G. frateurii strain CHM9. It can be concluded that the “cyclic alcohol dehydrogenase” may be GLDH in this bacterial strain. Because the structures of the closed-ring form of the sugars are similar to those of the cyclic alcohols, it might be unsurprising that GLDH oxidizes L-tagatose and L-lyxose as reported in this study.
GLDH-dependent oxidation products from L-tagatose and L-lyxose showed aniline-positive spots in the TLC analysis, suggesting production of ketones or aldehydes (Fig. 3). Moreover, the decline in pH in the reaction mixtures was observed to be similar between the wild-type and ∆sldBA membranes (until 6 h for L-tagatose), suggesting that acidification caused by L-lyxose oxidation may not be due to GLDH and thus the products are neutral, i.e., ketones or aldehydes. Based on the structural similarity of L-lyxose and L-tagatose, the first carbon on L-tagatose is not likely to be the site of oxidation. If that is the case, then the reaction products are limited to ketones. We suggest that the oxidation products are 3-keto-L-tagatose, 4-keto-L-tagatose, or 5-keto-L-tagatose from L-tagatose and 2-keto-L-lyxose, 3-keto-L-lyxose, or 4-keto-L-lyxose from L-lyxose (Fig. 1). We tried to isolate the oxidation product from L-tagatose to determine its structure, but have not yet completed it.
We successfully purified the oxidation product from L-ribose. HRMS and NMR analysis of the purified product strongly suggested that it is L-ribonate. This conclusion is consistent with other experimental results, i.e., the pH of the reaction mixture rapidly decreased over the course of the reactions. Additionally, no aniline-positive spots were detected in the TLC analysis of reaction products, suggesting the product is neither an aldehyde nor a ketone. In addition to those observations, time-lapse experiments clearly indicated that a decrease in the amount of L-ribose corresponds to the increase in L-ribonate. These lines of evidence suggest that the site specificity of L-ribose oxidation by GLDH is high, i.e., only the hydroxyl group on the anomeric carbon is oxidized. To our knowledge, this is the first study on GLDH on the substrate and site specificity and, as discussed later, the first oxidation product is L-ribonolactone.
It can be theoretically assumed that GLDH oxidizes the aldehyde group in the chain form of L-ribose to produce L-ribonic acid by a catalytic mechanism similar to that of aldehyde dehydrogenase. However, the experimental evidence of GLDH does not support such a possibility (Mientus et al. 2017; Moonmangmee et al. 2001; Peters et al. 2013). Rather, it is reasonable to conclude that GLDH oxidizes the anomeric hydroxyl group of L-ribopyranose. We anticipated a molecular event between the oxidation of L-ribopyranose and production of L-ribonate, i.e., hydrolysis and the ring opening reaction of L-ribonolactone. By using the reactivity of the reduced form of PMS to molecular oxygen, we tried to biotransform L-ribose with purified GLDH. It is noteworthy to mention that the product from reduced PMS can be assumed to be hydrogen peroxide rather than the superoxide radical because catalase but not SOD-regenerated molecular oxygen in our oxygen consumption assay. Although oxidation yields were much lower than our expectations, correlations between decreases in L-ribose and increases in L-ribonate were observed without occurrence of any intermediate-like compounds. Therefore, it is plausible to conclude that L-ribonolactone produced by the catalytic function of GLDH is hydrolyzed spontaneously to the open structure. Moreover, we suggest that D-lyxose is oxidized in a similar manner to L-ribose, because the behavior on D-lyxose oxidation with the membranes was similar to that on L-ribose oxidation, i.e., GLDH oxidize D-lyxose, likely to produce D-lyxonate (Fig. 1).
Not only the one with cell membranes, whole-cell biocatalysis also achieved a fair-yield transformation of L-ribose to L-ribonate (Fig. 5), which is also practically feasible. With the ∆sldBA cell suspension, some acidic compounds might be produced slowly (Fig. 5), although no strong signals were detected in the analytical HPLC. Additionally, a small amount of the 12-min compound, having similar absorption properties as of L-ribonate was produced by the ∆sldBA cells, which may be tentatively assigned as L-ribonate (Fig. 5c). If that is the case, some cytoplasmic, NAD(P)+-dependent aldose dehydrogenases and/or aldo-keto reductases might be responsible for the L-ribonate production (Adachi et al. 1980; Adachi et al. 1999; Liu et al. 2011; Moonmangmee et al. 2000). Finally, we compared the two biotransformation systems using the membranes and the purified GLDH. In the membrane system, total enzyme activity (oxygen consumption) in the 5-ml reaction mixture was 2.7 μmol of L-ribose oxidized/min, and the actual L-ribonate production rate calculated from the data for 0–3 h was 19 mM/h (Fig. 4b); thus, 1.6 μmol of L-ribonate was produced per minute in the 5-ml reaction. The ratio of actual production to expected enzyme activity was about 60%. However, in the purified GLDH system, total enzyme activity (oxygen consumption) in the 1-ml reaction mixture was 0.52 μmol of L-ribose oxidized/min, and the actual L-ribonate production rate calculated from the data of 0–3 h was 2.2 mM/h (Fig. 6b); thus, 0.037 of μmol L-ribonate produced/min in the 1-ml reaction. The ratio of actual production to expected enzyme activity was about 7.1%. The difference in the reaction efficiency between two biotransformation systems clearly explains the unexpectedly slow production by the purified GLDH system. Moreover, L-ribonate production ceased over the course of the reaction, particularly after 3-h. Catalase indeed increased L-ribonate production, but even in the presence of catalase, GLDH may be exposed to hydrogen peroxide to some extent, which is likely harmful to the enzyme. Moreover, the solubilized state of GLDH may not be suitable to maintain the enzyme activity. Such unfavorable circumstances might have inhibited enzyme activity in our biotransformation experiment.
References
Adachi O, Ano Y, Toyama H, Matsushita K (2006) Enzymatic preparation of metabolic intermediates, 3-dehydroquinate and 3-dehydroshikimate, in the shikimate pathway. Biosci Biotechnol Biochem 70:3081–3083 Epub 2006 Dec 7
Adachi O, Fujii Y, Ghaly MF, Toyama H, Shinagawa E, Matsushita K (2001) Membrane-bound quinoprotein D-arabitol dehydrogenase of Gluconobacter suboxydans IFO 3257: a versatile enzyme for the oxidative fermentation of various ketoses. Biosci Biotechnol Biochem 65:2755–2762
Adachi O, Matsushita K, Shinagawa E, Ameyama M (1980) Crystallization and characterization of NADP-dependent D-glucose dehydrogenase from Gluconobacter suboxydans. Agric Biol Chem 44:301–308. https://doi.org/10.1271/bbb1961.44.301
Adachi O, Toyama H, Matsushita K (1999) Crystalline NADP-dependent D-mannitol dehydrogenase from Gluconobacter suboxydans. Biosci Biotechnol Biochem 63:402–407. https://doi.org/10.1271/bbb.63.402
Ameyama M, Shinagawa E, Matsushita K, Adachi O (1985) Solubilization, purification and properties of membrane-bound glycerol dehydrogenase from Gluconobacter industrius. Agric Biol Chem 49:1001–1010. https://doi.org/10.1271/bbb1961.49.1001
Angyal SJ (1969) The composition and conformation of sugars in solution. Angew Chem Int Ed Engl 8:157–166. https://doi.org/10.1002/anie.196901571
Ano Y, Hours RA, Akakabe Y, Kataoka N, Yakushi T, Matsushita K, Adachi O (2017) Membrane-bound glycerol dehydrogenase catalyzes oxidation of D-pentonates to 4-keto-D-pentonates, D-fructose to 5-keto-D-fructose, and D-psicose to 5-keto-D-psicose. Biosci Biotechnol Biochem 81:411–418
Buchert J (1991) A xylose-oxidizing membrane-bound aldose dehydrogenase of Gluconobacter oxydans ATCC 621. J Biotechnol 18:103–113. https://doi.org/10.1016/0168-1656(91)90239-R
Dulley JR, Grieve PA (1975) A simple technique for eliminating interference by detergents in the Lowry method of protein determination. Anal Biochem 64:136–141
Hanahan D (1983) Studies on transformation of Escherichia coli with plasmids. J Mol Biol 166:557–580
Hann RM, Tilden EB, Hudson CS (1938) The oxidation of sugar alcohols by Acetobacter suboxydans. J Am Chem Soc 60:1201–1203. https://doi.org/10.1021/ja01272a058
Kearney EB, Singer TP (1956) Studies on succinic dehydrogenase: I. Preparation and assay of the soluble dehydrogenase. J Biol Chem 219:963–975
Kostner D, Peters B, Mientus M, Liebl W, Ehrenreich A (2013) Importance of codB for new codA-based markerless gene deletion in Gluconobacter strains. Appl Microbiol Biotechnol 97:8341–8349
Kovach ME, Elzer PH, Hill DS, Robertson GT, Farris MA, Roop RM 2nd, Peterson KM (1995) Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166:175–176
Kulhánek M (1989) Microbial dehydrogenations of monosaccharides. In: Neidleman SL (ed) Adv Appl Microbiol (vol 34). Academic Press, pp 141–182
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Liu X, Yuan Z, Adam Yuan Y, Lin J, Wei D (2011) Biochemical and structural analysis of Gox2181, a new member of the SDR superfamily from Gluconobacter oxydans. Biochem Biophys Res Commun 415:410–415. https://doi.org/10.1016/j.bbrc.2011.10.083
Matsushita K, Fujii Y, Ano Y, Toyama H, Shinjoh M, Tomiyama N, Miyazaki T, Sugisawa T, Hoshino T, Adachi O (2003) 5-keto-D-gluconate production is catalyzed by a quinoprotein glycerol dehydrogenase, major polyol dehydrogenase, in Gluconobacter species. Appl Environ Microbiol 69:1959–1966
Matsushita K, Toyama H, Adachi O (1994) Respiratory chains and bioenergetics of acetic acid bacteria. In: Rose AH, Tempest DW (eds) Adv Microb Physiol, vol vol 36. Academic Press, London, pp 247–301
Meyer M, Schweiger P, Deppenmeier U (2013) Effects of membrane-bound glucose dehydrogenase overproduction on the respiratory chain of Gluconobacter oxydans. Appl Microbiol Biotechnol 97:3457–3466
Mientus M, Kostner D, Peters B, Liebl W, Ehrenreich A (2017) Characterization of membrane-bound dehydrogenases of Gluconobacter oxydans 621H using a new system for their functional expression. Appl Microbiol Biotechnol 101:3189–3200. https://doi.org/10.1007/s00253-016-8069-4
Mitchell P, Moyle J, Mitchell R (1979) Measurement of translocation of H+/O in mitochondria and submitochondrial vesicles. Methods Enzymol 55:627–640
Miyazaki T, Tomiyama N, Shinjoh M, Hoshino T (2002) Molecular cloning and functional expression of D-sorbitol dehydrogenase from Gluconobacter suboxydans IFO3255, which requires pyrroloquinoline quinone and hydrophobic protein SldB for activity development in E. coli. Biosci Biotechnol Biochem 66:262–270
Moonmangmee D, Adachi O, Ano Y, Shinagawa E, Toyama H, Theeragool G, Lotong N, Matsushita K (2000) Isolation and characterization of thermotolerant Gluconobacter strains catalyzing oxidative fermentation at higher temperatures. Biosci Biotechnol Biochem 64:2306–2315
Moonmangmee D, Fujii Y, Toyama H, Theeragool G, Lotong N, Matsushita K, Adachi O (2001) Purification and characterization of membrane-bound quinoprotein cyclic alcohol dehydrogenase from Gluconobacter frateurii CHM 9. Biosci Biotechnol Biochem 65:2763–2772
Nakano S, Ebisuya H (2016) Physiology of Acetobacter and Komagataeibacter spp.: acetic acid resistance mechanism in acetic acid fermentation. In: Matsushita K, Toyama H, Tonouchi N, Okamoto-Kainuma A (eds) Acetic acid bacteria: ecology and physiology. Springer Japan, Tokyo, pp 223–234
Nishikimi M, Appaji Rao N, Yagi K (1972) The occurrence of superoxide anion in the reaction of reduced phenazine methosulfate and molecular oxygen. Biochem Biophys Res Commun 46:849–854. https://doi.org/10.1016/S0006-291X(72)80218-3
Peters B, Mientus M, Kostner D, Junker A, Liebl W, Ehrenreich A (2013) Characterization of membrane-bound dehydrogenases from Gluconobacter oxydans 621H via whole-cell activity assays using multideletion strains. Appl Microbiol Biotechnol 97:6397–6412
Que L Jr, Gray GR (1974) Carbon-13 nuclear magnetic resonance spectra and the tautomeric equilibriums of ketohexoses in solution. Biochemistry 13:146–153. https://doi.org/10.1021/bi00698a023
Salusjärvi T, Povelainen M, Hvorslev N, Eneyskaya EV, Kulminskaya AA, Shabalin KA, Neustroev KN, Kalkkinen N, Miasnikov AN (2004) Cloning of a gluconate/polyol dehydrogenase gene from Gluconobacter suboxydans IFO 12528, characterisation of the enzyme and its use for the production of 5-ketogluconate in a recombinant Escherichia coli strain. Appl Microbiol Biotechnol 65:306–314
Sambrook J, Russel DW (2001) Molecular cloning: a laboratory manual, 3rd edn. Cold SpringHarbor Laboratory Press, Cold Spring Harbor, NY
Shinjoh M, Tomiyama N, Miyazaki T, Hoshino T (2002) Main polyol dehydrogenase of Gluconobacter suboxydans IFO 3255, membrane-bound D-sorbitol dehydrogenase, that needs product of upstream gene, sldB, for activity. Biosci Biotechnol Biochem 66:2314–2322
Acknowledgements
We are grateful to Armin Ehrenreich (Technische Universität München, Germany) for kindly providing pKOS6b to us. We thank Yuka Narita, Takahiro Torikai, and Koichi Furuya (Yamaguchi University, Japan) for their technical assistances.
Funding
This study was funded by MEXT KAKENHI (grant numbers 17K07722 to TY; 2660068 to KM).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary material
ESM 1
(PDF 929 kb)
Rights and permissions
About this article

Cite this article
Yakushi, T., Terada, Y., Ozaki, S. et al. Aldopentoses as new substrates for the membrane-bound, pyrroloquinoline quinone-dependent glycerol (polyol) dehydrogenase of Gluconobacter sp.. Appl Microbiol Biotechnol 102, 3159–3171 (2018). https://doi.org/10.1007/s00253-018-8848-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-018-8848-1