
Abstract
Acetic acid bacteria oxidize a great number of substrates, such as alcohols and sugars, using different enzymes that are anchored to the membrane. In particular, Gluconacetobacter diazotrophicus is distinguished for its N2-fixing activity under high-aeration conditions. Ga. diazotrophicus is a true endophyte that also has membrane-bound enzymes to oxidize sugars and alcohols. Here we reported the purification and characterization of the membrane-bound glucose dehydrogenase (GDHm), an oxidoreductase of Ga. diazotrophicus. GDHm was solubilized and purified by chromatographic methods. Purified GDHm was monomeric, with a molecular mass of 86 kDa. We identified the prosthetic group as pyrroloquinoline quinone, whose redox state was reduced. GDHm showed an optimum pH of 7.2, and its isoelectric point was 6.0. This enzyme preferentially oxidized d-glucose, 2-deoxy-d-glucose, d-galactose and d-xylose; its affinity towards glucose was ten times greater than that of E. coli GDHm. Finally, Ga. diazotrophicus GDHm was capable of reducing quinones such as Q 1, Q 2, and decylubiquinone; this activity was entirely abolished in the presence of micromolar concentrations of the inhibitor, myxothiazol. Hence, our purification method yielded a highly purified GDHm whose molecular and kinetic parameters were determined. The possible implications of GDHm activity in the mechanism for reducing competitor microorganisms, as well as its participation in the respiratory system of Ga. diazotrophicus, are discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Gluconacetobacter diazotrophicus (Ga. diazotrophicus; formerly Acetobacter diazotrophicus) belongs to the acetic acid bacteria group. It is an aerobic Gram-negative bacterium that performs nitrogen fixation and is a true endophyte isolated from sugar cane [2, 10, 22, 36]. Among the diazotrophs, Ga. diazotrophicus is an extremely aerotolerant organism [52]; it has a respiratory system with unique properties associated with its lifestyle, specifically to its aerotolerant diazotrophic capacity [20]. Like many acetic acid bacteria, Ga. diazotrophicus contains a broad range of active membrane-bound dehydrogenases that donate electrons directly to the respiratory chain [20]. In addition, they are capable of partially oxidizing different alcohols and sugars in processes referred to as “oxidative fermentation”, accumulating large amounts of the oxidation products in the growth medium [43]. When Ga. diazotrophicus use glucose as a substrate, respiratory levels four times larger than those found in common aerobic bacteria have been reported [20]. This finding suggests particular kinetic characteristics for the entire system, specifically for dehydrogenases, such as membrane-bound glucose dehydrogenase (GDHm). Currently, the kinetic and molecular properties of this enzyme in Ga. diazotrophicus are unknown.
In general, dehydrogenases of acetic acid bacteria have their active site oriented towards the periplasmic space and can involve cofactors such as pyrroloquinoline quinone (PQQ) and flavin adenine dinucleotide (FAD). Other dehydrogenases can have associated c-type cytochromes [28]. Among the dehydrogenases with quinone cofactors are the cytosolic glucose dehydrogenases (GDHs). These GDHs were initially described in Bacterium anitratum [30] and later reported in many Gram-negative bacteria, including enteric bacteria, aerobic bacteria such as Pseudomonas, and acetic acid bacteria [43].
Currently, two types of glucose dehydrogenase are known: (1) the membrane-bound form (GDHm), which is the most ubiquitous in bacteria; and (2) the soluble form (GDHs), described in A. calcoaceticus [14]. The presence of a pyrroloquinoline quinone-dependent membrane-bounded glucose dehydrogenase (PQQ-GDH) was reported in A. diazotrophicus by Attwood et al. [6]; it is responsible for the periplasmic conversion of glucose to gluconate. Later, it was also identified in Acinetobacter calcoaceticus [40], Pseudomonas sp. [38], Gluconobacter suboxydans [3], Klebsiella aerogenes [33] and Rhizobium sp. [7], indicating a broad distribution among Gram-negative bacteria. Although GDHm and GDHs oxidize d-glucose, they slightly differ in properties such as substrate specificity, oligomeric composition and stability [28].
GDHm contains PQQ as a prosthetic group and catalyzes the direct oxidation of d-glucose to d-gluconate in the periplasm space, subsequently donating electrons to the respiratory chain via ubiquinone (Q10) [54]. GDHm has an N-terminal domain oriented towards the cytoplasm, besides to five transmembrane segments that serve as anchors for the protein, and a C-terminal domain oriented towards the periplasmic side of the membrane [54]. Furthermore, GDHm has a super barrel structure consisting of eight antiparallel β-sheets arranged with radial symmetry, similar to a propeller’s blades; it has a catalytic site in its interior that binds PQQ [12] as well as Ca2+ or Mg2+ [28].
GDHm is present in various acetic acid organisms, such as A. calcoaceticus [40] and G. suboxydans [41], in aerobic bacteria, such as Pseudomonas aeruginosa [48], and in enteric bacteria, such as Escherichia coli [18], where it is reported as a monomer of 82–88 kDa. In E. coli, GDHm is an apoprotein because it does not synthesize PQQ. However, the enzymatic activity can be reconstituted by the exogenous addition of PQQ [45]. GDHm can also react with quinone analogs, and its activity can be reconstituted in a proteoliposome system [41].
In this work, we report a method for the purification and characterization of Ga. diazotrophicus GDHm and have identified the presence of the organic cofactor PQQ using spectroscopic assays and high-performance liquid chromatography (HPLC). We determined the kinetic parameters for diverse sugars, optimum pH and thermostability. We also evaluated the effect of myxothiazol, an inhibitor of the mitochondrial bc 1 complex [35], on GDHm activity. Interestingly, myxothiazol inhibited quinone reductase activity, suggesting that it can affect the quinone binding site.
2 Materials and Methods
2.1 Chemicals
All chemical regents used in this work have been previously described [23, 24] and were purchased from Sigma-Aldrich, SUPELCO-Sigma-Aldrich (St. Louis MO, USA) and Bio-Rad Laboratories (Hercules, CA, USA).
2.2 Strain, Growth Conditions, Preparation of Membranes and Culture Methods
Gluconacetobacter diazotrophicus PAL5 (ATCC 49037), was grown under the conditions reported [50], using a LGIP modified medium (sucrose instead glucose) supplemented with 1 mM (NH4)2SO4 [20]. Four liters of an active culture were obtained growing aerobically at 30 °C for 24 h, and were used to inoculate a bioreactor Bioflow 5000 fermentor (New Brunswick Scientific), containing 60 L of medium. The cells were grown at 30 °C, 60 L of air per min−1 and stirred at 120 rpm. Next, the cells were harvested at the end of the logarithmic phase (42 h) with a continuous flow Sharples centrifuge (Sharples Stokes, S.A. de C.V.). Then, the cells were washed threefold with 50 mM K2HPO4, pH 6.5 containing 1 mM MgCl2 and 1 mM CaCl2. Procedures used for cell disruption, and membranes preparations have been described previously [19, 23, 24]. Membranes were instantly frozen in liquid N2 in order to preserve and store them.
2.3 Purification of the Membrane-Bound Ga. diazotrophicus GDHm
To obtain the solubilized GDHm, a differential solubilization with Triton X-100 was performed. We first removed the membrane-bound alcohol (ADH) and aldehyde (ALDH) dehydrogenases. For this aim, the membranes (10 mg protein mL−1) were suspended in 10 mM potassium phosphate buffer, pH 6.0 (KP buffer) supplemented with Triton X-100 to a final concentration of 0.3 % (v/v). Then, the sample was centrifuged at 144,000×g at 4 °C for 60 min. The supernatant containing the ethanol and aldehyde dehydrogenase activities was removed. The residual membrane particles were resuspended (10 mg protein mL−1) in KP buffer supplemented with 1 % Triton X-100. The suspension was incubated for 120 min at 4 °C under gentle shaking and then, centrifuged for 30 min (86,000×g) at 4 °C. The supernatant containing the GDHm activity was dialyzed extensively against KP buffer, pH 6.0 plus 0.1 % Triton X-100 (KP-T buffer). The sample was gotten into an anion-exchange column (QAE-Toyopearl; 5 × 20 cm) pre-equilibrated with PB-T buffer. The column was washed until the absorbance at 280 nm of the mobile phase decayed to zero. Then, the GDHm was eluted from column by using a linear gradient from 0 to 0.25 M NaCl in the mobile phase. The fractions containing GDHm activity were collected and dialyzed extensively against 10 mM sodium acetate buffer, pH 5.5 plus 0.1 % Triton X-100 (SA-T buffer). After that, the sample was applied to a cation-exchange column (CM-Toyopearl; 3 × 18 cm) pre-equilibrated with SA-T buffer. The GDHm activity was obtained in the washed volume. This fraction was collected, concentrated by ultrafiltration and applied to a gel filtration column (Sephacryl S-200; 3 × 120 cm) pre-equilibrated with KP-T buffer. The fractions with GDHm activity were pooled, concentrated and stored at 4 °C without appreciable loss of activity. All the process of purification was performed at 4 °C.
3 Analytical Procedures
3.1 Electrophoresis
The purity of GDHm was analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) by using 10 % polyacrylamide, following the method of Goodhew et al. [27]. For the zymography of the purified GDHm, a native PAGE was performed where SDS was replaced by 0.1 %Triton X-100 as reported [23]. The zymography activity for the GDHm was assayed employing a medium containing 0.34 mM nitro blue tetrazolium (NTB), 2 mM Phenazine methasulfate (PMS) and 100 mM d-glucose as substrate was used. For total protein, the gels were stained with silver or Coomassie Brilliant Blue R-250. The isoelectric point (pI) of the purified GDHm from Ga. diazotrophicus was performed according with the previously reported by Gomez-Manzo et al. [23, 25], in which a Phast system (Amersham Biosciences) and gels with a pH range of 3–9 were used.
3.2 Identification of PQQ
The presence of the PQQ prosthetic group into GDHm purified from Ga. diazotrophicus was determined by two different strategies. In the first strategy, we identified the PQQ through UV–visible analysis as reported previously for other preparations containing PQQ [23]. Briefly, purified protein (0.2 mg) was suspended in PB-T buffer and its spectra were recorded with an OLISSLM DW2000 spectrophotometer. The samples were reduced with sodium dithionite, whereas reference standards (exogenous PQQ purchased from Sigma Aldrich) were oxidized with ammonium persulfate.
In the second strategy, fluorescence experiments were performed according to the procedure previously reported [23]. For this approach, the prosthetic group was first extracted from the purified enzyme solution by mixing it with nine volumes of methanol. After 30 min of incubation at 25 °C, the extract was concentrated by evaporation; this extract was named as PQQ fraction. The PQQ fraction was scanned from 290 to 460 nm in a Perkin-Elmer LS-55 spectrofluorometer; an excitation wavelength of 370 nm was used for detecting PQQ as previously described by Matsushita et al. [44].
3.3 Determination of PQQ by Reverse-Phase HPLC (RP-HPLC)
To determine the redox properties of the prosthetic group of GDHm from Ga. diazotrophicus, the enzyme was subject to 90 % methanol to extract the PQQ as reported before [23, 44]. After that, the extracted PQQ and the standards quinones were analyzed by RP-HPLC (Waters model 996). The prosthetic group was separated and identified in a RP-analytical column (Waters C18 Spherisorb S5 OD52; 4.6 × 150 mm) as described [24–26]. The system was calibrated using the following commercial standards: methanol-extracted PQQ associated with the purified ADHa and ALDH from Ga. diazotrophicus (retention times: 4.5 and 6.9 min, respectively). Commercial PQQ reduced with dithionite (PQQH2) and PQQ oxidized with ammonium persulfate (PQQ), Q7 and Q10 were used as standards (RT: 4.1, 6.8, 9.28, and 11.19 min, respectively). The concentration of PQQ of the GDHm from Ga. diazotrophicus was calculated from the area under the peak at 275 nm using commercial PQQ as a standard.
3.4 Thermal Inactivation of Purified GDHm
Thermal inactivation curves of purified GDHm were performed with d-glucose as substrate. The residual activity was measured with PMS plus DCPIP as an electron carrier and an electron acceptor, respectively [1]. The enzyme (0.2 mg mL−1) was suspended in KP buffer, pH 6.0 plus 0.1 % Triton X-100, and was incubated at a constant temperature (40 °C), and every 2.5 min a sample was taken, and the enzymatic activity toward d-glucose measured at 25 °C. The data were fitted to a first-order decay equation.
3.5 Enzyme Activity Assay
The dehydrogenase activity of GDHm from Ga. diazotrophicus was determined spectrophotometrically following the decay of absorbance at 600 nm using PMS plus DCPIP as electron acceptors [1]. Reaction mixture contained 80 mM DCPIP, 0.06 mM PMS in a total volume of 1 mL of PB-T buffer. The reaction was started with 0.03 mg of purified GDHm enzyme. All substrates tested were used at a final concentration of 20 mM, except for D-galactose, which was used at 60 mM. The quinone reductase activity of GDHm from Ga. diazotrophicus was measured according to the standard method described before [42]. The quinone analogs used were decylubiquinone (DUQ), quinone with one (Q1) and two isoprene (Q2). The kinetic constants V max and Km were calculated by initial velocities data obtained by varying the respective substrate concentration from 0 to 5 mM. The kinetic parameters were calculated from double-reciprocal plots. Oxidase activities in the membrane samples of Ga. diazotrophicus were determined using a Clark oxygen electrode as reported [20].
The effect of myxothiazol and antimycin on the activity of GDHm was studied in both the GDHm purified and the membrane fraction from Ga. diazotrophicus. In both cases, the inhibitors myxothiazol and antimycin were dissolved in dimethylsulfoxide (DMSO); this latter was added in not more than 5 % of final concentration. Both the GDHm purified and the membrane fraction were incubated by 10 min with the respective inhibitors, and then, the residual oxidase and dehydrogenase activities were measured using 20 mM glucose as substrate. Protein concentration was determined as previously reported by Dulley and Grieve [17].
4 Results
4.1 Alignment of the Aminoacyl Sequence of Ga. diazotrophicus GDHm with that from Different Organisms
To obtain structural information of Ga. diazotrophicus GDHm, the comparison of its sequence with that of other organisms, such as E. coli, was performed by taking advantage of the existence of a tridimensional model for GDHm of the latter [12]. The amino acid sequences of E. coli GDHm and Ga. diazotrophicus GDHm presented significant sequence identity (59.6 %), we identified in the latter essential structural elements for its integration into the membrane, the residues associated with binding the prosthetic group, a sequence of eight tryptophan motifs (W), and a possible region of interaction with membrane-bound quinones (Figure S1).
In addition, the hydrophobic analysis of the primary structure of Ga. diazotrophicus GDHm (data not shown) (accession number: YP_001603508.1) allowed us to identify five transmembrane helices located in the N-terminal portion, similar to that previously determined in the PQQ-GDHs of A. calcoaceticus [40], Pseudomonas sp. [38], G. suboxydans [3], Klebsiella aerogenes [47] and Rhizobium sp. [7].
Hence, the alignment of the primary structure of GDHm from Ga diazotrophicus (accession number YP_001603508.1), along with the correlation of the tridimensional model of E. coli GDHm, predict the existence of all the elements required for its function. Furthermore, because of this study also predicted the presence of five transmembrane helices in the enzyme, we focused our efforts on membrane extracts as the source of the Ga. diazotrophicus GDHm.
4.2 Extraction and Separation of GDHm by Liquid Chromatography
To structurally and functionally characterize GDHm, we isolated and purified it from Ga. diazotrophicus membranes. Cells of this organism were lysed and solubilized in KP buffer plus 0.3 % Triton X-100. From the solubilized, dehydrogenase activities were assayed by using as substrates ethanol and acetaldehyde as previously reported by Gomez-Manzo et al. [23]; showing activities for both substrates (data not shown). The enzymatic activity for the substrate d-glucose was also measured; no activity was detected. The results suggest that the GDHm is retained inside the membranes. To attempt the extraction and solubilization of the enzyme, the membrane residues previously treated with detergent were resuspended in KP buffer supplemented with 1 % Triton X-100. We measured the activity of the supernatant using d-glucose as substrate, obtaining a two-fold increment of the specific activity (0.86 µmol min−1 mg−1) with respect to the activity achieved in the membrane fraction (Table 1).
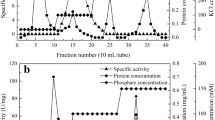
After obtaining a soluble fraction with GDHm activity, we purified the enzyme by chromatography. The fraction containing GDHm activity was added to a QAE-Toyopearl® column as described in the Sect. 2. We eluted the GDHm enzyme using a linear gradient of NaCl. The fractions that showed activity for the d-glucose substrate were obtained during approximately 70–145 mM NaCl of linear gradient; these fractions appeared before the protein maximum peak (Fig. 1a, b). This chromatographic step substantially increased GDHm purity by seven-fold. The fractions with activity were concentrated, dialyzed and applied to a CM-Toyopearl column. The fraction with activity was obtained from the volume that contains unbound proteins by the column; however, this step allowed us to increase the specific activity five-fold (Table 1), suggesting that a large number of contaminating proteins were removed. The sample with activity was concentrated and then applied to a Sephacryl S-200 column. The GDHm activity was obtained in the second protein peak (Fig. 1c).

Purification of GDHm by liquid chromatography. a Chromatogram of the fractions eluted from a QAE-Toyopearl column by a NaCl gradient. b Elution profile of the anionic-exchange chromatography followed by DCPIP reductase activities with d-glucose as a substrate. c Chromatogram of a sample applied to a Sephacryl S-200 column; this sample was obtained after pooled and concentrate the fractions with glucose activity obtained from the purification step by CM-Toyopearl column. The inset shows the electrophoretic analysis of purified GDHm; lane 1 SDS-PAGE of purified GDHm visualized by silver staining; lane 2 MW markers; lane 3 native PAGE analysis of purified GDHm visualized by Coomassie staining; lane 4 GDHm stained by zymography
Table 1 summarize the purification process of Ga. diazotrophicus GDHm; solubilization with 1 % Triton X-100 facilitated its extraction. Besides, it shows that the ionic exchange steps were crucial to increasing its purity. Overall, the addition of purification steps increased the purity of the enzyme 288-fold, with a total yield of 7.7 %. This result is significantly higher than the purification scheme reported for Erwinia sp. GDHm, where the enzyme could only be purified 33.4-fold [37]. Likewise, a total protein of 0.9 mg was obtained, which correlates with that previously reported by Meyer et al. [46], in which used an overexpression system of GDHm in G. oxydans to obtain 60 µg of total protein. It is important to mention that the specific activity obtained in Ga. diazotrophicus GDHm (112 IU mg−1) is in the range of the activities for those membrane-bound GDHs previously reported in P. fluorescens reported before [39], E. coli [32, 55] and G. oxydans [46], ranging from 140 to 300 IU mg−1.
4.3 Molecular Characterization of Ga. diazotrophicus GDHm
GDHm purity was corroborated by SDS-PAGE, where only one band was observed and corresponded to a protein of 86 kDa (inset Fig. 1c, lane 1). The molecular weight (MW) was confirmed by mass spectrometry (86,389 Da), corresponding to a protein with 805 amino acid residues. This value is comparable to the molecular mass of membrane-bound GDHs previously reported in the acetic acid bacteria G. oxydans (87 kDa) [3, 46] and A. calcoaceticus (83 kDa) [40] as well as with those obtained in Pseudomonas fluorescens (87 kDa) [39] and E. coli (88 kDa) [4]. In all of these organisms, membrane-bound GDH was reported as a monomer, with a MW of 80–87 kDa. Furthermore, we slice the protein band of 86 kDa from a SDS-PAGE in order to sequence internal peptides; the data showed that the peptides matched with regions of GDHm from Ga. diazotrophicus (YP_001603508.1) (data not shown).
After that, we analyzed the enzyme in a native gel (7.5 %) stained with Coomassie Brilliant Blue; only one band was observed (inset Fig. 1c, lane 3). The GDHm zymogram using d-glucose as a substrate, showed only one well-defined band (inset Fig. 1c, lane 4). Later, the isoelectric point (pI) for Ga. diazotrophicus GDHm was determined (pI = 6.0) using pI standards from a kit (see the Sect. 2). The result differs from those reported for G. suboxydans GDHm and A. calcoaceticus GDHs, whose pI values are 7.8 and 9.5, respectively [3, 14].
4.4 Spectroscopic Properties of Ga. diazotrophicus GDHm
Determination of PQQ. Previous studies have demonstrated the existence of PQQ in Ga. diazotrophicus membranes [26], but the entity containing this cofactor was not identified. Therefore, the possible presence of a pyrroloquinoline quinone (PQQ) prosthetic group in purified Ga. diazotrophicus GDHm was evaluated using several spectroscopic methods. Initially, the presence of the prosthetic group was observed using UV–visible spectroscopy, commercial PQQ was used as control. GDHm was reduced with d-glucose, while exogenous PQQ was reduced with dithionite. Figure 2a shows that both the control PQQ and the PQQ of the enzyme were analyzed in the 300–600 nm region at 25 °C. In both cases, a signal was observed at 357 nm, suggesting the presence of PQQ in Ga. diazotrophicus GDHm. The signal at 357 nm is a characteristic signal previously reported in diverse quinoproteins and quinohemoproteins [5, 23–25, 29].

Spectroscopic and chromatography properties of purified Ga. diazotrophicus GDHm. a Analysis of GDHm by UV–visible absorption spectrum. b The excitation/emission fluorescence spectra from isolated PQQ by methanol extraction. c RP-HPLC of PQQ extracted with methanol from GDHm of Ga. diazotrophicus. d Redox state determination of PQQ from GDHm; different PQQ’s obtained from several resources and conditions were used as standards and are described under the Sect. 2
The presence of PQQ was corroborated spectrofluorometrically of a methanol extract from GDHm (Fig. 2b). The excitation peak at 370 nm and the emission peak at 480 were observed; this emission signal is characteristic of authentic PQQ [15, 16, 23].
We corroborated the redox state of the PQQ prosthetic group to Ga. diazotrophicus GDHm by RP-HPLC, by analyzing the PQQ extracted by methanol. As observed in Fig. 2c, the PQQ presented a main peak with a retention time (RT) of 4.16 min. To confirm the identity of the PQQ, it was compared against reference standards, such as commercial PQQ oxidized with ammonium persulfate and reduced PQQ with dithionite. In addition, the PQQ extracted from membrane-bound alcohol dehydrogenase (PQQ-ADHa) was included; PQQ-ADHa contains a PQQ prosthetic group in the form of a semiquinone. The PQQ extracted from aldehyde dehydrogenase (PQQ-ALDH), which is present in the oxidized state [24, 25] was also included. The retention time of the PQQ of GDHm was the same as that of the commercial cofactor reduced with sodium dithionite (RT = 4.16 min), represented as PQQH2 in Fig. 2d. Based on these results, we proposed that the PQQ from GDHm reside in a reduced state. In contrast, the PQQ obtained from the PQQ-ADHa enzyme purified from the same organism presented a semiquinone state (RT = 4.5 min) [24], while in PQQ-ALDH, the prosthetic group was found in an oxidized state (RT = 6.9 min) [25]. The PQQ concentration was quantified as described under Materials and methods section. The calculated value of the PQQ in Ga. diazotrophicus GDHm was 7.0 nmol per mg protein, suggesting that stoichiometry is around of one molecule of PQQ per monomer of enzyme.
4.5 Optimum pH and Thermostability of Ga. diazotrophicus GDHm
To further explore the functional and structural characteristics, a titration curve of purified GDHm was performed to determine the optimum pH for enzyme activity. This determination was carried out subsequently to generate saturation curves of diverse substrates at the calculated optimum pH and to obtain kinetic parameters. The optimum pH of GDHm enzyme activity oscillate between 6.5 and 7.5, using PMS/DCPIP as an electron carrier and an acceptor, respectively (Fig. 3a); therefore, all of the subsequent tests were performed at pH 7.0. The optimum pH for GDHm of Ga. diazotrophicus is comparable to that previously reported for membrane-bound G. oxydans GDH [46]. However, our data differ from that reported for the GDHm of Erwinia sp. [37] and A. calcoaceticus [40]. In these two organisms, the catalytic activity is optimum at slightly basic pH values (i.e., 7.5–8.5 and 8–9, respectively).

Stability and functional studies of purified membrane-bound Ga. diazotrophicus GDHm. a pH profile of GDHm; the enzyme activity was assayed in a pH ranged from 2.0 to 10. b Thermal inactivation studies of GDHm. c Kinetics of both the quinone reductase and dehydrogenase activities in the presence of myxothiazol. d Quinone reductase activity of GDHm in the absence (open circles) or presence of 100 µM myxothiazol (closed squares)
Regarding the thermostability of Ga. diazotrophicus GDHm, a thermal denaturation curve was performed at different temperatures (30–60 °C) in a defined time of 10 min. The value of T1/2 calculated for Ga. diazotrophicus GDHm was 38.4 °C. This value is below the value obtained for E. coli GDHm; for its Apo form, the T1/2 was of 41 °C, while for the Holo-form a value of 46 °C was obtained.
To obtain more information on Ga. diazotrophicus GDHm, we evaluated its thermostability in a time course at a fixed temperature (40 °C); this temperature was selected based on the thermal denaturation results. As observe in Fig. 3b, the enzyme had an exponential decay of activity as the incubation time increased, showing a first-order inactivation constant value of 4.44 min−1. After 15 min of incubation, no remnant activity was detected toward the d-glucose substrate. This result is in contrast to that reported before in two membrane-bound GDH enzymes. In the A. calcoaceticus GDH, 30 % of the residual activity is conserved when the enzyme is incubated at 40 °C for 40 min [21], whereas in the Erwinia sp. GDHm [37], 80 % of the residual activity is maintained when the enzyme was incubated at 50 °C for 1 h. The latter enzyme is only inactivated at temperatures above 50 °C [37]. These results suggest that in contrast to that observed in periplasmic GDHs, which have better thermal stability, Ga. diazotrophicus GDHm presents higher thermosensitivity. This property is relevant because thermotolerance should be considered when such enzymes are used to develop biosensors.
4.6 Substrate Specificity and Kinetic Parameters of Ga. diazotrophicus GDHm
-
GDHm of other organisms reduces a broad spectrum of sugars. Therefore, we evaluated the specificity for different mono- and disaccharides as substrates of Ga. diazotrophicus GDHm. In agreement with the reported before for other organisms, Ga. diazotrophicus GDHm presented a broad specificity for substrates, preferentially oxidizing d-glucose, 2-deoxy-d-glucose, d-galactose and d-xylose (Table 2). In addition, this enzyme poorly oxidized disaccharides and their derivatives. Other d- and l-aldoses derived from aldoses and d-fructose were also assayed, and they showed very low activity. Ga. diazotrophicus GDHm presented a similar substrate specificity to those previously reported in E. coli [13] and G. oxydans [46]. In these organisms, membrane-bound GDHs preferentially oxidize the substrates d-glucose (100 %), 2-deoxy-d-glucose (96 %) and d-galactose (86 %). However, purified PQQ-GDH oxidized d-arabinose, d-fructose, d-maltose and d-lactose with different efficiencies, which, in general, were not suitable substrates [13, 37, 38, 46].
Table 2 Substrate specificity of purified Ga. diazotrophicus GDHm
The kinetic parameters for purified Ga. diazotrophicus GDHm were determined by plotting the values of the initial velocities by nonlinear regression and then fitting the data to the Michaelis–Menten equation. Table 3 shows the kinetic parameters calculated for the preferentially oxidized substrates. The Km values showed that there was an affinity for d-glucose 400 times greater than for d-galactose (0.1 and 40 mM for d-glucose and D-galactose, respectively). It is important to mention that the affinity for preferentially oxidize substrates was in the micromolar range. This characteristic is similar to that observed for soluble GDHs of A. calcoaceticus [31, 51] as well as for the membrane-bound GDH of P. aeruginosa [28], G. oxydans [46] and E. coli [32, 54].
4.7 Effect of the Myxothiazol on the Activity of Ga. diazotrophicus GDHm
In 1999, Flores-Encarnación and co-workers [20] showed that Ga. diazotrophicus does not incorporate any bc 1 complex within its respiratory chain. However, they observed that the inhibitors antimycin and myxothiazol affect glucose oxidase activity. These inhibitors classically affect dehydrogenases that notably contain bc1 complexes; however, because these types of inhibitors decrease GDH activity, we decided to explore the effect of myxothiazol. Hence, the isolated enzyme was incubated with different concentrations of myxothiazol for 10 min at 25 °C. We started the reaction by adding 20 mM d-glucose; the enzyme incubated with myxothiazol did not show any inactivation in the presence of PMS/DCPIP (Fig. 3c). This result suggests that the inhibitor did not affect electron transfer between the PQQ cofactor and the donor compounds. However, when we assayed the effect of the myxothiazol using decylubiquinone (DUQ) as an electron acceptor, a concentration-dependent inhibition was observed. The loss of half of the enzyme activity was obtained at 8.5 µM myxothiazol. The same result was obtained when inhibition was assayed without PMS/DCPIP. To explore the effect of myxothiazol on the ability of GDHm to transfer its electrons to other acceptors, we determined the reduction of quinone DUQ using a time course assay. After the purified enzyme had been incubated with 25 μM myxothiazol for 10 min, 20 mM of d-glucose was added. Then, the quinone DUQ was added as an electron acceptor, and DUQ-reductase activity was followed by the decrease of the absorbance at 275 nm. As shown in Fig. 3d, inhibition of enzyme by myxothiazol was time-dependent obtaining the total loss of activity after 60 min of incubation. Similar results were obtained when the assay was performed with 100 µM antimycin (data not shown). These data suggest that the site of action of myxothiazol and antimycin is located after the PQQ, probably close to the region where the membrane ubiquinone Q10 (UQ10), accepts electrons from the GDHm-complex.
5 Discussion
In this work, we present the purification and characterization of Ga. diazotrophicus GDHm. GDHm is localized to the plasma membrane of different Gram-negative bacteria, including the acetic acid bacterium Ga. diazotrophicus. The GDHm gene of this organism is reported in the NCBI database with the reference sequence: YP_001603508.1. The sequence alignment of different GDHm enzymes, particularly with that of E. coli, for which there is a tridimensional model, allowed us to identify in Ga. diazotrophicus the most characteristic structural elements of these groups of enzymes (Figure S1). In addition, the alignment also predicted the existence of five hydrophobic helices in the N-terminus that anchor the enzyme to the cellular membrane. Such a prediction correlates with our purification method in which extraction of Ga. diazotrophicus GDHm from membrane preparations required a treatment with high detergent concentration. This latter indicates that our enzyme is firmly anchored to the membrane. In contrast, membrane-bound dehydrogenases such as PQQ-alcohol dehydrogenase, FAD-gluconate dehydrogenase and PQQ-aldehyde dehydrogenase, need lower concentrations of detergent [23, 25].
Our SDS-PAGE (Inset of Fig. 1c, lane 1) and mass spectroscopy data show that the monomer of GDHm purified from Ga. diazotrophicus has a MW of 86 kDa and contain a sequence that match with the sequence reported as YP_001603508.1. Besides, the determined MW value of GDHm agrees with those obtained for a previously purified membrane-bound GDH from other species [13, 37, 38, 40, 41]. The electrophoresis of GDHm in a native gel showed a band with a molecular mass equivalent to the monomer with reactivity towards the NTB (see inset of Fig. 1c, lane 4). This result indicates that the native state of Ga. diazotrophicus GDHm is naturally monomeric. The UV–visible and fluorescence tests (Fig. 2a, b) unequivocally demonstrated the presence of PQQ associated with Ga. diazotrophicus GDHm. Likewise, RP-HPLC experiments showed that the PQQ redox state of the purified GDHm is reduced (Fig. 2c, d).
Gluconacetobacter diazotrophicus GDHm presents a broad specificity for different substrates, making it a versatile enzyme in the dehydrogenation of sugars located in the periplasm. The kinetic data suggest that Ga. diazotrophicus GDHm has similar properties to other dehydrogenases purified from acetic bacteria. These circumstances suggest that the conversion of such substrates to their gluconate forms imply a strategy for excluding potential competitors since other microorganisms do not readily metabolized these acid species.
On the other hand, the coupling of GDHm to the respiratory chain [20] and its possible implication in energy generation processes [9, 53], along with the increase in its synthesis during processes that involve a high energy demand [33], can represent a scenario that favors the aerotolerant diazotrophic process in Ga. diazotrophicus. It was proposed that this enzyme is probably an important component of the Ga. diazotrophicus respiratory system that, when it coordinates with the other chain transporters such as endogenous quinones and the ubiquinol oxidase Cyt-a1 [20], could contribute to the protection of nitrogenases. This kinetic gear could explain the high respiratory levels observed because of the glucose oxidase activity in the membrane. To understand the real contribution of this enzyme in the aerotolerant diazotrophic process in Ga. diazotrophicus, molecular genetics studies that involve a GDH-null mutant are required. Respect to the myxothiazol effect, we propose that this compounds inhibits the binding of the endogenous quinone (Q10) but not to the level of the active site (i.e., PQQ cofactor), according to the scheme represented in Figure S2.
In conclusion, we reported an isolation method for GDHm from Ga. diazotrophicus that rendered a highly purified enzyme; its behavior indicated that this enzyme is strongly anchored to the membrane, probably through of five transmembrane helices predicted by its aminoacyl sequence. Its molecular and kinetics parameters could determined, notwithstanding that large concentrations of detergent had to be used for its solubilization, indicating that GDHm contain indeed a robust structure.
Abbreviations
- ATCC:
-
American type culture collection
- Cyt-a1:
-
Cytochrome a1
- DMSO:
-
Dimethyl sulfoxide
- FAD:
-
Flavin adenine dinucleotide
- KP-T:
-
Phosphate buffer plus 0.1 % Triton X-100
- NTB:
-
Nitro blue tetrazolium
- Q7:
-
Quinone seven isoprene
References
Adachi O, Tayama K, Shinagawa E, Matsushita K, Ameyama M (1978) Purification and characterization of particulate alcohol dehydrogenase from Gluconobacter suboxydans. Agric Biol Chem 42:2045–2056
Alvarez B, Martínez-Drets G (1995) Metabolic characterization of Acetobacter diazotrophicus. Can J Microbiol 41:918–924
Ameyama M, Shinagawa E, Matsushita K, Adachi O (1981) d-glucose dehydrogenase from Gluconobacter suboxydans: solubilization, purification and characterization. Agric Biol Chem 45:851–861
Ameyama M, Nonobe M, Shinagawa E, Matsushita K, Takimoto K, Adachi O (1986) Purification and characterization of the quinoprotein. d-glucose dehydrogenase apoenzyme from Escherichia coli. Agric Biol Chem 50:49–57
Anthony C, Zatman LJ (1967) The microbial oxidation of methanol: the prosthetic group of alcohol dehydrogenase of Pseudomonas sp. M27; A new oxidoreductase prosthetic group. Biochem J 104:960–969
Attwod MM, Van Dijken JP, Pronk JT (1991) Glucose metabolism and gluconic acid production by Acetobacter diazotrophicus. J Ferment Bioeng 72:101–105
Bernardelli CE, Luna MF, Galar ML, Boiardi JL (2001) Periplasmic PQQ-dependent glucose oxidation in free-living and symbiotic rhizobia. Curr Microbiol 42:310–315
Bertalan M, Albano R, de Pádua V, Rouws L, Rojas C, Hemerly A, Teixeira K, Schwab S, Araujo J, Oliveira A, França L, Magalhães V, Alquéres S, Cardoso A, Almeida W, Loureiro MM, Nogueira E, Cidade D, Oliveira D, Simão T, Macedo J, Valadão A, Dreschsel M, Freitas F, Vidal M, Guedes H, Rodrigues E, Meneses C, Brioso P, Pozzer L, Figueiredo D, Montano H, Junior J, de Souza FG, Quintana Flores VM, Ferreira B, Branco A, Gonzalez P, Guillobel H, Lemos M, Seibel L, Macedo J, Alves-Ferreira M, Sachetto-Martins G, Coelho A, Santos E, Amaral G, Neves A, Pacheco AB, Carvalho D, Lery L, Bisch P, Rössle SC, Ürményi T, Pereira AR, Silva R, Rondinelli E, von Krüger W, Martins O, Baldani JI, Ferreira PCG (2009) Complete genome sequence of sugarcane nitrogen-fixing endophyte Gluconacetobacter diazotrophicus Pal5. BMC Genom 10:450
Bont JAM, Dokter P, van Schie BJ, van Dijken JP, Frank JZNJ, Duine J, Kuenen JG (1984) Role of the quinoprotein glucose dehydrogenase in gluconic acid production by Acinetobacter calcoaceticus. Ant van Leeuwenhoek 50:76–77
Cavalcante VA, Döbereiner J (1988) A new acid-tolerant nitrogen-fixing bacterium associated with sugarcane. Plant Soil 108(23):31
Cleton-Jansen AM, Goosen N, Odle G, van de Putte P (1988) Nucleotide sequence of the gene coding for quinoprotein glucose dehydrogenase from Acinetobacter calcoaceticus. Nucleic Acids Res 16:6228
Cozier GE, Anthony C (1995) Structure of quinoprotein glucose dehydrogenase of Escherichia coli modeled on that of methanol dehydrogenase from Methylobacterium extorquens. Biochem J 312:679–685
Cozier GE, Salleh RA, Anthony C (1999) Characterization of the membrane quinoprotein glucose dehydrogenase from Escherichia coli and characterization of a site-directed mutant in which histidine-262 has been changed to tyrosine. Biochem J 340:639–647
Dokter P, Frank J, Duine JA (1986) Purification and characterization of quinoprotein glucose dehydrogenase from Acinetobacter calcoaceticus L.M.D. 79.41. Biochem J 239:163–167
Duine JA, Frank JZNJ, Jongejan JA (1987) Enzymology of quinoproteins. Adv Enzym Relat Areas Mol Biol 59:169–212
Duine JA, Jongejan JA (1989) Quinoproteins, enzymes with pyrroloquinoline quinone as cofactor. Ann Rev Biochem 58:403–426
Dulley JR, Grieve PA (1975) A simple technique for eliminating interference by detergents in the Lowry method of protein determination. Anal Biochem 64:136–141
Elias MD, Nakamura S, Migita CT, Miyoshi H, Toyama H, Matsushita K, Adachi O, Yamada M (2004) Occurrence of a bound ubiquinone and its function in Escherichia coli membrane-bound quinoprotein glucose dehydrogenase. J Biol Chem 279:3078–3083
Escamilla JE, Ramirez R, Del Arenal IP, Zarzosa G, Linares V (1987) Expression of cytochrome oxidase in Bacillus cereus; effects of oxygen tension and carbon source. J Gen Microbiol 133:3549–3555
Flores-Encarnación M, Contreras-Zentella M, Soto-Urzua L, Aguilar RG, Baca BE, Escamilla JE (1999) The respiratory system and diazotrophic activity of Acetobacter diazotrophicus PAL5. J Bacteriol 181:6987–6995
Geiger O, Görisch H (1989) Reversible thermal inactivation of the quinoprotein glucose dehydrogenase from Acinetobacter calcoaceticus. Ca2+ ions are necessary for re-activation. Biochem J 261:415–421
Gillis M, Kersters K, Hoste B, Janssens D, Kroppenstedt RM, Stephan MP, Teixeira KRS, Dobereiner J, De Ley J (1989) Acetobacter diazotrophicus sp. nov., a Nitrogen-Fixing Acetic Acid Bacterium Associated with Sugarcane. Int J Syst Bact 39:361–364
Gómez-Manzo S, Contreras-Zentella ML, González-Valdez A, Sosa-Torres M, Arreguín-Espinoza R, Escamilla Marván E (2008) The PQQ-alcohol dehydrogenase of Gluconacetobacter diazotrophicus. Int J Food Microbiol 125:71–78
Gómez-Manzo S, Solano-Peralta A, Saucedo-Vázquez JP, Escamilla-Marván JE, Kroneck PMH, Sosa-Torres ME (2010) The membrane-bound quinohemoprotein alcohol dehydrogenase from Gluconacetobacter diazotrophicus PAL5 carries a [2Fe-2S] cluster. Biochem 49:2409–2415
Gómez-Manzo S, Chavez-Pacheco JL, Contreras-Zentella M, Sosa-Torres ME, Arreguín-Espinosa R, Perez de la Mora M, Membrillo-Hernández J, Escamilla JE (2010) Molecular and catalytic properties of the aldehyde dehydrogenase of Gluconacetobacter diazotrophicus, a quinoheme protein containing pyrroloquinoline quinone, cytochrome b, and cytochrome c. J Bact 192:5718–5724
Gonzalez B, Martinez S, Chavez JL, Lee S, Castro NA, Dominguez MA, Gomez S, Contreras ML, Kennedy C, Escamilla JE (2006) Respiratory system of Gluconacetobacter diazotrophicus PAL5 Evidence for a cyanide-sensitive cytochrome bb and cyanide-resistant cytochrome ba quinol oxidases. Biochim Biophys Acta 1757:1614–1622
Goodhew CF, Brown KR, Pettigrew GW (1986) Heme staining in gels, as useful tool in the study of bacterial c-type cytochromes. Biochim Biophys Acta 852:228–294
Goodwin PM, Anthony C (1998) The biochemistry, physiology and genetics of PQQ and PQQ-containing enzymes. Adv Microb Physiol 40:1–80
Groen BW, van Kleef MAG, Duine JA (1986) Quinohaemoprotein alcohol dehydrogenase apoenzyme from Pseudomonas testosteroni. Biochem J 234:611–615
Hauge JG (1964) Glucose dehydrogenase of Bacterium anitratum: an enzyme with a novel prosthetic group. J Biol Chem 239:3630–3639
Heuberger EHML, Poolman B (2000) A spectroscopic assay for the analysis of carbohydrate transport reactions. Eur J Biochem 267:228–234
Hommes RWJ, Postma PW, Neijssel OM, Tempest DW, Dokter P, Duine JA (1984) Evidence of a quinoprotein glucose dehydrogenase apoenzyme in several strains of Escherichia coli. FEMS Microbiol Lett 24:329–333
Hommes RWJ, van Hell B, Postma PW, Neijssel OM, Tempest DW (1985) The functional significance of glucose dehydrogenase in Klebsiella aerogenes. Arch Microbiol 143:163–168
Imanaga Y (1989) Investigations on the active site of glucose dehydrogenase from Pseudomonas fluorescens. In: PQQ and quinoprotein. Springer, Netherlands
von Jagow G, Ljungdahl PO, Graf P, Ohnishi T, Trumpower BL (1984) An inhibitor of mitochondrial respiration which binds to cytochrome b and displaces quinone from the iron-sulfur protein of the cytochrome bc1 complex. J Biol Chem 259:6318–6326
Jiménez-Salgado T, Fuentes-Ramirez LE, Tapia-Hernandez A, Mascarua-Esparza MA, Martinez-Romero E, Caballero-Mellado J (1997) Coffea arabica L., a new host plant for Acetobacter diazotrophicus, and isolation of other nitrogen-fixing Acetobacteria. Appl Environ Microbiol 63:3676–3683
Marcinkevičienė L, Bachmatova I, Semėnaitė R, Rudomanskis R, Bražėnas G, Meškiene R, Meškys R (1999) Purification and characterization of PQQ-dependent glucose dehydrogenase from Erwinia sp. Biotechnol Lett 21:187–192
Matshusita K, Ohno Y, Shinagawa E, Adachi O, Ameyama M (1980) Membrane-bound d-glucose dehydrogenase from Pseudomonas sp: solubilization, purification and characterization. Agric Biol Chem 44:1505–1512
Matsushita K, Ameyama M (1982) d-Glucose dehydrogenase from Pseudomonas fluorescens, membrane-bound. Meth Enzymol 89:149–154
Matsushita K, Shinagawa E, Adachi O, Ameyama M (1989) Quinoprotein d-glucose dehydrogenases in Acinetobacter calcoaceticus L.M.D. 79.41: purification and characterization of the membrane-bound enzyme distinct from the soluble enzyme. Antonie Van Leeuwenhoek 56:63–72
Matsushita K, Shinagawa E, Adachi O, Ameyama M (1989) Reactivity with ubiquinone of quinoprotein d-Glucose Dehydrogenase from Gluconobacter suboxydans. J Biochem 105:633–637
Matsushita K, Ebisuya H, Ameyama M, Adachi O (1992) Change of the terminal oxidase from cytochrome a 1 in shaking cultures to cytochrome o in static cultures of Acetobacter aceti. J Bact 174:122–129
Matshusita K, Toyama H, Adachi O (1994) Respiratory chains and bioenergetics of acetic acid bacteria. Adv Microbial Physiol 36:247–301
Matsushita K, Toyama H, Ameyama M, Adachi O, Dewanti A, Duine JA (1995) Soluble and membrane-bound quinoprotein d-glucose dehydrogenases of the Acinetobacter calcoaceticus : the binding process of PQQ to the apoenzymes. Biosci Biotechnol Biochem 59:1548–1555
Matshusita K, Arents JC, Bader R, Yamada M, Adachi O, Postma PW (1997) Escherichia coli is unable to produce pirroloquinoline quinone (PQQ). Microbiol 143:3149–3156
Meyer M, Schweiger P, Deppenmeier U (2013) Effects of membrane-bound glucose dehydrogenase overproduction on the respiratory chain of Gluconobacter oxydans. Appl Microbiol Biotechnol 97:3457–3466
Neijssel OM, Tempest DW, Postma PW, Duine JA, Frank JZNJ (1983) Glucose metabolism by K+-limited Klebsiella aerogenes: evidence for the involvement of a quinoprotein glucose dehydrogenase. FEMS Microbiol Lett 20:35–39
Ng FMW, Dawes EA (1973) Chemostat studies on the regulation on glocose metabolism in Pseudomona aeruginosa by citrate. Biochem J 132:129–137
Prust C, Hoffmeister M, Liesegang H, Wiezer A, Fricke WF, Ehrenreich A, Gottschalk G, Deppenmeier U (2005) Complete genome sequence of the acetic acid bacterium Gluconobacter oxydans. Nat Biotechnol 23:195–200
Reis Olivares FL, Döbereiner J (1994) Improved methodology for isolation of Acetobacter diazotrophicus and confirmation of its endophytic habitat, World. J Microbiol Biotech 10:401–405
Sode K, Ootera T, Shirahane M, Witarto AB, Igarashi S, Yoshida H (2000) Increasing the thermal stability of the water-soluble pyrroloquinoline quinone glucose dehydrogenase by single amino acid replacement. Enzyme Microb Technol 26:491–496
Stephan MP, Oliveira M, Teixeira KRS, Martinez-Drets G, Dobereiner J (1991) Physiology and dinitrogen fixation of Acetobacter diazotrophicus. FEMS Microbiol Lett 77:67–72
van Schie BJ, Hellingwerf KJ, van Dijken JP, Elfereink MGL, van Dijl JM, Kuenen JG, Konings WN (1985) Energy transduction by electron transfer via a pyrrolo-quinoline quinone-dependent glucose dehydrogenase in Escherichia coli, Pseudomonas aeruginosa, and Acinetobacter calcoaceticus (var. lwoffi). J Bact 163:493–499
Yamada M, Sumi K, Matsushita K, Adachi O, Yamada J (1993) Topological analysis of quinoprotein glucose dehydrogenase in Escherichia coli and its ubiquinone-binding site. J Biol Chem 268:12812–12817
Yamada M, Inbe H, Tanaka M, Sumi K, Matsushita K, Adachi O (1998) Mutant isolation of the Escherichia coli quinoprotein glucose dehydrogenase and analysis of crucial residues Asp-730 and His-775 for its function. J Biol Chem 273:22021–22027
Acknowledgments
This work is a requisite to obtain the PhD grade in Science; we appreciate to Posgrado en Ciencias Biológicas UNAM, for the academic training of Martín Sará Páez during its doctorate studies. We also appreciate to Dirección General de Estudios de Posgrado (DGEP), for the scholarship granted to MS-P during his studies. SG-M is supported by Consejo Nacional de Ciencia y Tecnología Grant 154570. The technical assistance of Javier Gallegos Infante (IFC/UNAM) for assistance in bibliographic materials is greatly appreciated. This work was started by the Prof. José Edgardo Escamilla in the Instituto de Fisiología Célular, UNAM; but unfortunately deceased, and was concluded by the Dr. Horacio Reyes Vivas from Instituto Nacional de Pediatría. The authors concluded this project that now is published in his honor and memory.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Sará-Páez, M., Contreras-Zentella, M., Gómez-Manzo, S. et al. Purification and Characterization of the Membrane-Bound Quinoprotein Glucose Dehydrogenase of Gluconacetobacter diazotrophicus PAL 5. Protein J 34, 48–59 (2015). https://doi.org/10.1007/s10930-014-9596-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10930-014-9596-4