
Abstract
For the detailed molecular analysis, genomic modification, and application of acetic acid bacteria such as Gluconobacter in biotechnological processes, a simple markerless deletion system is essential. The available methods have either low efficiencies or their applicability is restricted to strains containing an upp mutation. We now developed a method based on counterselection by cytosine deaminase, encoded by the codA gene from Escherichia coli, in the presence of the fluorinated pyrimidine analogue 5-fluorocytosine (FC). The codA-encoded enzyme converts nontoxic FC to toxic 5-fluorouracil, which is channeled into the metabolism by the uracil phosphoribosyltransferase, encoded by the chromosomal upp gene of Gluconobacter. We found that the presence of E. coli codB, encoding a cytosine permease, was needed for a high efficiency of gene deletion. The system is applicable in wild-type strains because no preceding deletions are required. Based on the fact that a codA gene is absent and an upp gene is present in almost all acetic acid bacteria sequenced so far, the method should also be applicable for other genera of the Acetobacteraceae.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The group of acetic acid bacteria contains many genera with interesting biochemical characteristics. They are widely used in biotechnology. An important representative of the family is Gluconobacter oxydans, which is employed in several biotechnological processes due to its ability to perform incomplete oxidations of a large variety of carbohydrates, alcohols, and sugar alcohols at relatively high substrate concentrations (Gupta et al. 2001). The oxidative fermentations allow the substitution of certain or multiple steps in industrial synthesis that cannot be achieved by organic chemistry with sufficient yield. For molecular analysis of the physiology of acetic acid bacteria and construction of strains tailored for biotechnological production purposes, an effective and easily applicable deletion system is important. So far, most of the mutants were constructed by inserting an antibiotic-resistance cassette into the genes of interest (Hölscher and Görisch 2006; Hölscher et al. 2007). Instead of a knockout system in which persisting resistance markers remain in the strain, accompanied by polar effects on the expression of downstream genes, there is a strong need for methods, which provide markerless in-frame deletions in acetic acid bacteria. This is also necessary because the available resistance cassettes that can be used for multiple knockouts are limited.
Typical markerless deletion systems are two-step methods, where a first step of vector integration at the target site is followed by a second step where a counter-selectable marker is employed to select for clones, which have lost the vector. Therefore, a good counterselection method is the key for an effective markerless deletion system. However, the sacB system that has been used in Gluconobacter for this purpose does not work satisfyingly (Gay et al. 1985; Hölscher et al. 2007). Commonly used approaches for counterselection in other organisms exploit genes involved in the purine or pyrimidine metabolism. For example, upp coding for a phosphoribosyltransferase, pyrE/ura5 coding for an oratate phosphoribosyltransferase, or hpt encoding a hypoxanthine phosphoribosyltransferase have been used (Boeke et al. 1984; Boeke et al. 1987; Fabret et al. 2002; Keller et al. 2009; Pritchett et al. 2004; Wagner et al. 2012). All these systems are based on the fact that purine or pyrimidine analogues are converted to toxic compounds. Only cells lacking the gene for the converting enzyme can survive the presence of the analogue. Plating of cells on media containing the analogue leads to a strong selection for clones, which have lost any chromosomally integrated copy of the gene for the converting enzyme. Therefore, parental strains used for genome modification must be devoid of the respective gene for purine or pyrimidine nucleotide biosynthesis. In a previous study by our group, the upp gene was utilized for the counterselection step (Peters et al. 2012). The uracil phosphoribosyltransferase encoded by this gene catalyzes the conversion of the pyrimidine analogue 5-fluorouracil (FU) to 5-fluorouridine-monophosphate (F-UMP) (Martinussen and Hammer 1994). F-UMP is then transformed to 5-fluorodesoxyuridine-monophosphate, which elicits a toxic effect by inhibition of thymidylate synthase, thereby blocking DNA repair and replication (Neuhard 1982). An upp-deficient strain of G. oxydans 621H and a deletion vector containing an upp gene were created for this system. The counterselection against this vector was done on media supplemented with FU. Despite its high efficiency, the need to work with a ∆upp strain is an important restriction that limits its application in the variety of acetic acid bacteria strains used in biotechnology.
In this study, we developed an alternative counter-selectable marker based on codA and codB in order to address this problem and describe its utilization in a convenient markerless in-frame deletion system.
Materials and methods
Chemicals
Yeast extract and peptone were obtained from Carl Roth (Karlsruhe, Germany), 5-fluorocytosine (FC) from TCI Europe N.V. (Zwijndrecht, Belgium), kanamycin (Km) from AppliChem (Darmstadt, Germany), mannitol, glucose, cefoxitin, FU, and other chemicals were purchased from Sigma-Aldrich (Steinheim, Germany).
Bacterial strains and culture methods
Escherichia coli Top10 was purchased from Invitrogen (Karslruhe, Germany) and grown in LB medium at 37 °C and 180 rpm on a rotary shaker (Sambrook et al. 1989). For plasmid selection, the medium was supplemented with 50 μg/ml Km. The strains G. oxydans DSM 2343 (ATCC 621H), DSM 3504, DSM 7145, Acidomonas methanolica DSM 5432, Gluconacetobacter diazotrophicus DSM 5601, Acidiphilium multivorum DSM 11245, Granulibacter bethesdensis DSM 17861 were obtained from Deutsche Sammlung von Mikroorganismen und Zellkulturen (Braunschweig, Germany), the strain Acetobacter pasteurianus LMG 1513 from BCCM/LMG Bacteria Collection (Gent, Belgium). The G. oxydans strains were grown in mannitol medium containing 5 g/L yeast extract, 3 g/L peptone, and 50 mM mannitol at pH 6. For G. diazotrophicus and A. pasteurianus, the mannitol level was increased to 250 mM. A. methanolica was grown in glucose medium containing 5 g/L yeast extract, 20 g/L glucose, 3 g/L (NH4)2SO4, 1 g/L KH2PO4, MgSO4 × 7 H2O 0.7 g/L, 0.5 g/L NaCl, 0.4 g/L Ca(NO3)2 × 4H2O at pH 6.8, G. bethesdensis in 50 g/L glucose, 12.5 g/L CaCO3, and 5 g/L yeast extract. A. multivorum was cultivated in 2 g/L (NH4)2SO4, 1 g/L glucose, 0.5 g/L K2HPO4, 0.5 g/L MgSO4 × 7 H20, 0.3 g/L yeast extract, and 0.1 g/L KCl at pH 3. The pH was adjusted using hydrochloric acid. The cultures were grown at 180 rpm on a rotary shaker at 30 °C. The selection conditions in growth media were 50 μg/μL for Km and 60 μg/μL for FC. Table 1 summarizes the strains used in this study.
DNA manipulations
Routine molecular biological procedures were done according to standard protocols (Sambrook et al. 1989). For preparation of plasmids, the AxyPrep (Union City, USA) plasmid miniprep kit was used. Genomic DNA from G. oxydans DSM 3504 and 621H was extracted with the Epicenter MasterPure DNA purification kit (Madison, USA). DNA purification was done with the Promega PCR clean-up system (Madison, USA). Restriction enzymes, DNA ligase, and alkaline phosphatase (FastAP) were obtained from Fermentas (Waltham, USA). PCR was done according to the manuals of the enzymes from Finnzyme (Vantaa, Finland). Phire polymerase was used for analytical reactions and Phusion polymerase for amplifications requiring proofreading. Oligonucleotides and sequencing services were provided by Eurofins MWG GmbH (Ebersberg, Germany). The fusion PCR technique was used to ligate the PCR products of flanking regions for the deletion vectors according to a long flanking homology (LFH) protocol (Peters et al. 2012; Wach 1996). Colony PCR was used to screen for mutants or to confirm integration of a deletion vector into the genome. Colonies were picked and dissolved in water. During PCR, the initial denaturation step was prolonged to 8 min. Enzyme-free cloning technique (Tillett and Neilan 1999) was used for the construction of the deletion vector pKOS6b. Table 2 summarizes the plasmids used for and constructed in this study.
Transformation of Gluconobacter
G. oxydans DSM 3504 was transformed via electroporation (Hall et al. 1992; Kallnik et al. 2010; Mostafa et al. 2002). Cells were grown to an OD600 of 0.9 in rich EP medium containing 80 g/L mannitol, 15 g/L yeast extract, 2.5 g/L MgSO4 × 7H20, 1.5 g/L CaCl2, and 0.5 g/L glycerol adjusted to pH 6. The culture was centrifuged at 4,000 rpm, 4 °C for 10 min, and washed three times in HEPES (1 mM, pH 7). Cells were shock frozen in liquid nitrogen in 50 μL aliquots. The electroporation was carried out in cuvettes with 1 mm electrode distance from Peqlab (Erlangen, Germany) with 2.5 kV, 25 μF, and 400 Ω by a Gene Pulser apparatus from Bio-Rad GmbH (München, Germany). Fresh EP medium was added after the pulse cells were incubated on a rotary shaker over night for a better regeneration and were plated on mannitol plates containing kanamycin.
G. oxydans 621H was transformed by conjugation using triparental mating. E. coli Top10 was used as donor together with helper strain E. coli HB101 carrying plasmid pRK2013. G. oxydans and E. coli cells were grown to an OD600 of 0.9; 1 mL of donor and helper strain were mixed and washed with fresh LB, then 4 mL of G. oxydans culture was added and centrifuged. The resulting pellet was resuspended in fresh G. oxydans medium and placed on a mannitol-containing agar plate in a drop. After 24 h incubation, the cells were washed from the plate with fresh mannitol medium and the resulting cell suspension was plated on mannitol plates containing kanamycin and cefoxitin.
Results
The codA gene of E. coli K12 was chosen as counter-selectable marker gene for the new strain-independent and markerless in-frame deletion system. To investigate the importance of codB, the second gene in the cod operon, we constructed one vector containing codAB and a second vector that includes only codA.
Construction of deletion vectors pKOS6a and pKOS6b
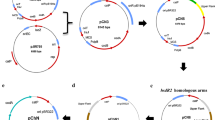
The construction of a cod-based deletion vector took pAJ63a as a starting point. Enzyme-free cloning techniques were used to remove the upp cassette and replace it with codAB genes, but retaining the upp promoter region. The primers pAJdUPP_F/F_ov and pAJdUPP_R/R_ov were used for the amplification of the vector and the primers codBA_F/codBA_F_ov and codBA_R/codBA_R_ov to amplify the codAB operon including 24 bp upstream sequence, using E. coli K12 chromosomal DNA as template (Fig. 1). The resulting vector pKOS6b was transformed in chemically competent E. coli Top10 cells and was completely sequenced. The sequence data was deposited in GenBank under the accession number JX885593.

Construction of deletion vector pKOS6b from pAJ63a. The upp cassette was replaced with the codAB operon amplified from E. coli K12 under maintenance of the upp promoter
A smaller vector, pKOS6a, lacking codB was constructed to investigate the role of codB for the functionality of the cod-based counterselection. The previously created pKOS6b served as a template for the amplification with the primers pKos6AmpFxho and pKos6aAmpRxho. The resulting PCR product was digested with XhoI and circularized with T4 ligase to pKOS6a. The vector was transformed in E. coli Top10 and verified by sequencing.
Construction of G. oxydans DSM 3504 ∆9570
A chromosomal deletion was constructed in strain G. oxydans DSM 3504 to examine the functionality of the generated vector carrying codBA as the counter-selectable marker. We choose GLS_C09570, a levansucrase, as the target gene. The flanking regions, covering approximately 900 base pairs (bp) upstream and downstream of the gene GLS_C09570, were amplified using the primers 09570_F_kpn/09570_FUSR and 09570_FUSF/09570_xba_R. Next, a LFH PCR was performed with primers 09570_F_kpn and 09570_xba_R to fuse the two fragments. The fused fragment and pKOS6b were digested with KpnI and XbaI and ligated in the corresponding sites of a dephosphorylated pKOS6b vector. The resulting vector pKOS6bΔ9570 was transformed in chemical competent E. coli Top10 cells and verified by colony PCR and sequencing using the primers pk18MCS_F and pk18MCS_R/pk18MCS_2R.
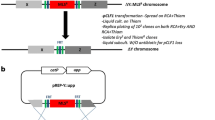
The transformation of pKOS6bΔ9570 in G. oxydans DSM 3504 ∆9570 was done by electroporation. The integration of the plasmid in the genome by a first recombination, which occurs randomly either up- or downstream of the target gene, resulted in several clones, which were verified using colony PCR with the genome-specific primer 09570check_F and the vector-specific primer pk18MCS_R (Fig. 2). All the clones were positive. They were grown in a medium, containing Km, and were plated on mannitol selection plates including 60 μg/mL FC. This results in around 50 clones per plate. Only those that had lost the vector by a second recombination were able to survive. This second recombination could either result in revertants or in the desired replacement of the target gene with the fused gene flanks of the deletion vector. After 3–5 days of incubation, the correct clones were identified by colony PCR and verified by sequencing, using the primers 09570check_F and 09570check_R. 9 out of 21 checked clones contained the desired deletion. The 1,317 bp deletion was also confirmed by Southern blot analysis using a probe targeting the upper flanking region (Fig. 3).

Deletion of GLS_c9570 by integration via double crossing over and selection of mutants on 5-FC. The integration process is displayed in (a). b Shows the resulting markerless deletion mutant with lacking GLS_c9570 between the two flanking regions

Southern-blot analysis of G. oxydans DSM 3504 ∆9570 mutant clones. EcoRI-digested chromosomal DNA from the wild type and three independent mutant clones were separated and hybridized with a probe consisting of 720 bp. The observed fragment sizes correspond nicely to the expected fragment sizes of 6,284 bp for the wild type and 4,967 for the mutant clones, confirming the markerless in-frame deletion
Construction of G. oxydans 621H Δ854Δ855
To prove the suitability of the method as a generalized deletion system that works for different G. oxydans strains, we created a Δ854Δ855 mutant lacking the polyol dehydrogenase in G. oxydans strain 621H. The construction and validation of the deletion vector was done as described above for pKOS6bΔ9570. The primers used are given in Table 3, and the insert was cloned in the EcoRI and XbaI sites of pKOS6b.
The deletion vector pKOS6bΔ854Δ855 was verified by PCR and sequencing and was transformed in strain 621H by conjugation using triparental mating. The rest of the procedure corresponds to the creation of the ∆9570 mutant in strain DSM 3504. The mutant had the expected phenotype of being unable to grow on mannitol due to its lack of polyol dehydrogenase. The validation of the in-frame deletion was done by PCR and Southern Blot (data not shown). The efficiency of gene deletion in strain 621H corresponded to the results for strain DSM 3504, demonstrating the suitability of the new deletion method for use in different G. oxydans strains.
Influence of codB on the efficiency of counterselection
Two vectors containing 1,094 bp of the downstream flanking region of the G. oxydans 621H gene GOX2567 were constructed. These constructs were able to integrate in the chromosome of G. oxydans 621H after transformation via a single recombination. The vector pKOS6a2567flank contained only the codA gene, while the vector pKOS6b2567flank contained the whole codAB operon. The minimal inhibitory concentration (MIC) of FC on both strains with chromosomally integrated codA and codAB, respectively, was determined in liquid cultures containing 50 μg/mL Km. After 3 days of growth, a MIC of 60 μg/mL FC was observed for the strain carrying both the codA and the codB gene, while a MIC of 500 μg/mL was observed for the vector lacking the codB gene (Table 4 below). This result clearly shows the importance of the cytosine permease gene codB for the efficiency of counterselection by FC.
Applicability in other strains of acetic acid bacteria
Due to the high effectiveness of the new deletion method in the two different G. oxydans strains DSM 3504 and 621H, we also examined the applicability in other species of acetic acid bacteria. We focused on strains with available genomic sequence data. In silico examination of sequence data revealed the presence of a uracil phosphoribosyltransferase gene (upp) in all of the regarded strains. A cytosine deaminase gene was identified by homology in A. multivorum, but the similarity to the E. coli K12 gene was low with an identity of 187 of 433 amino acids (43.2 %). Growth experiments were done to define the MIC of FC and FU for the different strains. The resistance of the organisms towards FC and the sensibility towards FU are required for the functionality of the method described in this study. The growth studies were carried out in test tubes containing 5 mL of strain-specific media. FC and FU were added in various concentrations. All tested strains were able to grow after 24 h incubation even in the presence of FC concentrations up to 500 μg/mL. A. multivorum DSM 11245 showed normal growth in the presence of FC after 2 days of incubation. Furthermore, we found inhibition of growth by FU for every tested strain at concentrations over 60 μg/mL (Tables 4 and 5).
Discussion
Despite its usefulness for the introduction of deletions in G. oxydans 621HΔupp, the system published by Peters et al. (2012) has a crucial drawback; it cannot be used in wild-type strains. The elementary step for the development of a new strain-independent markerless in-frame deletion system was the choice of a reliable counter-selectable marker gene. To avoid the need to first introduce deletions in wild-type strains, it was fundamentally necessary to find a gene that works as counter-selectable marker but is naturally absent in G. oxydans and other acetic acid bacteria of interest. In contrast to other potential marker genes taken into consideration, this was the case for the codA gene of E. coli K12. The gene codes for an E. coli cytosine deaminase and is arranged in an operon together with codB, which encodes a cytosine permease (Danielsen et al. 1992). The first attempt to use codA in a deletion system was made by Duraisingh et al. (2002) in Plasmodium falciparum. Unfortunately, however, the organisms did not develop any sensitivity towards FC, despite of the confirmed expression of codA in the cells. Several years later, a deletion system based on fungal cytosine deaminase delivered better results (Orr et al. 2012). Deletion systems based on E. coli codA were established in the genus Rhodococcus (van der Geize et al. 2008) and Streptomyces (Dubeau et al. 2009), indicating general applicability of the system for the Actinobacteria. Interestingly, however, the gene codB was never included in the deletion vectors, and its role was not investigated before this study. In our markerless deletion system for the acetic acid bacteria, the inclusion of codB was crucial. The codB-encoded cytosine permease obviously facilitates the entry of FC into the cell. The inhibition of growth in the presence of 500 μg/mL FC in G. oxydans 621H cells carrying the integrated vector pKOS6a2567 flank showed that with a vector containing codA, but lacking the cytosine permease codB gene, supplementation of high FC concentrations was needed to reach intracellular FC levels sufficient for growth inhibition. The presence of codB decreased the MIC to values under 60 μg/mL, and thus allows efficient counterselection. This suggests the general possibility to utilize FC counterselection in a broad range of bacteria by enhancing FC toxicity via functional expression of the codB in addition to codA, especially in microorganisms with a poor sensitivity for FC.
The parental vector pAJ63a was chosen because of its inability to stable replicate in G. oxydans strains (unpublished data). Such a suicide deletion vector is under strong pressure to integrate in the chromosome via recombination, if homologous regions are present as shown in other studies (van der Geize et al. 2008). This property ensures that clones with the ability to grow in the presence of Km are inevitably carrying the integrated vector.
In addition to the successful application in strain 621H, chromosomal deletions could also be achieved in G. oxydans DSM 3504. The deletion of GLS_C09570 in DSM 3504 was the first deletion ever described for this strain, for which previous deletion strategies did not work. The reliability of this new method was proven by numerous further deletions in strain DSM 3504 (data not shown).
The general presence of upp and the absence of an E. coli-like codA in almost all investigated acetic acid bacteria open a wide range of potential applications for this system. The growth analysis actually showed that the putative cytosine deaminase present in some strains, which differs at the sequence level substantially from E. coli codA, seems not able to convert the cytosine analogue efficiently.
In conclusion, we report the development of a new codA-based markerless deletion system for acetic acid bacteria by using the codAB genes from E. coli for counterselection. This offers the possibility to use this system for targeted genome modifications such as in-frame deletions, replacements, or insertions in wild type strains of different genera of acetic acid bacteria. Given the increasing number of genome sequences available for acetic acid bacteria and their increasing importance in biotechnology, this reliable deletion method will be instrumental to investigate and modify their genomes and to engineer genomes of acetic acid bacteria for improved biotechnological application.
References
Boeke JD, LaCroute F, Fink GR (1984) A positive selection for mutants lacking orotidine-5'-phosphate decarboxylase activity in yeast: 5-fluoro-orotic acid resistance. Mol Gen Genet 197(2):345–346
Boeke JD, Trueheart J, Natsoulis G, Fink GR (1987) 5-Fluoroorotic acid as a selective agent in yeast molecular genetics. Methods Enzymol 154:164–175
Boyer HW, Roulland-Dussoix D (1969) A complementation analysis of the restriction and modification of DNA in Escherichia coli. J Mol Biol 41 (3):459–472
Danielsen S, Kilstrup M, Barilla K, Jochimsen B, Neuhard J (1992) Characterization of the Escherichia coli codBA operon encoding cytosine permease and cytosine deaminase. Mol Microbiol 6(10):1335–1344
De Ley J (1961) Comparative Carbohydrate Metabolism and a Proposal for a Phylogenetic Relationship of the Acetic Acid Bacteria. J Gen Microbiol 24:31–50
De Ley J, Swings J, Gossele F (1984) The genus Gluconobacter. In: N Krieg and J Holt (ed) Bergey’s Manual of Systematic Bacteriology. Williams & Wilkins Co., Baltimore, pp 267–278
Dubeau MP, Ghinet MG, Jacques PE, Clermont N, Beaulieu C, Brzezinski R (2009) Cytosine deaminase as a negative selection marker for gene disruption and replacement in the genus Streptomyces and other Actinobacteria. Appl Environ Microbiol 75(4):1211–1214
Duraisingh MT, Triglia T, Cowman AF (2002) Negative selection of Plasmodium falciparum reveals targeted gene deletion by double crossover recombination. Int J Parasitol 32(1):81–89
Fabret C, Ehrlich SD, Noirot P (2002) A new mutation delivery system for genome-scale approaches in Bacillus subtilis. Mol Microbiol 46(1):25–36
Figurski, DH, Helinski, DR (1979) Replication of an origin-containing derivative of plasmid RK2 dependent on a plasmid function provided in trans. Proc Natl Acad Sci USA 76(4):1648–1652
Gay P, Le Coq D, Steinmetz M, Berkelman T, Kado CI (1985) Positive selection procedure for entrapment of insertion sequence elements in gram-negative bacteria. J Bacteriol 164(2):918–921
Gillis M, De Ley J (1980) Intra- and intergenic similarities of the ribosomal ribonucleic acid cistrons of Acetobacter and Gluconobacter. Int J Sys Bacteriol 30:7–27
Gossele F, Swings J, Kersters K, Pauwels P, De Ley J (1983) Numerical Analysis of Phenotypic Features and Protein Gel Electrophoregrams of a Wide Variety of Acetobacter strains. Proposal for the Improvement of the Taxonomy of the Genus Acetobacter Beijerinck 1898, 215. Syst Appl Microbiol 4(3):338–368
Greenberg DE, Porcella SF, Stock F, Wong A, Conville PS, Murray PR, Holland SM, Zelazny AM (2006) Granulibacter bethesdensis gen. nov., sp. nov., a distinctive pathogenic acetic acid bacterium in the family Acetobacteraceae. Int J Syst Evol Microbiol 56(Pt 11):2609–2016
Gupta A, Singh VK, Qazi GN, Kumar A (2001) Gluconobacter oxydans: its biotechnological applications. J Mol Microbiol Biotechnol 3(3):445–456
Hanahan D (1983) Studies on transformation of Escherichia coli with plasmids. J Mol Biol 166(4):557–580
Hall PE, Anderson SM, Johnston DM, Cannon RE (1992) Transformation of Acetobacter xylinum with plasmid DNA by electroporation. Plasmid 28(3):194–200
Hölscher T, Görisch H (2006) Knockout and overexpression of pyrroloquinoline quinone biosynthetic genes in Gluconobacter oxydans 621H. J Bacteriol 188(21):7668–7676
Hölscher T, Weinert-Sepalage D, Görisch H (2007) Identification of membrane-bound quinoprotein inositol dehydrogenase in Gluconobacter oxydans ATCC 621H. Microbiology 153(Pt 2):499–506
Kallnik V, Meyer M, Deppenmeier U, Schweiger P (2010) Construction of expression vectors for protein production in Gluconobacter oxydans. J Biotechnol 150(4):460–465
Keller KL, Bender KS, Wall JD (2009) Development of a markerless genetic exchange system for Desulfovibrio vulgaris Hilden borough and its use in generating a strain with increased transformation efficiency. Appl Environ Microbiol 75(24):7682–7691
Mason LM, Claus GW (1989) Phenotypic Characteristics Correlated with Deoxyribonucleic Acid Sequence Similarities for Three Species of Gluconobacter: G. oxydans (Henneberg 1897) De Ley 1961, G. frateurii sp. nov., and G. asaii sp. nov. Int J Syst Bacteriol 39:174–184
Martinussen J, Hammer K (1994) Cloning and characterization of upp, a gene encoding uracil phosphoribosyltransferase from Lactococcus lactis. J Bacteriol 176(21):6457–6463
Mostafa HE, Heller KJ, Geis A (2002) Cloning of Escherichia coli lacZ and lacY genes and their expression in Gluconobacter oxydans and Acetobacter liquefaciens. Appl Environ Microbiol 68(5):2619–2623
Motizuki K, Takarazuka I, Kanzaki T (1966) Method for producing 2-keto-L-gulonic acid. US Patent Office 3,234,105
Neuhard J (1982) Utilization of preformed pyrimidine bases and nucleosides. In: Munch-Peterson A (ed) Metabolism of nucleotides nucleosides and nucleobases in microorganisms. Academic, London, pp 95–148
Orr RY, Philip N, Waters AP (2012) Improved negative selection protocol for Plasmodium berghei in the rodent malarial model. Malar J 11:103
Peters B, Junker A, Brauer K, Muhlthaler B, Kostner D, Mientus M, Liebl W, Ehrenreich A (2013) Deletion of pyruvate decarboxylase by a new method for efficient markerless gene deletions in Gluconobacter oxydans. Appl Microbiol Biotechnol 97(6):2521–30
Pritchett MA, Zhang JK, Metcalf WW (2004) Development of a markerless genetic exchange method for Methanosarcina acetivorans C2A and its use in construction of new genetic tools for methanogenic archaea. Appl Environ Microbiol 70(3):1425–1433
Sambrook J, Fritsch E, Maniatis T (1989) Molecular cloning a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY
Tillett D, Neilan BA (1999) Enzyme-free cloning: a rapid method to clone PCR products independent of vector restriction enzyme sites. Nucleic Acids Res 27(19):e26
Urakami T, Tamakoa J, Suzuki KI, Komagata K (1989) Acidomonas gen. nov. Incorporating Acetobacter methanolicus as Acidomonas methanolica comb. nov. Int J Syst Bacteriol 39:50–55
van der Geize R, de Jong W, Hessels GI, Grommen AW, Jacobs AA, Dijkhuizen L (2008) A novel method to generate unmarked gene deletions in the intracellular pathogen Rhodococcus equi using 5-fluorocytosine conditional lethality. Nucleic Acids Res 36(22):e151
Wach A (1996) PCR-synthesis of marker cassettes with long flanking homology regions for gene disruptions in S. cerevisiae. Yeast 12(3):259–265
Wagner M, van Wolferen M, Wagner A, Lassak K, Meyer BH, Reimann J, Albers SV (2012) Versatile genetic tool box for the crenarchaeote Sulfolobus acidocaldarius. Front Microbiol 3:214
Wakao N, Nagasawa N, Matsuura T, Matsukura H, Matsumoto T, Hiraishi A, Sakurai Y, Shiota H (1994) Acidophilum multivorum sp. nov., an Acidophilic Chemoorganotrophic Bacterium from Pyritic Acid Mine Drainage. J Gen Appl Microbiol 40:143–159
Yamada Y, Hoshino K, Ishikawa T (1997) The phylogeny of acetic acid bacteria based on the partial sequences of 16S ribosomal RNA: the elevation of the subgenus Gluconoacetobacter to the generic level. Biosci Biotechnol Biochem 61(8):1244–1251
Yamashita S, Uchimura T, Komagata K (2004) Emendation of the genus Acidomonas Urakami, Tamaoka, Suzuki and Komagata 1989. Int J Syst Evol Microbiol 54(Pt 3):865–870
Acknowledgments
We thank the Bundesministerium für Bildung und Forschung (BMBF) for funding this work as part of the GenomikTransfer initiative (FKZ: 0315632C).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kostner, D., Peters, B., Mientus, M. et al. Importance of codB for new codA-based markerless gene deletion in Gluconobacter strains. Appl Microbiol Biotechnol 97, 8341–8349 (2013). https://doi.org/10.1007/s00253-013-5164-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-013-5164-7