
Abstract
Addition of MgCl2 to the culture medium has been found to dramatically increase the activity of Bacillus deramificans pullulanase expressed by Brevibacillus choshinensis. The specific activity of the pullulanase obtained from medium supplemented with MgCl2 was also higher than that obtained in culture medium without added magnesium ions. In this work, the mechanism of this increase was studied. When cultured in medium without added magnesium ions, B. choshinensis mainly produced a thermolabile, inactive form of pullulanase. The addition of magnesium ions led to the production of a thermostable, active form of pullulanase. Circular dichroism assays revealed considerable differences in secondary structure between the active and inactive pullulanase forms. Transmission electron microscopy suggested that magnesium ion addition inhibits the shedding of cell wall protein (HWP) layers from the cell surface. Quantitative real-time PCR showed that magnesium ion addition represses transcription of HWP. Because the pullulanase gene and HWP have identical promoters, pullulanase gene transcription was also inhibited. These results suggest that when pullulanase is expressed slowly, it tends to fold into an active form.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Pullulanase (EC 3.2.1.41), which efficiently cleaves the α-1,6 linkage in amylopectin and related branched dextrins, is generally used in combination with other amylolytic enzymes (α-amylase, β-amylase, glucoamylase, and so on) during starch processing (Talekar et al. 2013). Because pullulanase can remarkably increase product yield, improve raw material conversion rate, and reduce reaction time (Hii et al. 2012; Zhang et al. 2013), it has been used widely in the production of sugar syrups (Lin et al. 2013; Singh et al. 2010), resistant starch (Zhang and Jin 2011), fuel ethanol (Gohel and Duan 2012), and low-calorie beers (Blanco et al. 2014).
In order to meet the demand for pullulanase resulting from these industrial applications, current research has focused on the over-expression of pullulanase through recombinant expression. The Brevibacillus choshinensis (formerly Bacillus brevis HPD31) expression system has been developed for highly efficient production of heterologous proteins (Mizukami et al. 2010, 2014; Mu et al. 2013), due to its ease of culture, high-efficiency synthesis of active protein, and exceptional capacity for extracellular protein secretion. In our previous work (Zou et al. 2015), B. choshinensis was shown to be valuable for the extracellular production of Bacillus deramificans pullulanase. This pullulanase belongs to type I pullulanase and the glycoside hydrolase family 13. The structure of pullulanase (Duan et al. 2015) consists of a complex multi-domain architecture in which a catalytic module is appended to several carbohydrate-binding (CBM) domains, as well as many domains of unknown function (termed X modules). Under optimal culture conditions, the pullulanase activity reached 1005.8 U/mL in a 3-L fermentor. Interestingly, MgCl2 has been found to dramatically increase the production of recombinant pullulanase.
Previous reports have shown that supplementation of the medium with MgCl2 has a negative effect on recombinant α-amylase production in B. brevis 47 (Adachi et al. 1991). In the early stationary phase of growth, the cell wall protein layers, including the outer wall protein (OWP) and middle wall protein (MWP), are shed from the bacterial surface (Yamada et al. 1981). Loss of the cell wall protein layers from the cell surface derepresses transcription from P2, one of the major promoters of the cell wall proteins, permitting enhanced synthesis and secretion of cell wall proteins into the medium. The addition of magnesium ions to the medium inhibits this shedding of the cell wall proteins and subsequently represses the transcriptional activity of P2. Since P2 is also the promoter used for the production of recombinant α-amylase, α-amylase production is simultaneously remarkably reduced (Adachi et al. 1991).
B. choshinensis contains HWP, a cell wall protein that shares considerable sequence homology (78 %) with MWP from B. brevis 47. The regulatory region of the HWP gene also shows surprisingly high homology (>85 %) with the regulatory region of the MWP gene (Ebisu et al. 1990). As in recombinant α-amylase production by B. brevis 47, HWP and pullulanase share the P2 promoter. Despite these striking similarities, the effect of MgCl2 supplementation on pullulanase production by B. choshinensis is diametrically opposite to its effect on α-amylase production by B. brevis 47.
In this work, the mechanism responsible for the effects of magnesium ions on the expression of pullulanase by B. choshinensis was investigated. Activity analyses were conducted to assess the specific activities of the pullulanases expressed in the presence and absence of added magnesium ions, and circular dichroism studies were conducted to assess their secondary structures. The results suggest that the relative rates of expression may be responsible for the observed increase in activity. Changes in cell wall structure, investigated using transmission electron microscopy, may also play a role.
Materials and methods
Bacterial strains
B. choshinensis (Takara Bio Inc.) was employed for the expression of heterologous proteins. Plasmid pNCMO2 (Takara Bio Inc.) was used as the expression vector. B. choshinensis strains harboring the plasmids pNCMO2/pulA-d2 (Zou et al. 2015), pNCMO2/pal I (Cheng 2015), and pNCMO2/cgt (Wu et al. 2015) were kept in our laboratory and used for the expression of B. deramificans pullulanase (NCBI accession number KT897705), Serratia plymuthica sucrose isomerase (NCBI accession number YP_004505648.1), and Paenibacillus macerans α-cyclodextrin glycosyltransferase (α-CGTase; NCBI accession number P04830), respectively.
Cultivation medium and conditions
Seed cultures were grown in TM medium that contained 10.0 g/L glucose, 10.0 g/L polypeptone, 5.0 g/L meat extract, 2.0 g/L yeast extract, 10.0 mg/L FeSO4·7H2O, 10.0 mg/L MnSO4·4H2O, and 1.0 mg/L ZnSO4·7H2O. Shake flask studies were performed in an optimal fermentation medium that contained 27.6 g/L glucose, 28.0 g/L beef extract, 5.0 g/L cottonseed meal, and 12.2 g/L MgCl2·6H2O. The MgCl2·6H2O in this fermentation medium might be removed or replaced by other salts, depending on the experimental design.
Expression studies were carried out in 250-mL shake flasks containing 50 mL of medium on a rotary shaker operating at 200 rpm. After being grown in TM medium for 12 h at 37 °C, the seed culture (0.5 mL) was diluted into the fermentation medium at 30 °C.
Enzyme activity assay
Pullulanase activity was determined by measuring the enzymatic release of reducing sugars during incubation with pullulan (Zou et al. 2014). The reaction mixture, which contained 1 mL 1 % (w/v) pullulan, 0.9 mL sodium acetate buffer (100 mM; pH 4.5), and 0.1 mL appropriately diluted enzyme solution, was incubated at 60 °C for 10 min. The reaction was stopped by the addition of 3 mL of 3,5-dinitrosalicylic acid solution (6.5 g/L 3,5-dinitrosalicylic acid, 185.0 g/L potassium sodium tartrate, 20.0 g/L NaOH, 5.0 g/L redistilled phenol, and 5.0 g/L sodium sulfite), and then boiled for 7 min, and after which, the entire mixture was diluted to 15 mL with deionized water. The concentration of reducing sugars was determined by measuring the absorbance of the mixture at 540 nm. One unit of pullulanase activity was defined as the amount of enzyme required to release 1 μM reducing sugar per minute from pullulan under the conditions specified.
Sucrose isomerase activity was determined using 100 g/L sucrose, solubilized in 50 mM citrate buffer (pH 6.0), as the substrate (Li et al. 2013). The reaction was initiated by adding 100 μL appropriately diluted enzyme to 900 μL sucrose solution, and then incubated at 30 °C for 15 min. The reaction was terminated by placing it in a boiling water bath. The product was analyzed using high-performance liquid chromatography (HPLC), which was conducted using an Elite 2000 HPLC System (Hitachi High Technologies Inc., USA). Analyses were carried out using a Syncronis Amino 12156 column (4.6 × 250 mm, Thermo Fisher Scientific Inc., USA) at 40 °C and a refractive index detector. The mobile phase, which contained 80 % (v/v) acetonitrile and 20 % (v/v) water, was maintained at a flow rate of 0.8 mL/min. One unit of enzyme activity was defined as the amount of enzyme necessary to release of 1 μmol isomaltulose per minute.
Alpha-CGTase activity was measured as previously reported (Li et al. 2009). One unit of α-CGTase activity was defined as the amount of enzyme required to release 1 μmol of α-cyclodextrin per minute.
SDS-PAGE gel electrophoresis
Enzyme samples were stained with Coomassie Brilliant Blue R-250 dye by heating at 100 °C for 10 min before electrophoresis. Electrophoresis was performed on an SE 250 mini-vertical electrophoresis unit (GE Healthcare, USA) using 5 % stacking and 12 % running gels. After electrophoresis, the running gel was stained for 1 h using an aqueous solution containing 0.1 % Coomassie Brilliant Blue R-250, 25 % isopropanol, and 10 % acetic acid. The stained gel was then destained using an aqueous solution containing 5 % ethanol and 10 % acetic acid.
Ion exchange chromatography
Ion exchange chromatography was applied to purify pullulanase from the culture medium. The supernatant was dialyzed overnight against 20 mM sodium phosphate buffer (pH 7.0). This sample was loaded onto a DEAE-Sepharose column pre-equilibrated with 20 mM sodium phosphate buffer (pH 7.0) and eluted with a linear gradient of NaCl from 0 to 1 M in the same buffer at a flow rate of 1 mL/min. The contents of the fractions were confirmed using SDS-PAGE and the enzyme activity of each fraction was determined. Fractions containing the desired enzyme were pooled.
Size-exclusion chromatography
Size-exclusion chromatography was carried out with an AKTA avant 25 system (GE Healthcare, USA) equipped with a Superdex 200 10/300GL column (GE Healthcare, USA). The elution buffer was 50 mM sodium phosphate buffer (pH 7.0). Samples were eluted at a flow rate of 0.4 mL/min and the column effluent was monitored using UV spectrometry at 280 nm. Four peptides, including α-glucosidase, bovine serum albumin, chymotrypsinogen A, and chicken ovalbumin were used as molecular weight standards (Sigma-Aldrich, Shanghai, China). The calibration curve (Fig. S1) of molecular weight versus elution volume was obtained with the molecular weight standards.
Circular dichroism analysis
Circular dichroism (CD) analyses were conducted using a BioLogic Mos-450 spectropolarimeter (BioLogic Science Instrument, Grenoble, France) equipped with a 1 mm quartz cuvette. The samples were diluted to a protein concentration of 0.05–0.20 mg/mL in 20 mM sodium phosphate buffer (pH 7.0). Ellipticity data of the enzymes in the far-ultraviolet (195–250 nm) spectrum was continuously collected. Secondary structure content was calculated from the CD spectrum using the self-consistent method (Sreerama and Woody 1993).
Transmission electron microscopy
B. choshinensis cells withdrawn from the culture medium were fixed with 5 % glutaraldehyde in 100 mM sodium phosphate buffer (pH 7.0) at 4 °C for 3 h. After repeated dehydration with increasing concentrations of ethanol, the fixed cells were embedded in epoxy resin and sliced into sections that were approximately 50 nm thick. The sections were stained for morphological observation using uranyl acetate solution and lead citrate. Finally, the sections were examined using a Hitachi H-7650B (Hitachi, Japan) transmission electron microscope at an accelerating voltage of 80 kV.
Real-time quantitative PCR
Total RNA was extracted using the Simply P Total RNA Extraction Kit (BioFlux, Hangzhou, China), following the manufacturer’s instructions. After treatment with DNase I (Fermentas International Inc., Ontario, Canada) to eliminate genomic DNA, the extracted RNA was used as the template for cDNA synthesis. First-strand complementary DNA (cDNA) was synthesized using the RevertAid™ First-Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Massachusetts, USA), following the manufacturer’s instructions.
The expression levels of the two genes encoding pullulanase and HWP were measured. The 16S rRNA gene was used as an internal control to normalize the results. The sequences of the primers used during quantitative real-time PCR (qPCR) are listed in Table 1. The qPCR analyses were performed using an ABI StepOne Real-Time PCR System (Applied Biosystems, SanMateo, CA, USA) with the Real Master Mix Kit (Tiangen, Beijing, China). Amplification was carried out in a 20-μL mixture containing 9 μL 2.5 × RealMasterMix/20 × SYBR solution, 1 μL forward primer, 1 μL reverse primer, 1 μL cDNA, and 8 μL DEPC-treated water. The cycling program for qPCR included the following: 94 °C for 1 min; followed by 35 cycles of 94 °C for 10 s, 55 °C for 30 s, and 68 °C for 15 s. The data were analyzed by using the 2−ΔΔCT model (Livak and Schmittgen 2001).
Results
Effects of magnesium ions on the expression of pullulanase by B. choshinensis
In previous work, supplementation of the growth medium with 60 mM MgCl2 substantially stimulated the production of pullulanase. In this work, to determine the ion that specifically improved the pullulanase production, the recombinant strain was cultured in a series of media containing 60 mM concentrations of different salts (MgCl2, MgSO4, NaCl, or Na2SO4), using a medium without addition of these ions as the control. As shown in Fig. 1, MgCl2 and MgSO4 slightly stimulated the cell growth, while NaCl and Na2SO4 had no obvious effect on cell growth. The protein concentration and pullulanase activity obtained from the medium containing NaCl or Na2SO4 were almost identical to those of the control. When the medium was supplemented with MgCl2 or MgSO4, the protein concentration reached 2.40 and 2.51 mg/mL, which was lower than that of the control by 30.4 and 27.3 %, respectively. On the other hand, the pullulanase activity reached 543 and 534 U/mL, which represent 5.4- and 5.3-fold increases compared with that of the control, respectively.

Effects of different salts on the expression of pullulanase by B. choshinensis. Dry cell weight (DCW) (filled bar), protein concentration (open bar), pullulanase activity (slashed bar)
After the fermentation was complete, the supernatants obtained from the fermentations performed in control or magnesium ions-containing medium were incubated with 60 mM MgCl2 or 10 mM EDTA, respectively. Both treatments resulted in a negligible change in the pullulanase activity.
Effects of magnesium ions on the expression of other heterologous proteins by B. choshinensis
Three foreign proteins (sucrose isomerase and α-CGTase) were expressed by B. choshinensis in medium supplemented with 60 mM MgCl2 or a control medium without MgCl2 supplementation. The results are listed in Table 2. The addition of magnesium ions slightly stimulated cell growth, but inhibited protein expression. When fermentation was conducted in a medium supplemented with magnesium ions, the activities of sucrose isomerase and α-CGTase declined by 11.1 and 10.1 %, respectively, compared with those of the control. Interestingly, the specific activities of these two heterologous proteins changed only slightly when the medium was supplemented with magnesium ions, while the specific activity of pullulanase was significantly enhanced when the medium was supplemented with magnesium ions.
Detection of expressed pullulanase as active and inactive forms by heat treatment
Pullulanase has previously been expressed in Escherichia coli. This recombinant pullulanase showed excellent thermostability (a half-life of 150 h) at 60 °C (Duan and Wu 2015). Heat treatment was therefore carried out to detect the forms of pullulanase expressed in B. choshinensis.
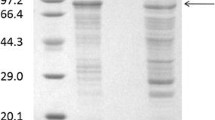
The supernatants obtained from fermentations conducted in medium supplemented with magnesium ions or in control medium were incubated at 60 °C for 30 min; the residual activities after heat treatment only decreased by 2.6 and 2.2 %, respectively. Although the pullulanase activities remained almost unchanged after heat treatment, the protein concentrations decreased by 75.9 and 51.3 %, respectively. The protein components of the supernatants before and after heat treatment were analyzed by SDS-PAGE. After heat treatment, the majority of the pullulanase (79 kD) obtained from the control medium precipitated as insoluble protein, while that obtained from the medium supplemented with magnesium ion addition remained soluble (Fig. 2). The thermolabile protein with negligible pullulanase activity will be referred to as the “inactive form,” while the thermostable protein with substantial pullulanase activity will be referred to as the “active form.”

SDS-PAGE analysis of protein components before and after heat treatment. Proteins obtained from the control medium (without magnesium ions) are shown in lanes 1 to 3: 1, culture supernatant before heat treatment; 2, soluble fraction of medium supernatant after heat treatment; 3, insoluble fraction of medium supernatant after heat treatment. Proteins obtained from the medium supplemented with magnesium ions are shown in lanes 4 to 6: 4, culture supernatant before heat treatment; 5, soluble fraction of medium supernatant after heat treatment; 6, insoluble fraction of medium supernatant after heat treatment
Purification, kinetic parameters, and thermostability of pullulanase
The recombinant proteins obtained from the medium with or without added magnesium ions were purified to homogeneity by ion exchange chromatography and size-exclusion chromatography (Fig. S2). The kinetic constants of the purified pullulanases with and without added magnesium ions were determined under identical conditions, using pullulan as substrate at 60 °C, pH 4.5. As shown in Table 3, both pullulanases showed similar K m values. The values of k cat and k cat/K m of pullulanase obtained from the medium without magnesium ions were 499 s−1 and 471 mL/mg·s−1, respectively, while those were 3.00 × 103 s−1 and 2.97 × 103 mL/mg·s−1 respectively for the pullulanase with magnesium ions added in the medium.
The purified enzymes were incubated at 60 °C, pH 4.5 to evaluate their thermostabilities. As shown in Fig. S3, the pullulanases expressed in the medium with or without magnesium ion addition showed similar thermostabilities (a half-life of 170 h).
Determination of aggregation state of pullulanase by size-exclusion chromatography
Pullulanase has a strong tendency to form aggregates when expressed in E. coli, which strongly influences its activity (Duan et al. 2015). Therefore, the effects of magnesium ion supplementation on the aggregation state of pullulanase were investigated. As shown in Fig. S4, the native molecular weight of the pullulanase protein expressed in medium-supplemented magnesium ions was found to be 150 kD by size-exclusion chromatography. The same result was obtained with pullulanase protein expressed in control medium. These results indicate that both pullulanase proteins form dimers in solution.
Structure analysis of pullulanase by circular dichroism
The effects of magnesium ion addition on the secondary structure of pullulanase were investigated using CD spectrophotometry. Figure 3 and Table 4 show the CD spectra and the content of secondary structures predicted by CD, respectively. The pullulanase expressed in medium supplemented with magnesium ions showed 12.6 % more α-helix and 11.3 % more β-sheet than the pullulanase expressed in medium without magnesium ions.

CD spectra of pullulanase samples. The CD spectrum of pullulanase obtained from the control medium (without magnesium ions) is shown by the solid line. The CD spectrum of pullulanase obtained from the medium supplemented with magnesium ions is shown by the dotted line
Morphological alterations of cell wall structure
Morphological alterations in cell wall structure were examined by transmission electron microscopy (Fig. 4). When cultured in control medium, the recombinant strain was surrounded by a rough outer layer, which may be composed of HWP (Ebisu et al. 1990) and a thin inner wall at 12 h (Fig. 4a). At 24 h, it was surrounded only by the thin inner wall (Fig. 4b), which may be composed of peptidoglycan (Yamada et al. 1981); the outer layer disappeared. However, when cultured in the medium supplemented with magnesium ions, the recombinant strain was surrounded by an intact outer layer, even at 48 h (Fig. 4c).

Transmission electron micrographs showing alteration of the cell wall structure of B. choshinensis. Cells were grown without magnesium ions for 12 h (a) and 24 h (b); cells were grown with magnesium ions for 48 h (c). The cells in (a) and (c) were surrounded by a rough outer layer and a thin inner wall. The cells in (b) were surrounded only by a thin inner wall
Transcription level of genes in B. choshinensis
The transcription levels of the pulA-d2 and HWP genes were investigated using qPCR. As shown in Fig. 5, the mRNA level of pulA-d2 and HWP genes were almost same at 12 h, whether the medium was supplemented with magnesium ions or not. The transcription level of these two genes markedly increased from 18 h when the recombinant strain was cultured in medium without magnesium ions. However, the transcription levels of these genes increased slowly, or even decreased, when the medium was supplemented with magnesium ions. When the recombinant strain was cultured in control medium, the mRNA levels of the pulA-d2 and HWP genes at 24 h were 4.8- and 2.7-fold greater than the levels at 12 h, respectively. When the recombinant strain was cultured in medium supplemented with magnesium ions, the transcription level of these two genes at 24 h were 1.8- and 0.9-fold of the levels at 12 h, respectively.

Transcription levels of B. deramificans pulA-d2 (a) and B. choshinensis HWP (b) genes in B. choshinensis cells cultured in the medium with (open) or without (filled) added magnesium ions
Discussion
In this study, supplementation of the medium with magnesium ions was observed to remarkably enhance the production of B. deramificans pullulanase by B. choshinensis. The structure of B. deramificans pullulanase was simulated by SWISS-MODEL. It was observed that there is no metal-ion-binding site in the inner structure of pullulanase. Therefore, magnesium ions are unlikely to tightly bind to the protein structure in the process of pullulanase biosynthesis. Addition of magnesium ions or EDTA to the supernatant collected from the fermentation broth made no change to the enzyme activity. This indicates that the increased pullulanase production did not result from the activation of pullulanase activity by magnesium ions. Protein concentration assays suggested that magnesium ions did not enhance protein production, either. On the contrary, addition of magnesium ions led to decreased protein production. Therefore, the increased enzyme activity must be the result of enhanced specific activity.
When cultured in control medium, B. choshinensis mainly produced a thermolabile, inactive form of pullulanase, while the pullulanase produced in medium supplemented with magnesium ions consisted mainly of a thermostable, active form. The values of k cat and k cat/K m of pullulanase without added magnesium ions were only 16.6 and 15.9 % of those with magnesium ion addition, respectively. The pullulanase expressed in medium supplemented with magnesium ions showed higher catalytic efficiency compared with that without magnesium ions. The native molecular weight of pullulanase was not influenced by the addition of magnesium ions, indicating that the quaternary structure of pullulanase was not affected by the addition of magnesium ions to the medium, despite the fact that the pullulanase from B. deramificans has a strong tendency to aggregate. The CD assay showed that the active and inactive forms of pullulanase had significantly different secondary structures. These results suggest that the addition of magnesium ions may facilitate the folding of pullulanase expressed in B. choshinensis.
When other foreign proteins (sucrose isomerase and α-CGTase) were expressed by B. choshinensis, addition of magnesium ions to the medium decreased both protein production and enzyme activity. These negative effects are consistent with effects seen previously when α-amylase was expressed by B. brevis 47 (Adachi et al. 1991). In that case, the mechanism of added magnesium ions was thought to inhibit protein production by inhibiting the shedding of OWP and MWP from the cell surface. Thus, the P2 promoter in B. brevis 47, which is shared by the cell wall proteins and the recombinant protein, was repressed. Considering the high homology between HWP and MWP, a similar mechanism may reduce protein production in B. choshinensis. The current morphology observations and mRNA quantitation results support this interpretation.
The enhancement of pullulanase specific activity that results from supplementation of the medium with magnesium ions, which is distinctive from other proteins, is probably dependent on the particular nature of pullulanase. Previous reports have shown that misfolded proteins and even insoluble inclusion bodies are normally generated during pullulanase expression, especially when it is synthesized at a high rate. This may be related to the high molecular weight and complicated structure of pullulanase (Chen et al. 2014; Duan et al. 2013). When the recombinant strain was cultured in control medium, a higher transcription level of the pulA-d2 gene was observed, which could lead to a rate of protein synthesis that is higher than the rate that occurs in the presence of magnesium ions. This accelerated rate of protein synthesis may result in hasty protein folding, which may be inadequate for pullulanase, which requires a meticulous folding process to achieve its active form. As a result, the pullulanase activity obtained without magnesium ions in the medium was lower than that obtained in the presence of magnesium ions.
The Sec-dependent secretion pathway was chosen for the secretion of recombinant proteins in B. choshinensis (Mizukami et al. 2010). In this pathway, the pre-proteins were transported through the cytoplasmic membrane and then folded to be mature proteins (Udaka and Yamagata 1993; Van Mellaert and Anné 2002). Morphological alterations showed that the addition of magnesium ions inhibited the shedding of HWP from the cell wall. This may strengthen the structure of the cell wall and act as an obstacle to the translocation of proteins into the medium (Sidhu and Olsen 1997; Udaka et al. 1989). Therefore, more time was allowed for the correct folding of pullulanase after translocation.
Notably, at 12 h, the protein concentration and the pullulanase activity obtained in the presence of added magnesium ions were almost the same as those in the absence of added magnesium ions (Fig. 6). Morphological examination and mRNA quantitation also suggest identical cell conditions and protein expression levels. The discrepancies appeared only after 12 h. This is consistent with our hypothesis that the presence of HWP on the cell wall favors the production of the active form of pullulanase. When HWP is shed from the cell wall after 24 h, most of the pullulanase synthesized was in the inactive form, as observed from the protein concentration and enzyme activity between 24 and 48 h.

Time profiles obtained from shake flask cultures. Symbols represent biomass (square), protein concentration (circle), and pullulanase activity (triangle) in the medium with (open) or without (filled) added magnesium ions
References
Adachi T, Yamagata H, Tsukagoshi N, Udaka S (1991) Repression of the cell wall protein gene operon in Bacillus brevis 47 by magnesium and calcium ions. J Bacteriol 173:4243–4245
Blanco CA, Caballero I, Barrios R, Rojas A (2014) Innovations in the brewing industry: light beer. Int J Food Sci Nutr 65(6):655–660
Chen A, Li YM, Liu XX, Long Q, Yang YK, Bai ZH (2014) Soluble expression of pullulanase from Bacillus acidopullulyticus in Escherichia coli by tightly controlling basal expression. J Ind Microbiol Biotechnol 41:1803–1810
Cheng S (2015) Expression, thermostability modification and application of sucrose isomerase from Serratia plymuthica [D]. Jiangnan University
Duan X, Wu J (2015) Enhancing the secretion efficiency and thermostability of a Bacillus deramificans pullulanase mutant (D437H/D503Y) by N-terminal domain truncation. Appl Environ Microb 81:1926–1931
Duan X, Chen J, Wu J (2013) Optimization of pullulanase production in Escherichia coli by regulation of process conditions and supplement with natural osmolytes. Bioresour Technol 146:379–385
Duan X, Zou C, Wu J (2015) Triton X-100 enhances the solubility and secretion ratio of aggregation-prone pullulanase produced in Escherichia coli. Bioresour Technol 194:137–143
Ebisu S, Tsuboi A, Takagi H, Naruse Y, Yamagata H, Tsukagoshi N, Udaka S (1990) Conserved structures of cell-wall protein genes among protein-producing Bacillus-brevis strains. J Bacteriol 172:1312–1320
Gohel V, Duan G (2012) Conventional process for ethanol production from Indian broken rice and pearl millet. Bioproc Biosyst Eng 35:1297–1308
Hii SL, Tan JS, Ling TC, Ariff AB (2012) Pullulanase: role in starch hydrolysis and potential industrial applications. Enzyme Res 2012:921362
Li ZF, Li B, Liu ZG, Wang M, Gu ZB, Du GC, Wu J, Chen J (2009) Calcium leads to further increase in glycine-enhanced extracellular secretion of recombinant alpha-cyclodextrin glycosyltransferase in Escherichia coli. J Agric Food Chem 57:6231–6237
Li S, Xu H, Yu J, Wang Y, Feng X, Ouyang P (2013) Enhancing isomaltulose production by recombinant Escherichia coli producing sucrose isomerase: culture medium optimization containing agricultural wastes and cell immobilization. Bioproc Biosyst Eng 36:1395–1405
Lin QL, Xiao HX, Liu GQ, Liu ZH, Li LH, Yu FX (2013) Production of maltose syrup by enzymatic conversion of rice starch. Food Bioprocess Tech 6:242–248
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 25(4):402–408
Mizukami M, Hanagata H, Miyauchi A (2010) Brevibacillus expression system: host-vector system for efficient production of secretory proteins. Curr Pharm Biotechnol 11:251–258
Mizukami M, Tokunaga H, Onishi H, Ueno Y, Hanagata H, Miyazaki N, Kiyose N, Ito Y, Ishibashi M, Hagihara Y, Arakawa T, Miyauchi A, Tokunaga M (2014) Highly efficient production of VHH antibody fragments in Brevibacillus choshinensis expression system. Protein Expr Purif 105:23–32
Mu T, Liang W, Ju Y, Wang Z, Wang Z, Roycik MD, Sang QX, Yu D, Xiang H, Fang X (2013) Efficient soluble expression of secreted matrix metalloproteinase 26 in Brevibacillus choshinensis. Protein Expr Purif 91:125–133
Sidhu MS, Olsen I (1997) S-layers of Bacillus species. Microbiol-Sgm 143:1039–1052
Singh RS, Saini GK, Kennedy JF (2010) Maltotriose syrup preparation from pullulan using pullulanase. Carbohydr Polym 80(2):401–407
Sreerama N, Woody RW (1993) A self-consistent method for the analysis of protein secondary structure from circular dichroism. Anal Biochem 209(1):32–44
Talekar S, Pandharbale A, Ladole M, Nadar S, Mulla M, Japhalekar K, Arage D (2013) Carrier free co-immobilization of alpha-amylase, glucoamylase and pullulanase as combined cross-linked enzyme aggregates (combi-CLEAs): a tri-enzyme biocatalyst with one pot starch hydrolytic activity. Bioresour Technol 147:269–275
Udaka S, Yamagata H (1993) Protein secretion in Bacillus brevis. Antonie Van Leeuwenhoek 64:137–143
Udaka S, Tsukagoshi N, Yamagata H (1989) Bacillus brevis, a host bacterium for efficient extracellular production of useful proteins. Biotechnol Genet Eng Rev 7:113–146
Van Mellaert L, Anné J (2002) Gram-positive bacteria as host cells for heterologous production of biopharmaceuticals[M]//novel frontiers in the production of compounds for biomedical use. Springer, Netherlands, pp. 277–300
Wu J, Duan X, Fang B (2015) A strain for α-cyclodextrin glycosyltransferase production and its application. China Patent CN201510056200.3
Yamada H, Tsukagoshi N, Udaka S (1981) Morphological alterations of cell wall concomitant with protein release in a protein-producing bacterium, Bacillus brevis 47. J Bacteriol 148:322–332
Zhang HX, Jin ZY (2011) Preparation of resistant starch by hydrolysis of maize starch with pullulanase. Carbohydr Polym 83:865–867
Zhang H, Tian Y, Bai Y, Xu X, Jin Z (2013) Structure and properties of maize starch processed with a combination of α-amylase and pullulanase. Int J Biol Macromol 52:38–44
Zou C, Duan X, Wu J (2014) Enhanced extracellular production of recombinant Bacillus deramificans pullulanase in Escherichia coli through induction mode optimization and a glycine feeding strategy. Bioresour Technol 172:174–179
Zou C, Duan XG, Wu J (2015) Efficient extracellular expression of Bacillus deramificans pullulanase in Brevibacillus choshinensis. J Ind Microbiol Biotechnol 1-10. doi:10.1007/s10295-015-1719-1
Acknowledgments
This work received financial support from the National Science Foundation for Distinguished Young Scholars (31425020), the National Natural Science Foundation of China (31271813, 31401636), the Project of Outstanding Scientific and Technological Innovation Group of Jiangsu Province, the Natural Science Foundation of Jiangsu Province (BK20140142), and the 111 Project (No. 111-2-06).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors confirm that they have no conflicts of interests.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary material
ESM 1
(PDF 264 kb)
Rights and permissions
About this article

Cite this article
Zou, C., Duan, X. & Wu, J. Magnesium ions increase the activity of Bacillus deramificans pullulanase expressed by Brevibacillus choshinensis . Appl Microbiol Biotechnol 100, 7115–7123 (2016). https://doi.org/10.1007/s00253-016-7386-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-016-7386-y