
Abstract
Objectives
To identify novel pullulanases from microorganisms and to investigate their biochemical characterizations.
Results
A novel pullulanase gene (BmPul) from Bacillus megaterium WW1210 was cloned and heterologously expressed in Escherichia coli. The gene has an ORF of 2814 bp encoding 937 amino acids. The recombinant pullulanase (BmPul) was purified to homogeneity and biochemically characterized. BmPul has an MW of approx. 112 kDa as indicated by SDS-PAGE. Optimum conditions were at 55 °C and pH 6.5. The enzyme was stable below 40 °C and from pH 6.5−8.5. The Km values of BmPul towards pullulan and amylopectin were 3.3 and 3.6 mg/ml, respectively. BmPul hydrolyzed pullulan to yield mainly maltotriose, indicating that it should be a type I pullulanase.
Conclusions
A novel type I pullulanase from Bacillus megaterium was identified, heterologously expressed and biochemically characterized. Its properties makes this enzyme as a good candidate for the food industry.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Pullulanases (pullulan-6-glucanohydrolase; EC 3.2.1.41) are usually defined as debranching enzymes which can specifically hydrolyze α-1,6-glycosidic bonds in pullulan, starch, amylopectin and related oligosaccharides (Hii et al. 2012). They are divided into two major categories on the basis of their substrate specificities and hydrolytic properties: (1) type I pullulanases specifically catalyze the hydrolysis of α-1,6-glycosidic linkages in pullulan and other branched oligosaccharides to form maltotriose or linear oligomers; and (2) type II pullulanases (amylopullulanases), not only attack the α-1,6-glycosidic linkages in pullulan but also cleave the α-1,4-glycosidic linkages in other polysaccharides, such as amylopectin, glycogen and starch (Li et al. 2012). Based on their amino acid sequence similarities, pullulanases have been assigned to glycoside hydrolase (GH) families 13 and 57 (Elleuche et al. 2015).
Pullulanases are widely used in food industry for the production of high-maltose corn syrup, high-fructose corn syrup, cyclodextrins (CDs) etc. (Song et al. 2016). A number of pullulanases have been identified and characterized from different microorganisms including Bacillus sp. (Kunamneni and Singh 2006), Geobacillus thermoleovorans (Ayadi et al. 2008), Bacillus sp. CICIM 263 (Li et al. 2012a), Lactococcus lactis (Wasko et al. 2011), Bacillus naganoensis (Nie et al. 2013; Song et al. 2016) and Klebsiella variicola (Chen et al. 2013). However, most pullulanases are type II enzymes (Kang et al. 2011; Li et al. 2012b). Type I pullulanases are preferable in industrial applications because they can release long glucan polymers and thus allow more efficient and rapid conversion capacity in starch processing industry. Only a few type I pullulanases have been studied, including pullulanases from G. thermoleovorans (Ayadi et al. 2008), Thermotoga neapolitana (Kang et al. 2011), Bacillus sp. (Li et al. 2012b), Bacillus cereus (Wei et al. 2014) and Shewanella arctica (Elleuche et al. 2015). No pullulanase from B. megaterium has been previously reported.
Bacillus megaterium WW1210 was isolated in our laboratory. Here, we describe gene cloning, heterologous expression and biochemical characterization of a novel type I pullulanase from B. megaterium WW1210. In addition, the potential application of the recombinant pullulanase in RS production from maize starch was evaluated.
Materials and methods
Strains and reagents
Bacillus megaterium WW1210 used in this study was previously isolated from soil in our laboratory. E. coli strains DH5α and BL21(DE3) (Novagen, Madison, USA) were used as hosts for gene cloning and protein expression, respectively. Plasmid pET-30a(+) (Novagen, Madison, USA) was used as cloning and expression vector. PrimeSTAR HS DNA polymerase and restriction endonucleases were purchased from TaKaRa (Tokyo, Japan). T4 DNA ligase was purchased from New England Biolabs. Chelating Sepharose (Ni-IDA) resin matrix was obtained from GE Life Sciences. Amylose (type Ш from potato) and amylopectin (from potato) were purchased from Sigma. α-Amylase and glucoamylase were obtained from Aoboxing Bio-Tech Company (Beijing, China). All other chemicals were of analytical grade unless otherwise stated.
Cloning of a pullulanase gene from B. megaterium WW1210
DNA manipulations were performed according to the recombinant DNA techniques. Genomic DNA of B. megaterium WW1210 was used as the template for amplification of the pullulanase-encoding gene with specific primers ( BmPulDF: 5′-(CATGCCATGGCAGACTCAACAAAAATCACGATTC-3′; BmPulDR: 5′- CCGCTCGAGTTATCTTACTAGCACAAATGTCGAAAGAG-3′) containing NcoI and XhoI restriction sites (underlined). The PCR conditions were as follows: a denaturation step at 94 °C for 5 min, followed by 30 cycles at 94 °C for 30 s, 60 °C for 30 s, and 72 °C for 3 min, with a final extension step at 72 °C for 10 min. The PCR product was cloned into the NcoI/XhoI site of the pET-30a(+) vector to produce pET30a-BmPul, which was then transformed into E. coli BL21 (DE3) for protein expression. For recombinant selection, kanamycin was used at 50 µg/ml.
Expression and purification of the recombinant pullulanase
E. coli BL21 (DE3) cells harboring the plasmid pET30a-BmPul was inoculated into lysogeny broth (LB) containing kanamycin (50 µg/ml) and incubated at 37 °C with shaking (200 rpm) until OD600 reached about 0.6−0.8. Expression was then induced by adding IPTG to 1 mM. Cultivation was continued at 25 °C overnight.
Recombinant cells were harvested from culture broth by centrifugation at 10,000×g for 10 min at 4 °C, re-suspended in lysis buffer A (50 mM phosphate buffer pH 7.4 containing 500 mM NaCl and 20 mM imidazole) and disrupted by ultrasonication. The debris was removed by centrifugation at 10,000×g for 10 min at 4 °C, and the supernatant was collected as the crude enzyme. The crude pullulanase solution was loaded onto a Ni-IDA column (1 × 5 cm) pre-equilibrated with buffer A at 0.5 ml/min. After binding for 30 min, the column was washed with 15 column volumes (CV) of buffer A, followed by 5 CV of buffer B (50 mM phosphate buffer pH 7.4 containing 500 mM NaCl and 50 mM imidazole) at 1 ml/min to remove unbounded proteins. Finally, the column was washed with buffer C (50 mM phosphate buffer pH 7.4 containing 500 mM NaCl and 200 mM imidazole). The fractions showing pullulanase activity were collected and checked for purity by SDS-PAGE.
SDS-PAGE and molecular mass determination
SDS-PAGE used 7.5% (w/v) separating gel and 4.5% (w/v) stacking gel. Protein bands were stained by Coomassie Brilliant Blue R-250. The denatured MW of the purified pullulanase was calculated by using a protein molecular mass calibration kit. The native molecular mass was determined on a Sephacryl S-200 gel filtration column by comparison with standard proteins: catalase (250 kDa), alcohol dehydrogenase (150 kDa), phosphorylase b (97.2 kDa), fetuin (68 kDa) and albumin from chicken egg white (45 kDa).
Enzyme assay and protein determination
Pullulanase activity was determined based on the amount of reducing sugars released from pullulan. Briefly, 500 μl appropriately diluted enzyme solution was added into 500 μl 1% (w/v) pullulan in 50 mM MES buffer (pH 6.5) and incubated at 55 °C for 10 min. The reaction was terminated by adding 500 µl 3,5-dinitrosalicylic acid reagent and the amount of released reducing sugars were determined spectrophotometrically. One unit (U) of pullulanase activity was defined as the amount of the enzyme that required to release 1 µmol glucose equivalent reducing sugar per min under the assay conditions. All assays were performed in triplicate.
Protein concentration was determined by the Lowry method using bovine serum albumin (BSA) as the standard. The specific activity was expressed as units per mg protein.
Biochemical characterization of the recombinant pullulanase
The optimal pH of BmPul was determined at 55 °C in 50 mM using: citrate buffer (pH 3–6), acetate buffer (pH 4–5.5), MES (pH 5–7), MOPS (pH 6.5–8.5) and CHES (pH 8–11). To determine the pH stability, BmPul was incubated in the above buffers at 40 °C for 30 min, and the residual activity was then determined using the standard assay.
Pullulanase activity was determined from 30 to 70 °C in 50 mM MES buffer (pH 6.5). For thermostability determination, BmPul was pre-incubated in 50 mM MES buffer (pH 6.5) from 30 to 70 °C for 30 min, followed by rapid cooling on ice, and the residual activity was then measured using the standard assay.
Substrate specificity and kinetic parameters of BmPul
The substrate specificity of BmPul was determined by measuring its activity in 50 mM MES buffer (pH 6.5) at 55 °C for 10 min using pullulan, amylopectin, amylose, maltoheptaose, maltohexaose, maltopentaose and maltotetraose at 10 mg/ml. The amount of released reducing sugars was determined by the DNS method. One unit (U) of pullulanase activity was defined as the amount of the enzyme that required releasing 1 µmol glucose equivalent reducing sugar per min under the assay conditions.
For determination of kinetic parameters, the enzyme reactions were carried out in 50 mM MES buffer (pH 6.5) at 55 °C for 5 min using different substrate concentrations. The apparent Michaelis constant (K m) and maximal velocity (V max) were calculated using the “GraFit” program.
Hydrolytic property of BmPul
The hydrolytic property of BmPul was investigated by analyzing the hydrolysis products of different substrates (pullulan and amylopectin) by the purified pullulanase. Briefly, 0.5 U/ml BmPul was added to pullulan and amylopectin at 10 mg/ml in 50 mM MES buffer (pH 6.5), separately, and incubated at 40 °C for 2 h. Samples were withdrawn and analyzed by TLC on a Kieselgel 60 plate (Merck) with 1-butanol/ethanol/water (5:3:2, by vol.). The hydrolysis products were visualized by heating for a few minutes at 130 °C after spraying with methanol/sulfuric acid (95:5, v/v). Maltotriose, maltotetraose, maltopentaose and maltohexaose were used as standards. The released products from pullulan and amylopectin were quantitatively determined by HPLC and high-performance anion exchange chromatography (HPAEC), respectively.
Results
Cloning of a pullulanase gene (BmPul) from B. megaterium WW1210 and its sequence analysis
A pullulanase gene (BmPul) was amplified by PCR using the genomic DNA of B. megaterium WW1210 as the template. The gene consists of an ORF of 2814 bp, encoding a protein of 937 amino acids with a predicted molecular mass of 106,063 Da and a theoretical pI of 7.7. The gene sequence has been submitted to GenBank under the accession number KX151777.
Multiple amino acid sequence alignment revealed that BmPul contained an NWGYDPKN motif that is involved in the hydrolysis of α-1,6 glycosidic bonds in pullulan, and it shared the highest identity of 63% with a type I pullulanses from Bacillus sp. CICIM 263 (Li et al. 2012a) (Fig. 1). In addition, four typical conserved regions [regions I (DVVYNH), II (DGFRFDLMGIHD), III (GEGWDL) and IV (EAHDN)] (Fig. 1) in GH13 α-amylases (Hatada et al. 2006) were found in the sequence. Asp631, Glu660 and Asp750 were presumed to be contributed to the catalytic triad of acidic residues.

Multiple amino acid sequence alignment of BmPul with other pullulanases. Numbers on the left are the residue numbers of the first amino acid in each line. Abbreviations and accession numbers of the pullulanases are as follows: Bacillus megaterium WSH-002 (B.m.YP005495430), Bacillus cereus (B.c.WP016108555), B.megaterium QM B1551 (B.m.YP003562500), Bacillus vireti (B.v.WP024029221), Bacillus sp. CICIM 263 (B.sp.AGA03915). Identical residues are shared in gray, and conserved residues are shaded in black. The three conservative catalytic amino acids (D631, E660, D750) are identified by asterisks. The four conserved regions (I, II, III and IV) and region (a) are shown by upper lines
Expression and purification of the recombinant pullulanase (BmPul)
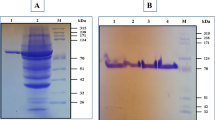
BmPul was expressed in E. coli as an intracellular soluble active protein. The recombinant enzyme was purified to homogeneity by Ni-IDA chromatography with a purification fold of 1.3 and a recovery yield of 48%. The specific activity was increased from 64.6 to 83.3 U/mg after purification. The purified enzyme showed a single protein band on SDS-PAGE with molecular mass of about 112 kDa (Fig. 2). The native molecular mass of BmPul was estimated as 114.5 kDa (data not shown).

SDS-PAGE analysis of BmPul from B. megaterium expressed in E. coli. Lane M broad molecular weight protein standards; lane 1 crude lysate; lane 2 purified BmPul
Biochemical characterization of BmPul
BmPul displayed an optimal pH of 6.5 in 50 mM MES buffer (Fig. 3a). It was stable from pH 6.5 to 8.5, retaining more than 85% of its original activity after being held in different buffers for 30 min (Fig. 3b). The enzyme was most active at 55 °C (Fig. 3c), and it was stable up to 40 °C (Fig. 3d).

Optimal pH (a) pH stability (b) optimal temperature (c) and thermostability (d) of the purified BmPul. The effect of pH on BmPul activity was determined at 50 °C using different buffers: filled square citrate buffer (pH 3.0−6.0), filled circle acetate buffer (pH 4.0−5.5), open diamond MES buffer (pH 5.0−7.0), filled triangle MOPS buffer (pH 6.5−8.5) and filled circle CHES buffer (pH 8.0−11.0). For pH stability, the enzyme was incubated in the above buffers at 40 °C for 30 min, and the residual activity was then measured. The optimal temperature was measured at different temperatures (30–70 °C) using 50 mM citrate buffer (pH 5.5). To determine thermostability, the enzyme was incubated in 50 mM MES buffer (pH 6.5) at different temperatures for 30 min, and the residual activity was then measured. The data are the means of three independent determinations, and the highest activity was treated as 100%
Substrate specificity, hydrolysis properties and kinetic parameters of BmPul
The purified BmPul showed the highest specific activity towards pullulan (83 U/mg), followed by amylopectin (23.2 U/mg), Table 1 but did not show any activity towards amylose, maltoheptaose, maltohexaose, maltopentaose and maltotetraose (Table 1). BmPul hydrolyzed pullulan to yield mainly maltotriose in 2 h (Fig. 4a), accounting for more than 95% (7 mg/ml) (data not shown) but hydrolyzed amylopectin to release mainly maltotriose, maltotetraose, Fig. 4 maltopentaose and maltohexaose (Fig. 4b), and their contents reached to 4.4, 6.8, 7.3, 25 and 35 μg/ml, respectively, after 2 h incubation (Fig. 4c).

TLC analysis of hydrolyzed products of pullulan a and amylopectin b by the purified BmPul and quantitatively analysis of the products released from amylopectin c 1% (w/v) of pullulan and amylopectin in 50 mM MES buffer (pH 6.5) were incubated with 0.5 U/ml of BmPul at 40 °C for 2 h, separately. The released products were analyzed by TLC. Substrates and incubation times were indicated. Lane M is the standard mixture containing maltotriose, maltotetraose, maltopentaose and maltohexaose. (filled square) Maltotriose, (open square) maltotetraose, (open triangle) maltopentaose, (open circle) maltohexaose, (filled triangle) maltoheptaose
The K m and V max values of BmPul towards pullulan and amylopectin were 3.3 ± 0.25 mg/ml and 249 ± 11 μmol/min/mg, and 3.6 ± 0.18 mg/ml and 195 ± 4.5 μmol/min/mg, respectively (Table 2).
Discussion
Pullulanases have considerable potential in industrial applications (Hii et al. 2012). Many microbial pullulanases have been reported (Wasko et al. 2011; Li et al. 2012a; Nie et al. 2013; Song et al. 2016), whereas there is still less information on type I pullulanases. Bacillus spp. produce pullulanases (Kunamneni and Singh 2006; Li et al. 2012b; Wei et al. 2014); however no type I pullulanase from B. megaterium has been reported. Here, for the first time, we described gene cloning, expression, characterization and application of a novel type I pullulanase from B. megaterium WW1210.
Multiple amino acid sequence alignment analysis showed that BmPul shared relative high similarities (~63%) with some type I pullulanases and contained four regions conserved in GH family 13 enzymes and a NWGYDPKN motif (Fig. 1). The five regions are involved in the hydrolysis of α-1,6 bonds in pullulan (Li et al. 2012b). Three amino acids, Asp631, Glu660 and Asp750, in the conserved regions may contribute to the active site architecture. Moreover, the molecular mass of BmPul with other type I pullulanases were compared. BmPul has a MW of 112 kDa (Fig. 2), which is a little lower than the type I pullulanases from K. variicola SHN-1 (118 kDa, Chen et al. 2013) and S. arctica (150 kDa, Elleuche et al. 2015) but higher than the type I pullulanases from Paenibacillus barengoltzii (75 kDa, Liu et al. 2016), B. cereus Nws-bc5 (81.4 kDa, Wei et al. 2014), T. neapolitana (93 kDa, Kang et al. 2011) and Bacillus sp. CICIM 263 (101 kDa, Li, Zhang et al. 2011). Thus, BmPul should be a novel type I pullulanase belonging to GH family 13.
Most microbial pullulanases have weakly acidic or neutral optimal pH (pH 6–7, Elleuche et al. 2015). In the present study, BmPul was most active at pH 6.5, which is similar to type I pullulanases from B. cereus (pH 6.0, Wei et al. 2014), Bacillus sp. (pH 6.0, Kunamneni and Singh 2006), G. thermoleovorans (pH 6.0, Ayadi et al. 2008), T. neapolitana (pH 6.5, Kang et al. 2011), Bacillus sp. (pH 6.5, Li et al. 2012b) and S. arctica (pH 7.0, Elleuche et al. 2015), but distinct from the type I pullulanase from L. lactis (Wasko et al. 2011) which has an optimal pH of 4.5. BmPul had an optimal temperature of 55 °C and this is higher than most of the cold or mesophilic type I pullulanases from S. arctica (35 °C, Elleuche et al. 2015), B. cereus (40 °C, Wei et al. 2014) and L. lactis (45 °C) (Wasko et al. 2011). However, it is lower than several thermostable type I pullulanases from thermophiles, such as Bacillus sp. (70 °C, Li et al. 2012), G. thermoleovorans (70 °C, Ayadi et al. 2008), T. neapolitana (80 °C, Kang et al. 2011) and Bacillus sp. AN-7 (90 °C, Kunamneni and Singh 2006) (Table 2).
Generally, type I pullulanases show broad substrate specificity towards polysaccharides such as pullulan, amylopectin and amylose (Liu et al. 2016), that have α-1,6-glycosidic bonds in their structures. BmPul showed strict substrate specificity towards pullulan (100%) and amylopectin (28%), but exhibited no activity towards amylose (Table 1), which is similar to that of the type I pullulanase from Bacillus subtilis (Wei et al. 2014). Although the pullulanase from Bacillus sp. CICIM 263 exhibited the same properties with BmPul, it had relatively high activity towards amylopectin, accounting up to 52% of its activities towards pullulan (Li, Zhang et al. 2012), but BmPul displayed a relative high specific activity of 83 U/mg (Table 1). This value is higher than those of most other reported type I pullulanases, such as the pullulanases from K. variicola SHN-1 (10 U/mg, Chen et al. 2013), G. thermoleovorans US105 (12 U/mg, Ayadi et al. 2008), T. neapolitana (28.7 U/mg, Kang et al. 2011), S. arctica (33.8 U/mg, Elleuche et al. 2015), G. thermoleovorans (36 U/mg, Ayadi et al. 2008), B. cereus (44.7 U/mg, Wei et al. 2014) and Bacillus sp. CICIM 263 (73.5 U/mg, Li et al. 2012b), representing the highest specific activity towards pullulan for type I pullulanaes ever reported.
BmPul hydrolyzed pullulan to yield mainly maltotriose (Fig. 4a), which is consistent with most other reported type I pullulanases (Kang et al. 2011; Li et al. 2012; Wei et al. 2014; Elleuche et al. 2015), further confirming that Bmpul is a type I pullulanaes. BmPul hydrolyzed amylopectin to release oligosaccharides with degree of polymerization of 3–7 as the major end products without formation of maltose (Fig. 4b and 4c), while other type I pullulanases exhibited different action manner. For example, the pullulanases from S. arctica (Elleuche et al. 2015) and T. neapolitana (Kang et al. 2011) degrade amylopectin to release mainly small oligosaccharides and maltose, while Bacillus sp. pullulanse hydrolyzed amylopectin to produce mainly maltose and maltotriose (Li et al. 2012).
In conclusion, a novel pullulanase (BmPul) from B. megaterium WW1210 was gene cloned, expressed and biochemically characterized. BmPul was a monomer with molecular mass of 112 kDa. It was most active at pH 6.5 and 55 °C, respectively. The enzyme showed strict substrate specificity towards pulullan and amylopectin. It hydrolyzed pullulan to produce mainly maltotriose, exhibiting typical action model of type I pullulanases. The excellent biochemical properties of this enzyme makes it valid candidate in food industry with potential applications.
References
Ashwar BA, Gani A, Wani IA, Shaha A, Masoodia FA, Saxena DC (2016) Production of resistant starch from rice by dual autoclaving-retrogradation treatment: in vitro digestibility, thermal and structural characterization. Food Hydrocoll 56:108–117
Ayadi DZ, Ali MB, Jemli S, Mabrouk SB, Mezghani M, Messaoud EB, Bejar S (2008) Heterologous expression, secretion and characterization of the Geobacillus thermoleovorans US105 type I pullulanase. Appl Microbiol Biotechnol 78:473–481
Chen WB, Nie Y, Xu Y (2013) Signal peptide-independent secretory expression and characterization of pullulanase from a newly isolated Klebsiella variicola SHN-1 in Escherichia coli. Appl Biochem Biotechnol 169:41–54
Elleuche S, Qoura FM, Lorenz U, Rehn T, Brück T, Antranikian G (2015) Cloning, expression and characterization of the recombinant cold-active type-I pullulanase from Shewanella arctica. J Mol Catal B Enzym 116:70–77
Hatada YJ, Masuda N, Akita M (2006) Oxidatively stable maltopentaose-producing α-amylase from a deep-sea Bacillus, isolate, and mechanism of its oxidative stability validated by site-directed mutagenesis. Enzyme Microb Technol 39:1333–1340
Hii SL, Tan JS, Ling TC, Ariff AB (2012) Pullulanase: role in starch hydrolysis and potential industrial applications. Enzyme Res 1:921362
Kang JH, Park KM, Choi KH, Park CS, Kim GE, Kim D, Cha J (2011) Molecular cloning and biochemical characterization of a heat-stable type I pullulanase from Thermotoga neapolitana. Enzyme Microb Technol 48:260–266
Kunamneni A, Singh S (2006) Improved high thermal stability of pullulanase from a newly isolated thermophilic Bacillus sp. AN-7. Enzyme Microb Technol 39:1399–1404
Li MR, Wang XB, Huang Y, Huang JL, Liang JY, Huang RL, Du QL, Wei YT (2012a) Gene expression and characterisation of three pullulanases from Bacillus cereus GXBC-3. Chin J Biotechnol 28:466–475
Li YR, Zhang L, Niu DD, Wang ZX, Shi G (2012b) Cloning, expression, characterization, and biocatalytic investigation of a novel bacilli thermostable type I pullulanase from Bacillus sp. CICIM 263. J Agric Food Chem 60:11164–11172
Liu JJ, Liu Y, Yan F, Jiang ZQ, Yang SQ, Yan QJ (2016) Gene cloning, functional expression and characterisation of a novel type I pullulanase from Paenibacillus barengoltzii and its application in resistant starch production. Protein Expr Purif 172:1562–1568
Nie Y, Yan W, Xu Y, Chen WB, Mu XQ, Wang XY, Xiao R (2013) High-level expression of Bacillus naganoensis pullulanase from recombinant Escherichia coli with auto-induction: effect of Iac operator. PLoS ONE 8:e78416
Song W, Nie Y, Mu XQ, Xu Y (2016) Enhancement of extracellular expression of Bacillus naganoensis pullulanase from recombinant Bacillus subtilis: effects of promoter and host. Protein Expr Purif 124:23–31
Wasko A, Polak-Berecka M, Targonski Z (2011) Purification and characterization of pullulanase from Lactococcus lactis. Prep Biochem Biotechnol 41:252–261
Wei W, Ma J, Guo S, Wei DZ (2014) A type I pullulanase of Bacillus cereus Nws-bc5 screening from stinky tofu brine: functional expression in Escherichia coli and Bacillus subtilis and enzyme characterization. Proc Biochem 49:1893–1902
Acknowledgements
This work was financially supported by the National Science Fund for Distinguished Young Scholars (No. 31325021) and the Program for Changjiang Scholars (No. T2014055).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Yang, S., Yan, Q., Bao, Q. et al. Expression and biochemical characterization of a novel type I pullulanase from Bacillus megaterium . Biotechnol Lett 39, 397–405 (2017). https://doi.org/10.1007/s10529-016-2255-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-016-2255-4