
Abstract
Data on glucose dehydrogenases (GDHs) are scarce and availability of these enzymes for application purposes is limited. This paper describes a new GDH from the fungus Pycnoporus cinnabarinus CIRM BRFM 137 that is the first reported GDH from a white-rot fungus belonging to the Basidiomycota. The enzyme was recombinantly produced in Aspergillus niger, a well-known fungal host producing an array of homologous or heterologous enzymes for industrial applications. The full-length gene that encodes GDH from P. cinnabarinus (PcGDH) consists of 2,425 bp and codes for a deduced protein of 620 amino acids with a calculated molecular mass of 62.5 kDa. The corresponding complementary DNA was cloned and placed under the control of the strong and constitutive glyceraldehyde-3-phosphate dehydrogenase promoter. The signal peptide of the glucoamylase prepro sequence of A. niger was used to target PcGDH secretion into the culture medium, achieving a yield of 640 mg L−1, which is tenfold higher than any other reported value. The recombinant PcGDH was purified twofold to homogeneity in a one-step procedure with a 41 % recovery using a Ni Sepharose column. The identity of the recombinant protein was further confirmed by immunodetection using western blot analysis and N-terminal sequencing. The molecular mass of the native PcGDH was 130 kDa, suggesting a homodimeric form. Optimal pH and temperature were found to be similar (5.5 and 60 °C, respectively) to those determined for the previously characterized GDH, i.e., from Glomerella cingulata. However PcGDH exhibits a lower catalytic efficiency of 67 M−1 s−1 toward glucose. This substrate is by far the preferred substrate, which constitutes an advantage over other sugar oxidases in the case of blood glucose monitoring. The substrate-binding domain of PcGDH turns out to be conserved as compared to other glucose-methanol-choline (GMCs) oxidoreductases. In addition, the ability of PcGDH to reduce oxidized quinones or radical intermediates was clearly demonstrated, which raises prospects for applying this enzyme to detoxify toxic compounds formed during the degradation of lignin.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Flavin adenine dinucleotide (FAD)-dependent glucose dehydrogenase (GDH, EC 1.1.99.10) is present in organisms of various origins and belongs to family AA3.2 of the CAZy carbohydrate-active enzymes database (Cantarel et al. 2009) that was recently expanded by oxidative enzyme families (Levasseur et al. 2013). GDH is found in bacterial periplasm and insect cell cytosol or extracellularly produced in fungi (Ferri et al. 2011). GDHs are glycosylated, monomeric to homo-oligomeric enzymes belonging to the large group of glucose-methanol-choline (GMCs) oxidoreductases, and closely related to AA3_2 glucose oxidases (GOX) (Sygmund et al. 2011a). GDH, a so-called d-glucose/acceptor 1-oxidoreductase, catalyzes the anomeric hydroxy group of glucose but is also reported to convert a pentose sugar, xylose. The cofactor FAD serves as the primary electron acceptor. In comparison with GOX, GDH does not use oxygen as an external electron acceptor but instead uses phenoxy radicals, quinones, redox dyes, and iron complexes such as ferricyanide and ferrocenium hexafluorophosphate. The first work on GDH appeared in 1931 describing an enzyme prepared from animal tissues (Harrison 1931). The first fungal GDH was reported in Aspergillus sp. (Ogura and Nagahisa 1937) and the enzyme characteristics were further studied by Bak (1967). A few other filamentous fungi GDH sources have since been discovered, i.e., from Aspergillus niger (Müller 1977), Aspergillus terreus (Tsujimura et al. 2006), Penicillium lilacinoechinulatum (Aiba and Tsugura-shi 2007), and more recently from Glomerella cingulata (Sygmund et al. 2011a, b). Even so, data on the physiological function of the fungal GDH is restricted to a few works and the availability of diverse enzyme GDHs for application purposes remains limited. Ogura (1951) showed that the substrate of GDH is glucose and not glucose-6-phosphate. Production of the Aspergillus oryzae GDH is induced by the addition of benzoquinone and hydroquinone (Bak and Sato 1967) and the GDH from the plant pathogen G. cingulata is able to efficiently reduce quinones and phenoxy compounds (Sygmund et al. 2011b). The authors also suggested two putative roles for GDH: (1) detoxification by a reduction of plant-generated quinones produced during fungal degradation and (2) reduction of the phenoxy radicals produced by the plant to protect the cell wall structure from fungal attack. GDH is thus thought to use the carbohydrates released from plant hydrolysis as an abundant cosubstrate. While there is still speculation over this putative function of GDH, its use in biotech applications has long been considered a possibility.
The closely related GOX was first utilized in a glucose sensor system to monitor glycemic levels using oxygen as electron acceptor (Clark and Lyons 1962). The second-generation sensors employed external electron acceptors to increase accuracy and reliability (Ferri et al. 2011). Fungal GDHs were used in some of these new sensors as they are not influenced by changing oxygen concentrations in blood and possess the narrow substrate specificity needed for accurate results to essentially measure glucose (Bai et al. 2012; Milutinovic et al. 2011; Saleh et al. 2012; Zafar et al. 2012). Another highly interesting application is as anode catalyst for miniaturized implantable biofuel cells in a process that uses glucose and oxygen as energy source (Wang et al. 2012; Zhang et al. 2012). In a very recent application, a recombinant bacterial GDH was used in an integrated system to detoxify industrial dye effluents in combination with a second enzyme, an azo-reductase, to catalyze azo bond cleavage and, more specifically, to degrade methyl red (Yang et al. 2013).
We recently sequenced the genome of the biotechnologically-relevant Pycnoporus cinnabarinus to study the specific enzyme machinery it produces to degrade plant lignocellulose (Levasseur et al. 2014). Among white-rot fungi, P. cinnabarinus is known to efficiently completely degrade lignin (see Lomascolo et al. 2011 for review). It belongs to the Polyporaceae family in the phylum of Basidiomycota. Its ligninolytic system features copper and iron-containing enzymes (metalloenzymes) that are strategic for targeted transformation of aromatic cell compounds (Lomascolo et al. 1999). Among the lignin-degrading enzymes, P. cinnabarinus is known to secrete a set of native laccases (AA1) (Eggert et al. 1996) at very high levels of up to 1 g l−1 (Lomascolo et al. 2003). P. cinnabarinus has also been studied for its original metabolic pathways involved in the functionalization of these cell wall aromatics to yield high-added-value compounds including aromas and antioxidants (Lesage-Meessen et al. 1996; Estrada Alvarado et al. 2001). In addition, the P. cinnabarinus cellobiose dehydrogenase (CDH) has recently been shown to enhance a Trichoderma reesei multienzyme cocktail in the microbial degradation of lignocellulosic biomass (Bey et al. 2011).
In the course of analyzing the P. cinnabarinus genome (Levasseur et al. 2014), we identified a new GMC family enzyme as another GDH enzyme closely related to GOX and CDH. This new enzyme was cloned and heterologously expressed in the industrial strain A. niger for possible large-scale production. The enzyme was fully characterized in terms of physicochemical properties, enzyme kinetics, substrate specificity, cofactor functionality, and putative reduction of oxidized compounds by P. cinnabarinus laccase. This study could open new perspectives for the detoxification of phenoxy groups during plant biomass degradation or pretreatment using the lignin-degrading system of P. cinnabarinus.
Materials and methods
Strain and culture media
Escherichia coli JM109 (Promega, Charbonnières, France) was used for vector construction and propagation. A. niger strain D15#26 (pyrG-) (Gordon et al. 2000) was used for heterologous expression of the GDH encoding complementary DNA (cDNA). After cotransformation with vectors containing the pyrG gene and the expression cassette, respectively, transformants of A. niger were grown for selection on solid minimal medium (without uridine) containing 70 mM NaNO3, 7 mM KCl, 11 mM KH2HPO4, 2 mM MgSO4, and 1 % (w/v) glucose and trace elements (1,000× stock; 76 mM ZnSO4, 178 mM H3BO3, 25 mM MnCl2, 18 mM FeSO4, 7.1 mM CoCl2, 6.4 mM CuSO4, 6.2 mM Na2MoO4, and 174 mM EDTA). For the procedure of screening the positive transformants, 100 mL of culture medium containing 70 mM NaNO3, 7 mM KCl, 200 mM Na2HPO4, 2 mM MgSO4, and 5 % (w/v) glucose and trace elements was inoculated with 2.106 spores mL−1 in a 250-mL baffled flask.
The P. cinnabarinus strain BRFM137 is available at the CIRM-CF (Centre International de Ressources Microbiennes-Champignons Filamenteux (https://www6.inra.fr/cirm_eng/Filamentous-Fungi).
Cloning and expression of glucose dehydrogenase encoding gene
The P. cinnabarinus genome strain BRFM137 was recently sequenced, and gene annotation resulted in selection of the GDH candidate genomic sequence scf184803.g17 (Levasseur et al. 2014). The gene sequence is available at the European Nucleotide Archive (ENA), EMBL-EBI, (accession number, [EMBL: PRJEB5237, scf184803.g17]). The open reading frame (ORF) was predicted from the genomic sequence by translating it in the six frames and detecting splicing sites following the general pattern GTRMGT…YAG. This prediction was corroborated by BLAST using the P. cinnabarinus cDNA library (Levasseur et al. 2014) with the Augustus gene prediction website (http://bioinf.uni-greifswald.de/augustus/). The ORF sequence was aligned against other organisms to check the splicing prediction (tblastx on NCBI). The synthesized cDNA was cloned in the pAN52.4 (EMBL accession: Z32699) expression vector using a restriction cloning approach with BssHII and HindIII enzymes. The 22 amino acids of the GDH signal peptide (MPHSRLLATIGAVALLARCVTA) were replaced by the 24 amino acid glucoamylase (GLA) prepro sequence from A. niger (MGFRSLLALSGLVCNGLANVISKR). Additional segments were incorporated: one encoding a His-Tag (CACCAT) × 3 which was integrated downstream and two restriction sites (MluI and HindIII) at the 5′ and 3′ ends, respectively, for cloning in the expression vector. After codon optimization for A. niger, the predicted cDNA sequence (accession number KJ934222) with all its additional fragments was synthesized (GeneArt, Carlsbad, USA), propagated in E. coli JM109 (Promega, Charbonnières, France), and sequence-checked (GATC Biotech, Konstanz, Germany). In the final expression cassette, the Aspergillus nidulans glyceraldehyde-3-phosphate dehydrogenase encoding gene (gpdA) promoter, the 5′ untranslated region of the gpdA mRNA and the A. nidulans trpC terminator were used to drive expression of the inserted encoding sequence.
Transformation, screening of transformants, and GDH assay
Co-transformation was carried out as described by Punt and van den Hondel (1992) using both the GDH expression vector and pAB4-1 (van Hartingsveldt et al. 1987) containing the pyrG selection marker in a 10:1 ratio. Transformants were selected for uridine prototrophy for growth on selective solid minimum medium (without uridine). In order to screen enzyme production in liquid medium, 50 mL of minimum culture medium (adjusted to pH 5.5 with 1 M citric acid) was inoculated with 2.106 spores mL−1 in a 250-mL baffled flask. The culture was monitored for 12 days at 30 °C in a shaker incubator (130 rpm). pH was daily adjusted to 5.5 with 1 M citric acid. From these liquid cultures, aliquots (1 mL) were collected daily and cells were pelleted by centrifugation (5 min at 10,000×g, ambient temperature). GDH standard activity was assayed spectrophotometrically by monitoring the decolorization of 0.4 mM 2,6-dichloroindophenol (DCIP, Ɛ M = 6,800 M−1 cm−1) at 520 nm in 50 mM citrate-phosphate buffer pH 5.5 and 0.6 M of d-glucose. Strongly dependence of DCIP Ɛ M to pH was not taking into account here since a single value of Ɛ M may be used for all pH values at which the dye is stable at 520 nm. (Armstrong 1964). When specified, 1,4-benzoquinone (Ɛ M = 2.24 mM−1 cm−1) was used as electron acceptor instead of DCIP. The reaction was followed for 60s at 30 °C in a Uvikon XS spectrophotometer (BioTek Instruments, Colmar, France). Activity is expressed in nkat, i.e., the amount of GDH that oxidizes 1 nmol of substrate per second.
Purification of the recombinant GDH
The higher-producing transformant was inoculated under the same conditions as in the screening procedure. After 10 days of growth in a shaking incubator (120 rpm), 3 L of culture containing 1,080 mg of protein was harvested clarified on GF/D and GF/F glass fiber filters (Whatman, Maidstone, UK), sterile-filtered (0.45 μm), and concentrated tenfold by ultrafiltration through a polyether-sulfone membrane with a 10 kDa molecular mass cutoff (Vivaflow crossflow cassette, Sartorius, Les Ulis, France). The retentate (911.6 mg protein) was adjusted to pH 7.8 and the His-tagged recombinant protein was purified on a Chelating Sepharose Fast Flow column (5 mL Ni Histrap, GE Healthcare, Velizy-Villacoublay, France) equilibrated with five column volumes of binding buffer (50 mM Tris-HCl, pH 7.8, 150 mM NaCl, 10 mM imidazole). After washing in five column volumes with binding buffer, bound proteins were eluted with six column volumes of 150 mM imidazole in binding buffer at a flow rate of 5 mL min−1 and collected in 1 mL fractions.
Characterization of the recombinant GDH
Protein analysis and western blot analysis
Protein concentration was determined according to Lowry et al. (1951) using bovine serum albumin as standard. Protein purification steps were followed by 12 % sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). The protein bands were stained with Coomassie Brilliant Blue R-250 and molecular mass was determined against reference proteins (pre-stained protein ladder, Euromedex, Souffelweyersheim, France). Electrophoresed proteins were electroblotted onto polyvinylidene difluoride membranes (iBlot, Life Technologies, Saint-Aubin France). Membranes were then incubated in tris-borate saline (TBS) blocking solution (10 mM Tris-HCl pH 7.4, 150 mM NaCl, and 5 % w/v milk powder) overnight at 4 °C, washed with TBS (10 mM Tris-HCl pH 7.4, 150 mM NaCl) and with TBS Tween (10 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.05 % Tween 20), and finally treated with blocking solution containing monoclonal antipolyhistidine antibody serum at a 1/2,000 dilution (Sigma-Aldrich, Saint-Quentin-Fallavier, France). Signal detection used the 5-bromo-4-chloro-3-indolyl phosphate-nitro blue tetrazolium assay (Roche Applied Science, Meylan, France) performed according to the manufacturer’s procedure.
N-terminal amino-acid sequence determination and enzyme deglycosylation
The N-terminal signal peptide was predicted with SignalP (http://www.cbs.dtu.dk/services/SignalP/ ) and the N-terminal amino acid sequence of the mature protein was determined according to Edman degradation from a GDH sample electroblotted onto a polyvinylidene difluoride membrane (iBlot, Life Technologies). Analyses were carried out on an Applied Biosystem 470A by the proteomics platform at the Institut de Microbiologie de la Méditerranée, CNRS–Aix-Marseille Université, Marseille, France. Recombinant GDH was deglycosylated using N-glycosidase F (PNGase F) according to the manufacturer’s procedure (New England Biolabs, Evry, France). Then, 20 μg of purified GDH was incubated with 2 μL (1,000 U) of PNGase F and 4 μL of G7 reaction buffer supplied by the manufacturer, in a final volume of 40 μL. The reaction was performed for 1 h at 37 °C. Deglycosylation efficiency was determined on SDS-PAGE. N-glycosylation sites were predicted using the CBS NetNGlyc 1.0 server at http://www.cbs.dtu.dk/services/NetNGlyc/.
The molecular mass of the native protein was estimated by loading of 100 μL of purified enzyme on a precalibrated Superdex 200 column (GE Healthcare, Vélizy, France).
Spectral characterization
Absorption spectra of the enzyme reduction process with D-glucose (300 mM) were recorded from 250 to 550 nm in 50 mM citrate-phosphate buffer, pH 5.5, at room temperature by using an Uvikon XS spectrophotometer (BioTek Instruments, Colmar, France).
Temperature and pH optima
To determine optimal temperature under the conditions used, aliquots of purified recombinant GDH (100 % refers to 9,000 nkat mL−1) were incubated at various temperatures from 20 to 70 °C and pH 5.5, and activity was assayed at various temperatures. For the pH profile, GDH activity (100 % refers to 9,000 nkatal mL−1) was determined in 50 mM citrate-phosphate buffer from pH 2.5 to 7.5 at 30 °C, all other conditions being equal to standard conditions. Each measurement was taken in triplicate.
Temperature and pH stability
Thermostability of the purified recombinant enzyme was studied by incubating pure enzyme (100 % refers to 9,000 nkat mL−1) at time points from 15 to 120 min and at temperatures ranging from 30 to 60 °C. Thermal inactivation was stopped by immediately cooling the treated protein aliquot on ice. pH stability was determined by incubating the purified enzyme in 50 mM citrate-phosphate at pH values ranging from 2.5 to 7.5 at 30 °C for up to 24 h. In both cases, residual enzyme activity was assayed by the standard DCIP assay.
Kinetics measurements and substrate specificity
The Michaelis constants for D-glucose, lactose, D-galactose, maltotriose, D-xylose, and maltose were measured under standard conditions (DCIP) and calculated from a Lineweaver-Burk plot using GraFit4 software (Erithacus Sofware, Horley, UK). Catalytic constants for the electron acceptors DCIP and 1,4-benzoquinone were measured using D-glucose as substrate. The relative activity of GDH for other carbohydrates, i.e., lactose, D-galactose, maltotriose, xylose, maltose, arabinose, saccharose, sorbose, mannose, methyl-α-D-glucopyranoside, methyl-α-D-mannopyranoside, fructose, mannitol, 2 deoxyribose, α-L-rhamnose, avicel, cellobiose, and carboxymethylcellulose, was measured by the standard DCIP assay and reported relative to D-glucose as baseline reference.
The inhibition of the laccase-catalyzed oxidation of phenols to quinones and phenoxy radicals by GDH was assayed spectrophotometrically using 0.1 mM of 2,6-dimethoxyphenol, guaiacol, coumaric acid, and sinapic acid as substrates and 2.8 to 70 nkat of P. cinnabarinus laccase (Sigoillot et al. 2004) in 1 mL final volume buffered at pH 4.5 with 50 mM tartrate buffer. The oxidation reaction with or without GDH (from 0.13 to 5.32 nkat) was performed at 30 °C for 1 min and quantified.
Analysis of reaction products
For electrospray ionization (ESI) tandem mass spectrometry measurements, samples were introduced at a flow rate of 5 μL min−1 into an LTQ Orbitrap VELOS system (Thermo Fischer Scientific, USA). Measurements were made in the ion trap part in negative ion mode. Tandem mass spectrometry spectra were recorded during 1 min. Collision energy was adapted based on the signal/noise ratio observed for fragments.
Structure modeling
The automated protein structure homology-modeling SWISS-MODEL server (accessible via the ExPASy web server: http://swissmodel.expasy.org/interactive) and Phyre2 (http://www.sbg.bio.ic.ac.uk/phyre2) were used to determine which GMC member was the most structurally similar to the novel GDH.
Sequence comparison of GMC oxidoreductases
The P. cinnabarinus GDH (PcGDH) sequence was aligned with the biochemically characterized G. cingulata GDH AER13600.1 (GcGDH) (Sygmund et al. 2011a) and two three-dimensional structures of GOX− one from A. niger (1cf3) (AnGOX) (Hecht et al. 1993) and one from Penicillium amagasakiense (1gpe) (PaGOX) (Wohlfahrt et al. 1999). The results were obtained using the Clustalw multiple sequence alignment program (http://www.ebi.ac.uk/Tools/msa/clustalw2/).
Results
Genome screening for candidate GDHs, A. niger transformation and screening
Annotation of the P. cinnabarinus genome revealed a gene encoding a protein related to a GDH (scf184803.g17) and its signal peptide. This gene was demonstrated to be functional, as the corresponding protein was identified in previously studied secretomes of P. cinnabarinus grown on maltose, birchwood, and maize bran induced media (Levasseur et al. 2014). The candidate PcGDH belongs to the Auxiliary Activities AA3_2 (Levasseur et al. 2013) corresponding to flavoproteins containing a FAD-binding domain. The genomic sequence of 2,425 bases is composed of 11 exons. The deduced cDNA counts 1,866 bases and the corresponding protein 620 amino acids. In order to produce the corresponding protein, the coding sequence corresponding to the first 23 amino acids of the signal peptide was removed and replaced by the coding sequence of the A. niger glucoamylase signal peptide (24 amino acids) followed by a KEX2-like cleavage site in the expression vector pAN52.4 (1 in Fig. 1a). In a cotransformation experiment, protoplasts of A. niger D15#26 were transformed with a mixture of plasmid pAB4-1 and the expression vector, and transformants were selected for their ability to grow without uridine supplementation. Approximately 100 uridine prototrophic transformants were obtained per microgram of expression vector.

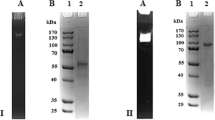
Cloning, production, purification, and characterization of P. cinnabarinus GDH in A. niger D15-PcGDH2. a 1 GDH gene expression cassette. See experimental procedures for more information. a 2 Extracellular production of the GDH in strain D15-PcGDH2 over time. GDH activity (filled squares) and mycelial dry weight (filled circles). a 3 Purification of GDH. b 1 SDS-PAGE analysis of the culture medium (lane 1 in 1), dialyzed proteins (lane 2 in 1), and purified protein (lane 3 in 1). SD are molecular mass standards. Proteins stained with Coomassie blue. b 2 N-deglycosylation of GDH, lane 4 is deglycosylated GDH, lane 5 is purified GDH. N-glycosidase F has a molecular weight of 36 kDa and is arrowed in lane 4. SD are molecular mass standards. Proteins stained with Coomassie blue
A total of 34 positive clones were cultured in standard liquid cultures and checked daily for GDH activity. Approximately 15 % of the tested transformants produced a GDH activity in the culture media. No GDH activity was detected in a negative control (transformed by pAB4-1 only). The best-producing PcGDH2 was selected, and the time-course of extracellular GDH production was determined in standard liquid cultures. PcGDH2 transformant reached a peak of 170 nkat mL−1 at 13 days (2 in Fig. 1a). GDH production appeared to plateau from days 10 to 18, suggesting that for the purification step, production could be stopped at day 10.
Purification procedure and study of the biochemical-physical characteristics
The recombinant GDH was purified from the PcGDH2 culture medium in three successive steps (3 in Fig. 1a). The culture medium was concentrated 30-fold by ultrafiltration with a recovery of 90 %. The resulting GDH was loaded onto a Ni-HIS trap column and purified to homogeneity with a recovery of 41 %, yielding approximately 220 mg of GDH from 3 L of culture broth.
The obtained N-terminal sequence of the GDH corresponded to ASSGITSD. Alignment of the N-terminal sequence with the predicted putative sequence confirmed that the 24-amino-acid GLA prepro sequence from A. niger was correctly processed. In addition, it was deduced that the first seven amino acids (VPHVQNR) of the mature GDH were truncated, as already described for the Piromyces equi EstA heterologously produced in T. reesei (Poidevin et al. 2009). Molecular mass of GDH was checked on SDS-PAGE by electrophoretic mobility of total secreted proteins in the culture medium, the dialyzed protein, and the purified protein (1 in Fig. 1b). The culture medium showed a predominant large band below 70 kDa indicating that the recombinant GDH is produced as the main enzyme in the A. niger secretome. This molecular mass was confirmed by mass spectrometric analysis. Immunodetection of the GDH was performed using antibodies raised against the HIS tag. Western blotting showed a unique band corresponding to the 70 kDa protein, demonstrating that this protein is the recombinant GDH and that it was correctly processed at the C-terminus. Concerning the N-glycosylation sites, three sites (amino acids 67, 217, and 468, counting starting from the Met) were predicted following the consensus sequence (Asn-Xaa-Ser/Thr). To confirm the presence of glycosylation, the recombinant GDH was treated with PNGase and the resulting protein showed a molecular mass of approximately 60 kDa, which corresponds to the calculated molecular mass of 62.5 kDa (2 in Fig. 1b). In addition, the molecular mass of the native protein estimated by gel filtration was around 130 kDa, indicating that the protein probably exists in a homodimeric form. The purified GDH is of yellow color and displays a characteristic flavoprotein spectrum with the typical FAD absorption maxima at 385 and 449 nm. Addition of 0.3 M glucose resulted in reduction of the enzyme and the disappearance of both peaks (Fig. 2).

Spectral characterization of GDH showing both the oxidized (grey) and reduced (black) spectra. Glucose was used to reduce the enzyme. Maxima are indicated
PcGDH was stable for 2 h at 40 °C and retained approximately 50 % of its activity after 40 min at 50 °C. Nearly all activity was lost after 5 min at 60 °C (Fig. 3). The recombinant GDH was stable for 60 min across the entire pH range (pH 2.5 to 7.5). After a 120-min incubation, the remaining activity started to decrease to 95 % at pH 2.5. After 24 h of incubation in acidic conditions, the recombinant GDH retained 83 and 95 % of its activity at pH 2.5 and 3.5, respectively. Up to the maximum tested pH 7.5, the recombinant protein was almost stable with less than 5 % loss of activity. Further studies on the effect of temperature revealed that the optimum temperature of PcGDH was 60 °C, with activity decreasing sharply above this temperature, probably in relation to its thermal stability. Optimum pH was determined to be between pH 5.0 and 6.5, with a peak at pH 5.5. At more acidic pH between pH 2.5 and 4.5, the remaining activity is reduced by 20 %, and at alkaline pH, i.e., 7.5, activity decreases to 20 %.

Purification of GDH. Effects of temperature (a) and pH (b) on the stability of the purified protein. The selected temperatures were 40 °C (filled squares), 50 °C (filled diamonds), and 60 °C (filled triangles), and the selected pH values were 2.50 (filled squares), 3.50 (filled diamonds), 4.50 (plus sign), 5.50 (black dots), 6.50 (filled triangles), and 7.50 (stars). Effects of temperature (c) and pH (d) on the activity of the purified protein. Various temperatures (20 to 70 °C) and pH values (2.5 to 7.5) were tested under standard conditions. All assays were performed with glucose as substrate
Substrate specificity of GDH
Analysis of the substrate specificity of PcGDH using a set of typical sugars under standard screening conditions with DCIP as electron acceptor revealed that glucose is the preferred substrate (Table 1). Kinetic constants were determined for the best sugar substrates (Table 2). PcGDH showed a Michaelis-Menten-type kinetics over the full substrate concentration range from 120 to 1,245 mM. K m and V max for glucose as substrate were 270 mM and 293 nkat mg−1, respectively, as calculated by curve fitting using nonlinear least-squares regression. Catalytic efficiency k cat/K m was by far the highest for glucose, i.e., 17- to 67-fold higher compared to other substrates. All the experiments were performed with DCIP as electron acceptor compared to 1,4-benzoquinone as an alternative cosubstrate. The K m and k cat values for the 1,4-benzoquinone, 0.172 M, and 46 M−1 s−1, respectively, were similar to DCIP, with a slightly higher catalytic efficiency for DCIP.
The reaction products of GDH with d-glucose were analyzed by ESI-Trap-MS. Glucose is present on the spectrum as an unprotonated species (m/z 179.4), a dimeric form (m/z 358.9) and with HPO3 adducts (m/z 276.8 and 456.8) induced by enzymatic buffer. Gluconic acid is found at m/z 194.9 [M-H]− (see Fig. 4a). The fragmentation ESI-Trap-MS/MS spectrum of this peak showed the presence of three fragments assigned to gluconic acid (m/z of 177, 159, and 129), confirming the formation of gluconic acid (see Fig. 4b).

a ESI-trap-MS spectrum. Glucose is present on the spectrum as unprotonated species (m/z 179.4), dimer form (m/z 358.9), and with HPO3 adducts (m/z 276.8 and 456.8) induced by enzymatic buffer. Gluconic acid is found at m/z 194.9 [M-H]−. b ESI-trap-MSMS spectrum of precursor ions corresponding to the [M_H]− of gluconic acid. The fragmentation spectrum validates the nature of gluconic acid. Loss of one water molecule corresponds to the formation of one double bond ([HO–CH2–C(OH)=CH–CHOH–CHOH–COO]− and isomers). Loss of two water molecules corresponds to the formation of two double bonds ([HO–CH2–C(OH)=CH–C(OH)=CH–COO−] and isomers). Loss of 66 kDa (m/z 128.7) corresponds to the structure CHOH=CH–C(OH)=CH–COO−
Inhibition of laccase-catalyzed oxidation of phenols and phenoxy radicals
The reduction of the phenoxy lignin precursors was studied by a first oxidation step with a P. cinnabarinus laccase replacing the plant laccases, then PcGDH was added to test its capacity to inhibit the laccase reaction, which would signify a protective effect to hinder exposure of the fungal cell to harmful oxidized monolignols and related monophenols. For all the four compounds tested, i.e., 2,6-dimethoxyphenol, guaiacol, p-coumaric acid, and sinapic acid, addition of glucose and a concomitantly increased concentration of GDH drastically reduced the amount of laccase-oxidized products. As observed by Sygmund et al. (2011a), the reduction rate was more marked for guaiacol and 2,6-dimethoxyphenol than for p-coumaric acid and sinapic acid (Table 3).
Sequence analysis
G. cingulata glucose dehydrogenase AER13600.1 (GcGDH) and two GOX, one from A. niger (1cf3) (AnGOX) and one from P. amagasakiense (1gpe) (PaGOX), showed 37, 36, and 36 % of identity and 54, 50, and 51 % similarity, respectively, against PcGDH. The GMC member most structurally similar to GDH returned by the SWISS-MODEL server is aryl-alcohol oxidase (AAO) from Pleurotus eryngii (PDB code 3fimB). With Phyre2, the best model is the GOX from P. amagasakiense 1gpeA.
The PcGDH sequence was aligned against GcGDH, AnGOX, and PaGOX sequences (Fig. 5). The overall alignment could be divided into the five structurally conserved regions already described for GMC family members (Kiess et al. 1998): (1) a FAD-binding domain (the highest similar region), (2) a flavin attachment loop and intermediate region (second most similar region), (3) an FAD covering lid, (4) an extended FAD-binding domain, and (5) a substrate-binding domain (the least similar region). Highly conserved residues are highlighted in the alignment, making it possible to delimit the domains in PcGDH and determine the main conserved residues for this enzyme: (1) the FAD-binding domain is divided into four regions corresponding to residues 12–50, 236–305, 541–565, and 572–591 (Fig. 5, in red); (2) the flavin attachment loop (residues 87–104) and intermediate region (residues 105–221) are two contiguous sequences regions (Fig. 5, in green); (3) the FAD covering lid is a short region formed by residues 71–86 (Fig. 5, in purple); (4) the extended FAD-binding domain is split into six separate short sequence regions adjoining the FAD-binding domain corresponding to residues 1-11, 51-70, 222-234, 306-318, 533-540 and 566-571 (Fig. 5, in yellow); (5) the substrate-binding domain is made up of a contiguous sequence region of residues 319–532 (Fig. 5, in light blue). The complete description of the first four domains is available in the supplementary data. The substrate-binding domain is described and discussed below.

Comparison of amino acid sequences of P. cinnabarinus GDH with other GOXs and GDHs. G. cingulata GDH (GcGDH) (Sygmund et al. 2011a) and the tertiary structures of glucose oxidases from P. amagasakiense (PaGOX_1gpe) (Wohlfahrt et al. 1999) and A. niger (AnGOX_1cf3) (Hecht et al. 1993) were aligned against the P. cinnabarinus GDH (PcGDH). The five distinct regions described for members of the GMC family were color coded as in Kiess et al. (1998)—FAD-binding domain (red), flavin attachment loop and intermediate region (green), FAD covering lid (purple), extended FAD-binding (yellow), and substrate-binding domain (light blue). Conserved residues are highlighted in black
Discussion
This work presents the first study of a glucose dehydrogenase (GDH, family AA3_2) from a basidiomycete, the white-rot fungus P. cinnabarinus. The characterization of this new enzyme brings additional biochemical insights into AA3_2 members (Levasseur et al. 2013) and their potential for use in biotechnological applications. GDH belongs to the GMC oxidoreductase family, which is represented by a wide variety of catalytically diverse enzymes of both prokaryotic and eukaryotic origin and contains, among others, CAZy family AA3_2 AAO, AA3_2 GOX, methanol oxidase, choline dehydrogenase, AA3_1 CDH, AA3_4 pyranose dehydrogenase (PDH), and pyranose oxidase (POX). All the members of this family are involved in the modification/breakdown of lignocellulosic biomass and are classified as auxiliary activities in the new version of CAZy (Levasseur et al. 2013).
The P. cinnabarinus gdh gene was demonstrated to be functional as the corresponding protein was identified in secretomes produced by P. cinnabarinus grown on maltose, birchwood, and maize bran-induced medium (Levasseur et al. 2014). Comparison studies of fungi-derived FAD-GDHs is limited as information on them is scarce. They are produced in some ascomycetes like the plant pathogens G. cingulata (Sygmund et al. 2011a) and Aspergillus species, i.e., A. niger, A. oryzae, A. flavus, A. terreus, and others (Mori et al. 2011; Tsujimura et al. 2006). In comparison, the closely-related GOX is a well-characterized enzyme in several fungi species, mostly in ascomycetes, and has attracted great attention due to its broad biotech applications (Wong et al. 2008). Other sugar oxidoreductases like PDH and POX have been more extensively studied and possess a much wider spectrum of action. However, in some cases, this lack of specificity could turn into a disadvantage (Kujawa et al. 2007).
A. niger has been used in previous work to produce fungal oxidases, i.e., laccases, at relatively high yield (Record et al. 2002; Mekmouche et al. 2014) compared to yeast systems such as Pichia pastoris or Yarrowia lipolytica (Otterbein et al. 2000; Madzak et al. 2005). High protein yield was also obtained for the production in A. niger of cellobiose dehydrogenase from Coprinopsis cinerea (Turbe-Doan et al. 2013), reaching about 100 mg L−1 at lab scale. Under our conditions, A. niger was able to secrete 640 mg L−1 of PcGDH in non-optimized 13-day cultures with a specific activity of 266 nkat mg−1. The well-characterized GDH from G. cingulata was mainly produced in inclusion bodies in E. coli and in non-optimized cultures of P. pastoris at a concentration of 3 mg L−1. This yield was greatly improved in a 7-L bioreactor, reaching 57 mg L−1 (Sygmund et al. 2011b). Other fungal GDHs from Aspergillus species were produced in E. coli in soluble form, but it is difficult to compare the volumetric yield as the soluble proteins were processed from sonicated bacterial cells to recover the recombinant proteins (Mori et al. 2011). Fungal hosts allow efficient secretion and high concentrations of GDH in the culture medium, which is consistent with a fast and efficient purification of the recombinant proteins using the affinity His-tagged sequence. This process of high-yield production and one-step purification allows us to produce more than 200 mg of purified GDH from 3 L of culture medium. This is by far the highest protein yield found to date for a GDH. The purified and deglycosylated PcGDH, using a N-glycosidase, had a molecular weight of approximately 60 kDa, which corresponds to the calculated mass (62.5 kDa), indicating that deglycosylation was probably complete and that glycosylation should be N-linked. Positions of glycosylation were predicted at positions 67, 217, and 468, and glycosylation accounts for approximately 10 % of the total mass of the protein. In contrast, G. cingulata GDH produced in P. pastoris was described as highly glycosylated at approximately 65 and to 70 % for the native protein (Sygmund et al. 2011b). The authors suggested that this degree of glycosylation was probably overestimated by SDS-PAGE due to the presence of band smears on the gel. As other GDHs were produced in the aglycone form in E. coli, we have no other references against which to compare our data. In contrast, in the glucose oxidases from the basidiomycota P. ostreatus (Shin et al. 1993) and P. chrysosporium (Forney et al. 1982), no glycosylation was demonstrated. For instance, in P. chrysosporium the GOX was suggested to be localized in the periplasmatic space in which the majority of proteins are not glycosylated. A. niger GOX is glycosylated at 10–16 % of its molecular weight and carbohydrate moieties are also found N-linked (Leskovac et al. 2005; Takegawa et al. 1989). PcGDH holds only three N-glycosylation sites (N38, N188, and N439) compared to the seven sites of PaGOX (N93, N165, N171, N317, N357, N392, and N502). N-glycosylation sites in glucose oxidase do not appear to be conserved in the other members of the GMC oxidoreductase family. However, two asparagines are found in PcGDH (N356 and N500) at the same position as PaGOX N-glycosylation sites (N357 and N501), but were not experimentally validated.
The protein was characterized for its main physicochemical properties in order to study its mechanisms of action and compare them to other experimentally characterized GDH. We suggested that the native form of the PcGDH exists in a homodimeric form. The molecular mass of the G. cingulata protein is slightly higher, with isoforms from 95 to 135 kDa, and exists as a monomer (Sygmund et al. 2011a). P. chrysosporium GOX has a molecular mass of 180 and is composed of two identical subunits with a molecular mass of 80 kDa (Kelley and Reddy 1986). In addition, P. amagasakiense and A. niger GOX are homodimeric glycoproteins which have a non-reduced molecular weight of 160 kDa with one tightly but noncovalently bound FAD cofactor per monomer (Swoboda and Massey 1965; Wohlfahrt et al. 1999). In contrast, the 290 kDa P. ostreatus GOX is composed of four subunits of 70 kDa (Shin et al. 1993), demonstrating that, in general, with the exception of C. cingulata GDH, these proteins have adopted a multidomain organization.
Other characteristics of the recombinant GDH, including temperature and pH optima, were determined and compared to other GDH. The temperature optimum is 60 °C, which is roughly 10 °C higher than G. cingulata GDH in recombinant (46 °C) and native (48 °C) form (Sygmund et al. 2011b), and A. terreus GDH (50 °C) (Tsujimura et al. 2006). The activity of P. cinnabarinus GDH is stable at 40 °C for at least 2 h but completely lost at higher temperatures. No information is available for other fungal GDHs. pH-dependent activity is highest at pH 5.5 and relatively stable between pH 2.5 and 6.5, which is in the range of other GDHs found to date.
Concerning the substrate specificity of P. cinnabarinus, D-glucose was by far the preferred substrate, ahead of lactose that reached just 14 % of glucose activity. This result was confirmed by the higher catalytic efficiency (67 M−1 s−1) for D-glucose compared to other tested substrates. Maltose had a similar apparent K m to D-glucose, but with a far lower (36-fold lower) k cat number resulting in very low catalytic efficiency (1 M−1 s−1). There was a lower catalytic efficiency for xylose, whereas G. cingulata GDH showed equivalent k cat/K m for glucose and xylose (Sygmund et al. 2011a). However, although G. cingulata GDH showed broad specificity against sugars, it showed a higher catalytic efficiency of 19,600 M−1 s−1 for glucose.
ESI-Trap-MS analysis of hydrolysis products found that oxidation takes place only at the C1 position and that the main reaction product is gluconic acid, consistent with previous reports for G. cingulata GDH (Sygmund et al. 2011a) and A. niger GOX (Ferri et al. 2011). Substrate hydrolysis studies seem to suggest that glucose epimers in the C2 and C3 position as well as a substituent of the hydroxy group at C1 (as in the methyl-d-glucosides) hinder activity, thus highlighting the importance of orientation and substitution of the hydroxy groups at these positions for specificity and reactivity in PcGDH. On the other hand, epimers of glucose at the C4 position and the presence of the exocyclic CH2OH group at the C5 position do allow some minor activity.
The PcGDH sequence was aligned against GcGDH and the two close AnGOX and PaGOX sequences to compare the conserved domains (Fig. 5 and Supplementary data). In particular, the substrate-binding domain turns out to be structurally conserved in all GMC oxidoreductases. The active site of glucose oxidase is set up by residues Y73, S114, R516, N518, H520, and H563 in PaGOX and Y68, T110, R512, N514, H516, and H559 in AnGOX forming 12 hydrogen bonds with the D-glucose (Wohlfahrt et al. 1999). In PcGDH, only the tyrosine and the two histidines are conserved (Y64, A100, A524, V526, H528, and H571), unlike in GcGDH where only the PaGOX S114 (T110 in AnGOX) changes into an alanine (Y62, A105, R517, N519, H521, and H564). Hydrophobic interactions are also formed with the substrate and with PaGOX F418 and W430 (AnGOX F414 and W426). These residues are all conserved between GOX and PcGDH (F416 and W430; Fig. 6) but not in the other GDHs (GcGDH W431 and L418). These differences are generally observed among GMC oxidoreductases, making the active site the most heterogeneous region (Kiess et al. 1998). H528 in PcGDH is highly conserved in all sequences and is known to participate in the enzyme catalytic mechanism by accepting the proton from the substrate. Highly conserved Y64 and H571 in PcGDH are involved in substrate binding but belong to extended FAD-binding domains 2 and 6, respectively. Surprisingly, H571 which stabilizes the hydroxy group from the glucose C1, is conserved in all the GOX and GDH sequences, unlike in previous studies reporting that this residue was not conserved across the members of the GMC family (Wohlfahrt et al. 1999). Asparagine and arginine residues participating in substrate binding at positions C2 and C3-C4, respectively, in GOX and presumably in GcGDH, are replaced by valine and alanine in PcGDH. This would probably explain the lack of reaction of glucose epimers at these positions in PcGDH and its narrow specificity for D-glucose. Valine is a nonpolar amino acid that hinders a H-bond with the substrate. In AnGOX, a mutant R512A lost the two H bonds to the glucose O3 and O4, thus strongly increasing K m . PcGDH exhibits the same change in amino acid yet presents a relatively high K m . In addition, β-D-galactose (epimer of β-D-glucose on the C4) and β-D-xylose (which lacks the exocyclic CH2OH) are also poor substrates for PcGDH. In comparison to β-D-glucose, AnGOX is missing the H bonds between Y68, R512, and the O4 of the β-D-galactose and between T110, FAD O4, a water molecule, and β-D-xylose, respectively. This therefore results in unfavorable contacts between W426 and β-D-galactose and with H559 and β-D-xylose that are conserved in PcGDH (W430 and H571).

View of the active site for PcGDH (a, model) and PaGOX (b, PDB entry 1gpe) drawn with PyMOL. Residues of the catalytic site forming hydroxyl interactions with the substrate are colored green and those forming hydrophobic interactions with the substrate are colored purple. The FAD cofactor molecule is colored yellow
Concerning the biological function of the GDH, there is limited knowledge on this aspect. One proposed role would be to reduce quinones or radical intermediates formed during lignin degradation, where GDH would prevent repolymerization of the released aromatic compounds or shield the fungus from exposure to these potentially toxic compounds, which would need an extracellular secretion mechanism to eliminate potentially toxic quinones from the cell (Sygmund et al. 2011a). A similar role has been proposed for other GMC oxidoreductases like CDH or POX for wood-degrading fungi and PDH for litter-decomposing fungi (Tan et al. 2013). Here, the monophenols—phenoxy radicals—oxidized by laccase were reduced with PcGDH at similar rates as with the G. cingulata GDH (Sygmund et al. 2011a). PcGDH also showed a higher reduction rate for guaiacol and 2,6-dimethoxyphenol than for the structural analogs p-coumaric acid and sinapic acid, again as with GcGDH. A possible explanation for a reduced GDH activity for monolignols would be the presence of a propene chain with the terminal carboxyl group or a lower polymerization rate than for the relatively stable phenoxy radicals of guaiacol and 2,6-dimethoxyphenol. Nonlinearity of the observed GDH inhibition by increasing GDH concentrations could also be due to a fast dimerization of monolignol radicals (Sygmund et al. 2011a).
The PcGDH also showed promising characteristics for biotech applications. It is inactive with molecular oxygen as electron acceptor compared to the GOX, which rules out the formation of H2O2 and thus potential oxidative damage to the enzyme. The PcGDH also demonstrated good stability and low glycosylation. D-glucose is by far the preferred substrate, which is a great advantage over other sugar oxidases since this property is vital for monitoring blood glucose, which is currently the most popular application for GDHs (FDA standards request no activity towards maltose; Mori et al. 2011).
References
Aiba H, Tsugura-shi J (2007) Novel glucose dehydrogenase. US Patent 2007/0105174
Armstrong JM (1964) The molar extinction coefficient of 2,6-dichlorophenol indophenol. Biochim Biophys Acta 86:194–197
Bai L, Wen D, Yin J, Deng L, Zhu C, Dong S (2012) Carbon nanotubes-ionic liquid nanocomposites sensing platform for NADH oxidation and oxygen, glucose detection in blood. Talanta 91:110–115
Bak TG (1967) Studies on glucose dehydrogenase of Aspergillus oryzae. Purification and physical and chemical properties. Biochim Biophys Acta 139:277–293
Bak TG, Sato R (1967) Studies on the glucose dehydrogenase of Aspergillus oryzae. Induction of its synthesis by ρ-benzoquinone and hydroquinone. Biochim Biophys Acta 139:265–276
Bey M, Berrin JG, Poidevin L, Sigoillot JC (2011) Heterologous expression of Pycnoporus cinnabarinus cellobiose dehydrogenase in Pichia pastoris and involvement in saccharification processes. Microb Cell Factories 10:113–127
Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B (2009) The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res 37:D233–D238
Clark LC Jr, Lyons C (1962) Electrode systems for continuous monitoring in cardiovascular surgery. Ann N Y Acad Sci 102:29–45
Eggert C, Temp U, Eriksson KE (1996) The ligninolytic system of the white rot fungus Pycnoporus cinnabarinus: purification and characterization of the laccase. Appl Environ Microbiol 62:1151–1158
Estrada Alvarado I, Lomascolo A, Navarro D, Delattre M, Asther M, Lesage-Meessen L (2001) Evidence of a new biotransformation pathway of p-coumaric acid into p-hydroxybenzaldehyde in Pycnoporus cinnabarinus. Appl Microbiol Biotechnol 57:725–730
Ferri S, Kojima K, Sode K (2011) Review of glucose oxidases and glucose dehydrogenases: a bird’s eye view of glucose sensing enzymes. J Diabetes Sci Technol 5:1068–1076
Forney LJ, Reddy CA, Pankratz HS (1982) Ultrastructural localization of hydrogen peroxide production in ligninolytic Phanerochaete chrysosporium cells. Appl Environ Microbiol 44:732–736
Gordon CL, Khalaj V, Ram AFJ, Archer DB, Brookman JL, Trinci APJ, Jeenes DJ, Doonan JH, Wells B, Punt PJ, van den Hondel CAMJJ, Robson GD (2000) Glucoamylase: fluorescence protein fusions to monitor protein secretion in Aspergillus niger. Microbiology 146:415–426
Harrison DC (1931) Glucose dehydrogenase: a new oxidising enzyme from animal tissues. Biochem J 4:1016–1027
Hecht HJ, Kalisz HM, Hendle J, Schmid RD, Schomburg D (1993) Crystal structure of glucose oxidase from Aspergillus niger refined at 2.3 A resolution. J Mol Biol 1:153–172
Kelley RL, Reddy CA (1986) Purification and characterization of glucose oxidase from ligninolytic cultures of Phanerochaete chrysosporium. J Bacteriol 166:269–274
Kiess M, Hecht HJ, Kalisz HM (1998) Glucose oxidase from Penicillium amagasakiense. Primary structure and comparison with other glucose-methanol-choline (GMC) oxidoreductases. Eur J Biochem 252:90–99
Kujawa M, Volc J, Halada P, Sedmera P, Divne C, Sygmund C, Leitner C, Peterbauer C, Haltrich D (2007) Properties of pyranose dehydrogenase purified from the litter-degrading fungus Agaricus xanthoderma. FEBS J 274:879–894
Lesage-Meessen L, Delattre M, Haon M, Thibault JF, Ceccaldi BC, Brunerie P, Asther M (1996) A two-step bioconversion process for vanillin production from ferulic acid combining Aspergillus niger and Pycnoporus cinnabarinus. J Biotechnol 50:107–113
Leskovac V, Trivić S, Wohlfahrt G, Kandrac J, Pericin D (2005) Glucose oxidase from Aspergillus niger: the mechanism of action with molecular oxygen, quinones, and one-electron acceptors. Int J Biochem Cell Biol 37:731–750
Levasseur A, Drula E, Lombard V, Coutinho PM, Henrissat B (2013) Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol Biofuels 6:41–54
Levasseur A, Lomascolo A, Chabrol O, Ruiz-Dueñas FJ, Boukhris-Uzan E, Piumi F, Kües U, Ram AFJ, Murat C, Haon M, Benoit I, Arfi Y, Chevret D, Drula E, Kwon MJ, Gouret P, Lesage-Meessen L, Lombard V, Mariette J, Noirot C, Park J, Patyshakuliyeva A, Sigoillot JCS, Wiebenga A, Wösten HAB, Martin F, Coutinho PM, de Vries RP, Martínez AT, Klopp C, Pontarotti P, Henrissat B, Record E (2014) The genome of the white-rot fungus Pycnoporus cinnabarinus: a basidiomycete model with a versatile arsenal for lignocellulosic biomass breakdown. BMC Genomics In press
Lomascolo A, Stentelaire C, Asther M, Lesage-Meessen L (1999) Basidiomycetes as new biotechnological tools to generate natural aromatic flavours for the food industry. Trends Biotechnol 17:282–289
Lomascolo A, Record E, Herpoël-Gimbert I, Delattre M, Robert JL, Georis J, Dauvrin T, Sigoillot JC, Asther M (2003) Overproduction of laccase by a monokaryotic strain of Pycnoporus cinnabarinus using ethanol as inducer. J Appl Microbiol 94:618–624
Lomascolo A, Uzan-Boukhris E, Herpoël-Gimbert I, Sigoillot JC, Lesage-Meessen L (2011) Peculiarities of Pycnoporus species for applications in biotechnology. Appl Microbiol Biotechnol 92:1129–1149
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193:265–275
Madzak C, Otterbein L, Chamkha M, Moukha S, Asther M, Gaillardin C, Beckerich JM (2005) Heterologous production of a laccase from the basidiomycete Pycnoporus cinnabarinus in the dimorphic yeast Yarrowia lipolytica. FEMS Yeast Res 5:635–646
Mekmouche Y, Zhou S, Cusano AM, Record E, Lomascolo A, Robert V, Simaan AJ, Rousselot-Pailley P, Ullah S, Chaspoul F, Tron T (2014) Gram-scale production of a basidiomycetous laccase in Aspergillus niger. J Biosci Bioeng 117:25–27
Milutinovic M, Sallard S, Manojlovic D, Mano N, Sojic N (2011) Glucose sensing by electrogenerated chemiluminescence of glucose-dehydrogenase produced NADH on electrodeposited redox hydrogel. Bioelectrochemistry 82:63–68
Mori K, Nakajima M, Kojima K, Murakami K, Ferri S, Sode K (2011) Screening of Aspergillus-derived FAD-glucose dehydrogenases from fungal genome database. Biotechnol Lett 33:2255–2263
Müller HM (1977) Gluconic acid forming enzymes in Aspergillus niger. Zentralbl Bakteriol Parasitenkd Infektionskr Hyg 132:14–24
Ogura Y (1951) Studies on the glucose dehydrogenase of Aspergillus oryzae. J Biochem 38:75–84
Ogura Y, Nagahisa M (1937) Untersuchungen über die Atmung und die Dehydrasesysteme von Aspergillus oryzae. Bot Mag Tokyo 51:597–612
Otterbein L, Record E, Longhi S, Asther M, Moukha S (2000) Molecular cloning of the cDNA encoding laccase from Pycnoporus cinnabarinus I-937 and expression in Pichia pastoris. Eur J Biochem 267:1619–1625
Poidevin L, Levasseur A, Paës G, Navarro D, Heiss-Blanquet S, Asther M, Record E (2009) Heterologous production of the Piromyces equi cinnamoyl esterase in Trichoderma reesei for biotechnological applications. Lett Appl Microbiol 49:673–678
Punt PJ, van den Hondel CA (1992) Transformation of filamentous fungi based on hygromycin B and phleomycine resistance markers. Methods Enzymol 216:447–457
Record E, Punt PJ, Chamkha M, Labat M, van Den Hondel CA, Asther M (2002) Expression of the Pycnoporus cinnabarinus laccase gene in Aspergillus niger and characterization of the recombinant enzyme. Eur J Biochem 269:602–609
Saleh FS, Mao L, Ohsaka T (2012) A promising dehydrogenase-based bioanode for a glucose biosensor and glucose/O2 biofuel cell. Analyst 137:2233–2238
Shin KS, Youn HD, Han YH, Kang SO, Hah YC (1993) Purification and characterisation of D-glucose oxidase from white-rot fungus Pleurotus ostreatus. Eur J Biochem 215:747–752
Sigoillot C, Record E, Belle V, Robert JL, Levasseur A, Punt PJ, van den Hondel CA, Fournel A, Sigoillot JC, Asther M (2004) Natural and recombinant fungal laccases for paper pulp bleaching. Appl Microbiol Biotechnol 64:346–352
Swoboda BE, Massey V (1965) Purification and properties of the glucose oxidase from Aspergillus niger. J Biol Chem 240:2209–2215
Sygmund C, Klausberger M, Felice AK, Ludwig R (2011a) Reduction of quinones and phenoxy radicals by extracellular glucose dehydrogenase from Glomerella cingulata suggests a role in plant pathogenicity. Microbiology 157:3203–3212
Sygmund C, Staudigl P, Klausberger M, Pinotsis N, Djinović-Carugo K, Gorton L, Haltrich D, Ludwig R (2011b) Heterologous overexpression of Glomerella cingulata FAD-dependent glucose dehydrogenase in Escherichia coli and Pichia pastoris. Microb Cell Factories 10:106–114
Takegawa K, Fujiwara K, Iwahara S, Yamamoto K, Tochikura T (1989) Effect of deglycosylation of N-linked sugar chains on glucose oxidase from Aspergillus niger. Biochem Cell Biol 67:460–464
Tan TC, Spadiut O, Wongnate T, Sucharitakul J, Krondorfer I, Sygmund C, Haltrich D, Chaiyen P, Peterbauer CK, Divne C (2013) The 1.6 Å crystal structure of pyranose dehydrogenase from Agaricus meleagris rationalizes substrate specificity and reveals a flavin intermediate. PLoS ONE 8:1–14
Tsujimura S, Kojima S, Kano K, Ikeda T, Sato M, Sanada H, Omura H (2006) Novel FAD-dependent glucose dehydrogenase for a dioxygen-insensitive glucose biosensor. Biosci Biotechnol Biochem 70:654–659
Turbe-Doan A, Arfi Y, Record E, Estrada-Alvarado I, Levasseur (2013) A Heterologous production of cellobiose dehydrogenases from the basidiomycete Coprinopsis cinerea and the ascomycete Podospora anserina and their effect on saccharification of wheat straw. Appl Microbiol Biotechnol 97:4873–4885
van Hartingsveldt W, Mattern IE, van Zeijl CM, Pouwells PH, van den Hondel CA (1987) Development of a homologous transformation system for Aspergillus niger based on the pyrG gene. Mol Gen Genet 206:71–75
Wang JY, Nien PC, Chen CH, Chen LC, Ho KC (2012) A glucose bio-battery prototype based on a GDH/poly(methylene blue) bioanode and a graphite cathode with an iodide/tri-iodide redox couple. Bioresour Technol 116:502–506
Wohlfahrt G, Witt S, Hendle J, Schomburg D, Kalisz HM, Hecht HJ (1999) 1.8 and 1.9 A resolution structures of the Penicillium amagasakiense and Aspergillus niger glucose oxidases as a basis for modelling substrate complexes. Acta Crystallogr D Biol Crystallogr 55:969–977
Wong CM, Wong KH, Chen XD (2008) Glucose oxidase: natural occurrence, function, properties and industrial applications. Appl Microbiol Biotechnol 78:927–938
Yang Y, Wei B, Zhao Y, Wang J (2013) Construction of an integrated enzyme system consisting azoreductase and glucose 1-dehydrogenase for dye removal. Bioresour Technol 130:517–521
Zafar MN, Wang X, Sygmund C, Ludwig R, Leech D, Gorton L (2012) Electron-transfer studies with a new flavin adenine dinucleotide dependent glucose dehydrogenase and osmium polymers of different redox potentials. Anal Chem 84:334–341
Zhang L, Zhou M, Wen D, Bai L, Lou B, Dong S (2012) Small-size biofuel cell on paper. Biosens Bioelectron 35:155–159
Acknowledgments
This work was partly funded by the European Commission under the INDOX project (KBBE-2013-7-613549). The authors thank Régine Lebrun, from the proteomics platform of the Institut de Microbiologie de la Méditerranée, CNRS-AMU, Marseille, France, for protein identification by mass spectrometry, and Christophe Boyer and Marie-Pierre Forquin-Gomez for the graphics.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 136 kb)
Rights and permissions
About this article

Cite this article
Piumi, F., Levasseur, A., Navarro, D. et al. A novel glucose dehydrogenase from the white-rot fungus Pycnoporus cinnabarinus: production in Aspergillus niger and physicochemical characterization of the recombinant enzyme. Appl Microbiol Biotechnol 98, 10105–10118 (2014). https://doi.org/10.1007/s00253-014-5891-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-014-5891-4