
Abstract
An extracellular β-glucosidase (BGL) from Fusarium oxysporum was purified to homogeneity by a single chromatography step on a gel filtration column. The optimum activity of BGL on cellobiose was observed at pH 5.0 and 60 °C. Under the same conditions, the K m and V max values for p-nitrophenyl β-d-glucopyranoside and cellobiose were 2.53 mM, 268 U mg protein−1 and 20.3 mM, 193 U mg protein−1, respectively. The F. oxysporum BGL enzyme was highly stable at acidic pH (t 1/2 = 470 min at pH 3). A commercial BGL Novo188 (Novozymes) and F. oxysporum BGL were compared in their ability to supplement Celluclast 1.5 L (Novozymes). In comparison with the commercial Novo188 (267 mg g substrate−1), F. oxysporum BGL supplementation released more reducing sugars (330 mg g substrate−1) from cellulose under simulated gastric conditions. These properties make F. oxysporum BGL a good candidate as a new commercial BGL to improve the nutrient bioavailability of animal feed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cellulases bring about the hydrolysis of cellulose (a homopolymer of β-1,4 linked glucose units) that comprises amorphous and crystalline regions by the synergistic action of its constituent enzymes. These enzymes include (a) β-1,4-endoglucanase (1,4-β-d-glucan 4-glucanohydrolase; EC 3.2.1.4, cellulase), which cleaves internal β-1,4-glycosidic bonds; (b) cellobiohydrolase (1,4-β-d-glucan cellobiohydrolase; EC 3.2.1.91, cellulose 1,4-β-cellobiosidase), an exo-acting enzyme which releases cellobiose from reducing and nonreducing ends of cellulose; and (c) β-glucosidase (BGL; β-d-glucoside glucohydrolase; EC 3.2.1.21, cellulase 1,4-β-glucosidase), which hydrolyzes cellobiose to glucose (Sateesh et al. 2012). BGLs catalyze the hydrolysis of alkyl-β-glucosides and aryl-β-glucosides as well as diglucosides and oligosaccharides.
Slow or incomplete digestion of fibrous substrates often limits the overall digestive process in the rumen and can significantly influence animal performance in livestock production systems that use forage as a major dietary component. As a result, many strategies have been developed to stimulate the digestion of the fibrous components in ruminant feeds. These have included the use of fibrolytic enzymes, such as cellulase, which stimulate fiber digestion and feed processing to increase the rate and extent of digestion. Cellulase preparations can be used to drive specific metabolic and digestive processes in the gastrointestinal tract and may augment natural digestive processes to increase nutrient availability and feed intake (McAllister et al. 2001) Adding cellulase enzyme preparations to the diets of ruminant animals has been the topic of many recent studies. A number of in vitro studies have demonstrated that it is possible to use cellulase enzyme preparations to enhance the processes associated with fiber digestion in the rumen (Azzaz 2009; Murad et al. 2009; Rodrigues et al. 2008; Sharma et al. 2012). The response is generally measured as an increase in the initial rate of dry matter and organic matter disappearance, increase in the rate of neutral detergent fiber and acid detergent fiber disappearance, altered ruminal pH, increase in volatile fatty acid production, reduced lag phase and improved efficiency of fermentation, increase in ruminal microbial growth, and increase in microbial protein synthesis in batch cultures of ruminal bacteria that have been supplemented with cellulase preparations (Abdel Gawad et al. 2007; Giraldo et al. 2008; Krueger et al. 2008; Nguyen et al. 2010).
Dong et al. (1999) demonstrated that cellulase activity may begin when the enzyme is in contact with the substrate, indicating the importance of enzyme–feed interaction. Giraldo et al. (2004) confirmed that a pre-ingestive enzyme–feed interaction is necessary for any significant beneficial effects on ruminal digestion. Others have also noted that a pre-feeding enzyme–feed interaction period is necessary for cellulase enzyme-mediated increases in digestion (Krueger et al. 2008; McAllister et al. 2001). The addition of enzymes to feeds may create a stable enzyme–feed complex that protects free enzymes from proteolysis in the rumen (Kung et al. 2000).
In the present study, an acid-stable BGL from Fusarium oxysporum, with high hydrolytic activity towards cellobiose at low pH, was purified and characterized. Compared with cellulose degradation by a commercial cellulase (Celluclast® 1.5 L) alone and a commercial BGL (Novo188) with Celluclast, the efficiency of cellulose hydrolysis was substantially increased by the supplementation of F. oxysporum BGL under all simulated gastric conditions.
Materials and methods
Culture conditions
F. oxysporum (KCTC 16909) obtained from the Korean Biological Resource Center (Korean Collection for Type Cultures) was used for BGL production. The fungal strain was subcultured every 3 weeks and stored at 4 °C in potato dextrose agar plates (Sarria-Alfonso et al. 2013). A 500-ml flask containing 50 ml of potato dextrose broth was used for seeding the culture. After a 4- to 5-day incubation period, 5 ml of this pre-culture was inoculated into 50 ml of the standard media with the following composition (in grams per liter): peptone, 8; yeast extract, 2; KH2PO4, 5; K2HPO4, 5; MgSO4·7H2O, 3; thiamine hydrochloride, 0.02; and cellulose, 20 with pH adjusted to 5.0. The inoculated flasks were shaken at 150 rpm and 25 °C.
Enzyme assays
BGL activity was determined by monitoring the release of p-nitrophenol from p-nitrophenyl β-d-glucopyranoside (pNPG). The enzyme solution (100 μl) was incubated with 10 mM pNPG (100 μl) in 800 μl of 100 mM sodium acetate buffer (pH 5.0) for 15 min at 50 °C. The reaction was stopped by adding 100 μl of 2 M sodium carbonate. The color developed was measured at 415 nm using a GENESYS 10S UV-visible spectrophotometer (Thermo Scientific, West Palm Beach, FL, USA). One unit of BGL activity was determined as the amount of enzyme catalyzing the hydrolysis of 1 μmol pNPG or cellobiose in 1 min (Jeya et al. 2010).
One-step purification of BGL
The culture broth was harvested by centrifugation at 10,000×g for 30 min. The supernatant was concentrated and desalted by ultrafiltration using a 10-kDa cutoff membrane. The concentrated enzyme solution was loaded onto a Hiload 16/60 Superdex 200 pg, equilibrated with 20 mM sodium acetate, containing 0.5 M NaCl at pH 5.0. Proteins were eluted with the same buffer at a flow rate of 0.5 ml min−1. BGL activity of the fractions was assessed. Protein concentration of the fractions was measured by the Bradford method, using bovine serum albumin as the standard. The purification procedures were performed at 4 °C. The concentration of the protein in the column effluents was monitored by measuring the absorbance at 280 nm. Chromatographic separation was performed by using a BioLogic FPLC system (Bio-Rad, Hercules, CA, USA).
Determination of optimal pH and temperature
The optimal pH for BGL activity was determined by incubating the purified enzyme with pNPG at 50 °C for 15 min in different buffers: citrate (100 mM, pH 3–4), sodium acetate (100 mM, pH 4–6), and phosphate (100 mM, pH 6–8). To determine the optimal temperature, the enzyme was incubated with pNPG in sodium acetate buffer (100 mM, pH 5) for 15 min at different temperatures ranging from 40 to 80 °C. To determine the thermostability of BGL activity, the purified enzyme was incubated at different temperatures (ranging from 40 to 80 °C) in the absence of substrate. After incubating them for various periods (0–72 h), the residual BGL activity was determined as described previously.
Determination of molecular mass
To determine subunit molecular mass, sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) was performed with 10 % gels. Protein bands were visualized with Coomassie brilliant blue R-250 (Sigma). Native PAGE was performed with 10 % polyacrylamide gels without SDS. BGL activity was tested on the polyacrylamide gel containing 50 μg of 4-methylumbelliferyl-β-d-glucopyranoside (MUG) by the zymogram method (Bahia and Ali 2006). The ensemble was incubated for 30 min at 50 °C for MUGase activity. Hydrolysis of MUG by BGL released fluorescent methylumbelliferone, which was visualized under ultraviolet light.
The molecular mass of the purified enzyme was determined by size exclusion chromatography using a Sephacryl S-300 high-resolution (HR) (Amersham Pharmacia Biotech, Uppsala, Sweden) column attached to a BioLogic FPLC system (Bio-Rad, Hercules, CA, USA). The enzyme was eluted with 20 mM sodium acetate (pH 5.0) at a flow rate of 0.5 ml min−1.
Substrate specificity
Substrate specificity of BGL was determined by using pNPG, o-nitrophenyl-β-d-glucopyranoside (oNPG), pNP-α-d-glucopyranoside, pNP-β-galactopyranoside (pNPgal), pNP-β-d-mannopyranoside (pNPM), pNP-β-cellobioside (pNPC), oNP-β-cellobioside (oNPC), cellobiose, cellotriose, cellotetraose, and cellopentaose as substrates at 10 mM concentration. BGL activities towards polysaccharides carboxymethyl cellulose, xylan, laminarin, and avicel (2 %) were also tested. The p-nitrophenol released was determined under standard enzyme assay conditions. The amount of released glucose was measured using the 3,5-dinitrosalicylic acid (DNS) method and confirmed by the GOD-POD method (Nguyen et al. 2010).
Determination of kinetic parameters
The values of the Michaelis constant (K m) and maximum velocity (V max) were determined for BGL by incubating mixtures of 100 mM sodium acetate buffer (pH 5) at 50 °C with pNPG and cellobiose at concentrations ranging from 0.5 to 50 mM. Inhibition of BGL by glucose was determined in the presence of pNPG as the substrate. Values for K m and V max were determined from Lineweaver–Burk plots using standard linear regression techniques.
Effect of metal ions and chemicals
The effects of various metal ions (Ca2+, Co2+, Cu2+, Fe3+, Hg2+, Mg2+, Mn2+, and Zn2+) and chemicals on BGL activity were determined by preincubating the enzyme with the individual reagents in 100 mM sodium acetate buffer (pH 5) at 30 °C for 30 min. Activities were then measured at 60 °C for 15 min in the presence of the metal ions or reagents. The activity assayed in the absence of metal ions or reagents was set as 100 %.
Cloning of F. oxysporum BGL gene (bgl)
The degenerate primers BGLF1 and BGLR1 (Supplementary Table S1) were designed according to the conserved amino acid sequence regions I (YRFSLCW) and II (LDNFEW) and partial peptide sequences obtained from nano-liquid chromatography–tandem mass spectrometry sequencing. The presence of the 1,077-bp polymerase chain reaction (PCR) fragment containing the specific sequence for the BGL gene was confirmed by DNA sequencing. In order to obtain the complete structural gene encoding BGL, thermal asymmetric interlaced PCR (TAIL-PCR) (Jeya et al. 2010) was performed to amplify the 5′ and 3′ flanking sequences of the known partial sequence (1,077 bp) of the BGL gene, using the genomic DNA of F. oxysporum as the template. Three interlaced specific primers, namely, SPR1, SPR2, SPR3 and SPF1, SPF2, SPF3 were used for flanking the 5′ and 3′ regions, respectively (Supplementary Table S1). Five random degenerate primers AD1, AD2, AD3, AD4, and AD5, in which N represents A/G/C/T, were used for flanking the 5′ and 3′ regions. The TAIL-PCR product of the 1,528-bp complete structural gene encoding BGL was cloned into pGEM-T Easy Vector (Promega) for DNA sequencing and designated as bgl.
Isolation and sequencing of bgl cDNA
Total RNAs were extracted by using the QIAGEN RNeasy Plant Kit (QIAGEN, Hilden, Germany) by following the manufacturer’s instructions. The primers BGLFox-F (designed to match the start codon ATG region) and BGLFox-R (designed to match the sequence immediately downstream of the stop codon) were designed according to the known 5′-end and 3′-end sequences of the BGL structural gene (Supplementary Table S1). The amplified cDNA fragment was then eluted and cloned, and its identity was confirmed by sequencing.
The nucleotide and amino acid sequences were analyzed by using the respective BLAST programs (http://www.ncbi.nlm.nih.gov/BLAST). The molecular mass of the mature protein was predicted using Vector NTI 10.0 software. Multiple alignments of protein sequences were performed using the ClustalW program (http://www.ebi.ac.uk/clustalW/).
Combination of F. oxysporum BGL with Celluclast under simulated gastric conditions to enhance cellulose hydrolysis
Cellulose hydrolysis under simulated gastric conditions was performed as described previously (Wang et al. 2001) with some modifications. Reactions (50 ml) containing 2.0 mg ml−1 NaCl and 0.5 % cellulose in 0.2 M glycine were initially adjusted to pH 2.0 with 1.0 M HCl on ice. Celluclast (0.05 FPU) or the enzyme combination (0.05 FPU Celluclast and 0.15 U F. oxysporum BGL or Novo188) was added, and the reactions were incubated for 20 min at 37 °C. Samples were placed on ice, and a pH gradient was applied using 1.0 M NaOH/HCl followed by incubation at 37 °C as follows: pH 5.5, 10 min; pH 4.6, 10 min; pH 3.8, 10 min; pH 2.8, 20 min; and pH 2.0, 40 min. Pepsin (0.5 mg ml−1) was added to the simulated gastric fluid (SGF) reactions at the beginning of each pH decrease. All procedures were conducted in a rotary incubator set at 220 rpm. Triplicate aliquots (500 μl) were collected at the end of each incubation period, and the amount of sugar released was determined using the DNS method (540 nm). Reactions with Celluclast enzyme were performed as negative controls.
Sequence submission
The nucleotide sequence of F. oxysporum bgl was deposited in the GenBank database under accession no. KC174716.
Results
Purification of BGL from F. oxysporum culture
An extracellular BGL from F. oxysporum was purified as described in the “Materials and methods” section, and the results are summarized in Table 1. Fractionation by an ultrafiltration membrane with a molecular weight cutoff of 10 kDa increased the specific activity about 1.85-fold, with 51.3 % recovery of BGL activity. The active fractions were subjected to gel filtration chromatography and yielded three main peaks. The first peak showed a single band on SDS-PAGE with high BGL activity. About 30.6-fold purification of BGL, with a recovery of 11.7 % was achieved. As shown in Fig. 1a, the purified enzyme appeared as a single band on PAGE, both in the presence and absence of SDS. Activity staining of the nondenaturing gel showed a single activity band with the same mobility as that of the protein band (Fig. 1a). Purified BGL from F. oxysporum was homogenous, with a molecular mass of 55 kDa (Fig. 1a). Size exclusion chromatography on a Sephacryl S-300 HR column resulted in the elution of the enzyme activity as a symmetrical peak corresponding to an M r of approximately 270 kDa (Fig. 1b), indicating that the enzyme is a pentamer.

Determination of the molecular mass of F. oxysporum BGL by SDS-PAGE and gel filtration chromatography. a Determination of subunit molecular mass by SDS-PAGE. Lane 1 crude fraction, lane 2 purified F. oxysporum BGL, lane 3 zymogram of purified BGL. b Determination of native molecular mass of F. oxysporum BGL by gel filtration chromatography on a Sephacryl S-300 HR column from Amersham
Identification of the partial peptide fragment
The BGL was partially digested with trypsin, then separated by 12.5 % SDS-PAGE and blotted onto a polyvinylidene difluoride membrane. The fragments were sequenced in an automated protein sequencer. The obtained peptide fragments of F. oxysporum BGL, KKASFHWFKDVIATRG and KYNGWLDPQIIKD, were identical to those of the BGLs of Vibrio fischeri MJ11 and Vitis vinifera belonging to GH1, respectively. Amino acid sequence similarity, enzymology, and bioinformatics studies strongly suggest that F. oxysporum BGL should be classified as a member of the GH1 family.
Optimum pH and temperature
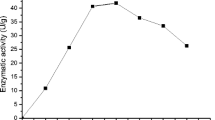
The optimum pH for the purified BGL was 5 (Fig. 2a). The pH stability was investigated by measuring the residual activity after incubation at 50 °C, at pH values ranging from 2.0 to 8.0. The enzyme was stable at acidic pH values ranging from 3.0 to 6.0. About 82.1 and 94.2 % of activity remained after incubation at pH 2.5 and 3.0, respectively (Fig. 2b). The optimum temperature for the hydrolysis reaction was 60 °C (Fig. 3). Under optimum conditions (pH 5, 60 °C), the enzyme has a half-life of 540 min.

a Effect of pH on the activity of purified F. oxysporum BGL. Enzyme activity was determined by the standard assay method by changing the buffer to obtain the desired pH. The buffers used were citrate (filled circles), sodium acetate (open circles), and phosphate (filled inverted triangles). b The stability of purified enzyme at different pH values. Each value represents the mean of triplicate measurements and it varied from the mean by not more than 15 %

Effect of temperature on the activity of purified F. oxysporum BGL. Enzyme activity was determined by the standard assay method at different temperatures ranging from 40 to 80 °C
Substrate specificity
The activities of F. oxysporum BGL with various substrates are shown in Table 2. oNPG showed 61.4 % enzyme activity when compared with pNPG, while pNPC showed slight activity (4.7 % compared with pNPG). Other compounds did not serve as substrates. F. oxysporum BGL was almost completely inactive with polymeric cellulose and xylan substrates.
Kinetics
The activity of F. oxysporum BGL towards cellobiose increased rapidly with increasing substrate concentrations up to 75 mM and then slightly decreased thereafter. Initial velocities were determined using the standard assay mixture. All of the substrates tested had hyperbolic saturation curves, and the corresponding double-reciprocal plots were linear. Figure 4 shows typical Michaelis–Menten-type kinetics for BGL activity with increasing pNPG and cellobiose concentrations. The Lineweaver–Burk plot obtained for the conversion of pNPG under standard assay conditions showed a V max of 268 U mg protein−1 and a K m of 2.53 mM. The V max and K m values for cellobiose were 193 U mg protein−1 and 20.3 mM, respectively. K m and V max values were also obtained from a nonlinear regression plot using the GraphPad Prism program (version 5.00, GraphPad Software, San Diego, CA, USA). There was no significant difference in kinetic parameters between the two methods.

a Effect of pNPG concentration on the activity of F. oxysporum BGL. b Effect of cellobiose concentration on the activity of F. oxysporum BGL. F. oxysporum BGL activity was measured in the presence of indicated concentrations of pNPG/cellobiose at pH 5. Each value represents the mean of triplicate measurements and it varied from the mean by not more than 15 %
Effects of metal ions and various compounds
The effect of metal ions and some chemical reagents on the activity of the enzyme are shown in Table 3. Ca2+, Co2+, Mg2+, and Zn2+ increased the activity by about 40 %. Fe3+ at 1 mM enhanced enzyme activity by 78 %. However, Mn2+ upgraded BGL activity by 236 %. Manganese ions at 10 mM exerted an approximate twofold enzyme activity enhancement of BGLs from Clostridium cellulovorans and termite Neotermes koshunensis (Kosugi et al. 2006; Tokuda et al. 2002). However, Hg2+ ions caused significant inhibition that appeared to be competitive with respect to the substrate pNPG or cellobiose. Addition of β-mercaptoethanol or cysteine did not result in a significant increase or decrease in activity, suggesting that thiol groups are not essential for catalytic activity (Yan and Lin 1997). N-ethylmaleimide (1 mM), which is often used to inactivate enzymes (presumably by reacting with the thiol group of cysteine residues), had no effect on the activity of F. oxysporum BGL. Ethanol and methanol at optimal concentration (5 %, v/v) increased BGL activity by 30 %.
Cloning and characterization of F. oxysporum bgl gene
Degenerate PCR was performed using the genomic DNA of F. oxysporum as the template and a 1,077-bp PCR fragment was obtained. After obtaining the 5′ and 3′ flanking sequences of the known 1,077-bp BGL gene, reverse transcription PCR was performed to amplify the full-length cDNA. F. oxysporum BGL encodes a protein composed of 490 amino acid residues. A homology search revealed that the deduced BGL3 had 74, 90, 69, 70, and 69 % amino acid identity with the BGLs of Verticillium albo-atrum VaMs.102 (GenBank accession no. XP_003000876.1), Nectria haematococca mpVI 77-13-4 (GenBank accession no. XP_003051841.1), Aspergillus oryzae RIB40 (GenBank accession no. XP_001819673.2), Aspergillus clavatus NRRL 1 (GenBank accession no. XP_001268785.1), and Aspergillus fumigatus Af293 (GenBank accession no. XP_752840.1), respectively (Fig. 5). All of the homologous sequences belong to GH1, which suggests that F. oxysporum BGL is also a member of the GH1 family.

Multiple sequence alignment of F. oxysporum BGL with other BGLs using ClustalW. Sequences used for alignment are from A. oryzae, A. fumigatus, A. clavatus, N. haematococca, and V. albo-atrum. The residues in black and gray backgrounds are identical and similar, respectively
Cellulose hydrolysis by the synergistic action of Celluclast and BGLs under simulated gastric conditions
The efficacy of cellulose hydrolysis in SGF for each combination (F. oxysporum BGL and Celluclast, Novo188 BGL and Celluclast, and Celluclast alone) is shown in Fig. 6. When the pH of SGF was adjusted according to the gastric pH gradient (pH 2.0–5.5) after food ingestion (Koseki et al. 2011), a significant difference in activity was observed between the Novo188 and F. oxysporum BGL. F. oxysporum BGL showed the highest activity under acidic pH conditions. F. oxysporum BGL had excellent synergistic efficacy with Celluclast in cellulose hydrolysis over a broad range of gastric pH values, releasing up to 336 mg sugar g substrate−1. These values were significantly higher than that (~270 mg sugar g substrate−1) obtained by the commercial BGL Novo188 with Celluclast.

Cellulose hydrolysis by Celluclast alone or by Celluclast combined with BGLs (F. oxysporum BGL or Novo188 BGL) under simulated gastric conditions. Cellulose hydrolysis was determined by the DNS method, following stepwise incubation of the enzyme(s) at pH 2.0–5.5, in the indicated buffer at 37 °C for 10–40 min (totally 110 min). The x-axis represents the accumulated incubation time under simulated gastric conditions. The black bar represents data from Celluclast alone; the gray bar represents data from Celluclast in combination with F. oxysporum BGL; and the dark gray bar represents data from Celluclast in combination with Novo188 BGL. Data reflect the means ± SD (n = 3)
Discussion
In the present study, we report a high BGL-producing strain F. oxysporum KCTC 16909. The BGL obtained using this strain showed high activity and stability at pH 3. Although an extracellular BGL from F. oxysporum with exo-glucosidase and transglycosylation activities was reported previously (Christakopoulos et al. 1994), this is the first report on an acid-stable BGL from F. oxysporum. The F. oxysporum BGL characterized in this study is distinguished from other BGLs by its molecular weight, acid-stable nature, and high catalytic efficiency. Some BGLs, including BGL from Fomitopsis pinicola (Joo et al. 2009), have higher catalytic efficiency; however, there was no activity at pH 3. Stereum hirsutum BGL, the most efficient fungal BGL described so far (Nguyen et al. 2010), was also less stable (t 1/2 = 20 min) at pH 3. On the other hand, F. oxysporum BGL exhibited stability for a long period (t 1/2 = 470 min). Thus, F. oxysporum BGL is one of the most acid-stable BGLs characterized so far and showed excellent stability over a broad range of acidic pH.
Table 4 shows a comparison of the properties of BGLs from various sources. On the basis of amino acid sequence similarities, glycosidases have been classified into several families, with most BGLs belonging to either family 1 or family 3 (Liu et al. 2012). Cellulases are generally composed of two or more discrete domains: a core, which contains the catalytic domain; a highly conserved cellulose-binding domain, which is involved in binding to crystalline cellulose; and connecting these two domains, a hinge, which tends to be glycosylated. When F. oxysporum BGL was compared with other related enzymes, its catalytic domain showed the highest identity with that of N. haematococca BGL (90 %) belonging to the GH1 family. The evidence from enzymology and bioinformatics studies strongly suggests that F. oxysporum BGL should be classified as a member of GH1.
A commercial cellulase preparation, Celluclast® 1.5 L, contains numerous endo-1,4-β-glucanases and cellobiohydrolases, but very little BGL. It has been reported that the level of BGL present in Celluclast® 1.5 L is likely to be insufficient to relieve the inhibitory effect of cellobiose. When used in combination with Celluclast® 1.5 L, F. oxysporum BGL hydrolyzed cellulose 25 % more efficiently than did a commercial BGL Novo188. This could be partly because F. oxysporum BGL has higher efficiency in the removal of cellobiose than Novozyme 188 under gastric conditions. This ability of F. oxysporum BGL to hydrolyze cellobiose more efficiently is likely to reduce the cellobiose concentration significantly and, thus, greatly facilitate the hydrolysis of cellulose under gastric conditions. F. oxysporum BGL can be a good candidate for cellulase enzyme preparations to enhance fiber digestion in the rumen.
In conclusion, a highly efficient BGL from F. oxysporum KCTC 16909 was purified to homogeneity from its culture filtrate. The successful purification of BGL from F. oxysporum allowed us to characterize this enzyme that was found to be one of the most acid-stable BGLs ever reported. F. oxysporum BGL has high pH stability in acidic conditions and exhibits excellent stability and activity under simulated gastric conditions. Our data suggest that the commercial cellulase Celluclast in combination with F. oxysporum BGL is a promising candidate for industrial applications requiring degradation of cellulose at acidic pH. Its properties suggest that F. oxysporum BGL has great potential to improve food and feed quality and increase nutrient uptake. In addition to the animal feed industry, the potential application of this enzyme in other fields requiring cellulose hydrolysis under acidic conditions will necessitate further investigation.
References
Abdel Gawad MH, Gad SM, El-Sabaawy EH, Ali HM, El-Bedawy TM (2007) In vitro and in vivo digestibility of some low quality roughages supplemented with fibrolytic enzyme for sheep. Egypt J Nutr Feeds 10:663–677
Azzaz HH (2009) Effect of cellulolytic enzymes addition to diets on the productive performance of lactating goats. M.Sc. thesis, Faculty of Agriculture, Cairo University, Egypt, pp 141–150
Bahia A, Ali G (2006) Characterization of novel beta-glucosidase from a Stachybotrys strain. Biotech Eng J 32:191–197
Chen M, Qin Y, Liu Z, Liu K, Wang F, Qu Y (2010) Isolation and characterization of a beta-glucosidase from Penicillium decumbens and improving hydrolysis of corncob residue by using it as cellulase supplementation. Enzyme Microb Technol 46:444–449
Christakopoulos P, Goodenough PW, Kekos D, Macris BJ, Claeyssens M, Bhat MK (1994) Purification and characterisation of an extracellular β-glucosidase with transglycosylation and exo-glucosidase activities from Fusarium oxysporum. Eur J Biochem 224(2):379–385
Dong Y, Bae HD, McAllister TA, Mathison GW, Cheng KJ (1999) Effects of exogenous fibrolytic enzymes, α-bromoethanesulfonate and monensin on fermentation in a rumen simulation (RUSITEC) system. Can J Anim Sci 79:491–498
Giraldo LA, Ranilla MJ, Tejido ML, Carro MD (2004) Effects of cellulase application form on the in vitro rumen fermentation of tropical forages. J Anim Feed Sci 13:63–66
Giraldo LA, Tejido ML, Ranilla MJ, Carro MD (2008) Effects of exogenous fibrolytic enzymes on in vitro ruminal fermentation of substrates with different forage: concentrate ratios. Anim Feed Sci Technol 141:306–325
Jeya M, Joo AR, Lee KM, Tiwari MK, Kim SH, Lee JK (2010) Characterization of β-glucosidase from a strain of Penicillium purpurogenum KJS506. Appl Microbiol Biotechnol 86(5):1473–1484
Joo AR, Jeya M, Lee KM, Sim WI, Kim JS, Kim IW, Kim YS, Oh DK, Gunasekaran P, Lee JK (2009) Purification and characterization of a β-1,4-glucosidase from a newly isolated strain of Fomitopsis pinicola. Appl Microbiol Biotechnol 83(2):285–294
Karnchanatat A, Petsom A, Sangvanich P, Piaphukiew J, Whalley AJ, Reynolds CD, Sihanonth P (2007) Purification and biochemical characterization of an extracellular β-glucosidase from the wood-decaying fungus Daldinia eschscholzii (Ehrenb.:Fr.) Rehm. FEMS Microbiol Lett 270(1):162–170
Kaur J, Bhupinder SC, Badhan AK, Ghatora K, Kaur S (2007) Purification and characterization of β-glucosidase from Melanocarpus sp. MTCC 3922. Electron J Biotechnol 10:261–270
Kim HJ, Park AR, Lee JK, Oh DK (2009) Characterization of an acid-labile, thermostable b-glycosidase from Thermoplasma acidophilum. Biotechnol Lett 31(9):1457–1462
Koseki S, Mizuno Y, Sotome I (2011) Modeling of pathogen survival during simulated gastric digestion. Appl Environ Microbiol 77(3):1021–1032
Kosugi A, Arai T, Doi RH (2006) Degradation of cellulosome-produced cello-oligosaccharides by an extracellular non-cellulosomal beta-glucan glucohydrolase, BglA, from Clostridium cellulovorans. Biochem Biophys Res Commun 349(1):20–23
Krueger NA, Adesogan AT, Staples CR, Krueger WK, Kim SC, Littell RC, Sollenberger LE (2008) Effect of method of applying fibrolytic enzymes or ammonia to Bermudagrass hay on feed intake, digestion, and growth of beef steers. J Anim Sci 86(4):882–889
Kung L Jr, Treacher RJ, Nauman GA, Smagala AM, Endres KM, Cohen MA (2000) The effect of treating forages with fibrolytic enzymes on its nutritive value and lactation performance of dairy cows. J Dairy Sci 83(1):115–122
Liu D, Zhang R, Yang X, Zhang Z, Song S, Miao Y, Shen Q (2012) Characterization of a thermostable β-glucosidase from Aspergillus fumigatus Z5, and its functional expression in Pichia pastoris X33. Microb Cell Fact 11:25. doi:10.1186/1475-2859-11-25
McAllister TA, Hristov AN, Beauchemin KA, Rode LM, Cheng KJ (2001) Enzymes in ruminant diets. In: Bedford MR, Patridge GG (eds) Enzymes in farm animal nutrition. CABI, Marlborough, pp 273–298
Murad HH, Hanfy MA, Kholif AM, Adbdel Gawad MH, Murad HA (2009) Effect of cellulases supplementation to some low quality roughages on digestion and milk production by lactating goats. J Biol Chem Environ Sci 4:791–809
Nguyen NP, Lee KM, Kim IW, Kim YS, Jeya M, Lee JK (2010) One-step purification and characterization of a β-1,4-glucosidase from a newly isolated strain of Stereum hirsutum. Appl Microbiol Biotechnol 87(6):2107–2116
Pontoh J, Low NH (2002) Purification and characterization of beta-glucosidase from honey bees (Apis mellifera). Insect Biochem Mol Biol 32(6):679–690
Rodrigues MAM, Pinto P, Bezerra RMF, Dias AA, Guedes CVM, Cardoso VMG, Cone JW, Ferreira LMM, Colaco J, Sequeira CA (2008) Effect of enzyme extracts isolated from white-rot fungi on chemical composition and in vitro digestibility of wheat straw. Animal Feed Sci Technol 141:326–338
Sarria-Alfonso V, Sánchez-Sierra J, Aguirre-Morales M, Gutiérrez-Rojas I, Moreno-Sarmiento N, Poutou-Piñales R (2013) Culture media statistical optimization for biomass production of a ligninolytic fungus for future rice straw degradation. Indian J Microbiol. doi:10.1007/s12088-013-0358-3
Sateesh L, Rodhe A, Naseeruddin S, Yadav K, Prasad Y, Rao L (2012) Simultaneous cellulase production, saccharification and detoxification using dilute acid hydrolysate of S. spontaneum with Trichoderma reesei NCIM 992 and Aspergillus niger. Indian J Microbiol 52(2):258–262
Sharma KK, Shrivastava B, Nandal P, Sehgal N, Sastry VRB, Kalra A, Kuhad RC (2012) Nutritional and toxicological assessment of white-rot fermented animal feed. Indian J Microbiol 52(2):185–190
Sue M, Ishihara A, Iwamura H (2000) Purification and characterization of a beta-glucosidase from rye (Secale cereale L.) seedlings. Plant Sci 155(1):67–74
Tokuda G, Saito H, Watanabe H (2002) A digestive β-glucosidase from the salivary glands of the termite, Neotermes koshunensis (Shiraki): distribution, characterization and isolation of its precursor cDNA by 5′- and 3′-RACE amplifications with degenerate primers. Insect Biochem Mol Biol 32(12):1681–1689
Valaskova V, Baldrian P (2006) Degradation of cellulose and hemicelluloses by the brown rot fungus Piptoporus betulinus-production of extracellular enzymes and characterization of the major cellulases. Microbiology 152(Pt 12):3613–3622
Wang Y, McAllister TA, Rode LM, Beauchemin KA, Morgavi DP, Nsereko VL, Iwaasa AD, Yang W (2001) Effects of an exogenous enzyme preparation on microbial protein synthesis, enzyme activity and attachment to feed in the Rumen Simulation Technique (Rusitec). Br J Nutr 85(3):325–332
Xie Y, Gao Y, Chen Z (2004) Purification and characterization of an extracellular β-glucosidase with high transglucosylation activity and stability from Aspergillus niger no. 5.1. Appl Biochem Biotechnol 119(3):229–240
Yan TR, Lin CL (1997) Purification and characterization of a glucose-tolerant β-glucosidase from Aspergillus niger CCRC 31494. Biosci Biotechnol Biochem 61(6):965–970
Acknowledgments
This research was supported by the Converging Research Center Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2011-50210).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Zongpei Zhao and Priyadharsini Ramachandran contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 39 kb)
Rights and permissions
About this article
Cite this article
Zhao, Z., Ramachandran, P., Kim, TS. et al. Characterization of an acid-tolerant β-1,4-glucosidase from Fusarium oxysporum and its potential as an animal feed additive. Appl Microbiol Biotechnol 97, 10003–10011 (2013). https://doi.org/10.1007/s00253-013-4767-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-013-4767-3