
Abstract
Conotruncal heart defect is a complex form of congenital heart disease and usually has a poor prognosis. Although previous studies have identified several missense variants in GATA4 gene that may cause CTD, it remains unclear whether they are involved in CTD pathogenesis because the study population was limited. The aim of the study was to investigate the mutations of GATA4 gene in isolated CTD Chinese Han patients and identify the pathomechanism of the missense mutations. In this report, the coding exons and exon–intron boundaries of the GATA4 gene were sequenced in 600 CTD patients and 300 controls. Functional significance of the novel GATA4 gene mutation (p.A167D) was analyzed using PolyPhen 2 and SIFT. And, the functional characteristics of the mutant GATA4 gene were assayed in contrast to its wild-type counterpart using a luciferase reporter assay system as well as Western blot. Eight heterozygous nonsynonymous variants (V380M, G64E, A167D, V267M, S377G, P163S, P407Q, A66T) were found in 22 patients, of which one (A167D) was reported here for the first time and five (G64E, A167D, S377G, P163S, A66T) were only found in CTD patients when compared with 300 controls. The PolyPhen 2 and SIFT programs predicted that the A167D substitution was expected to influence protein function. Subsequent functional analyses revealed that the transcriptional activity and Western blot of A167D mutant GATA4 protein were not altered. These variants may be involved in other mechanisms underlying CTD or may be unrelated to CTD occurrence.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Conotruncal heart defect (CTD), a subtype of congenital heart disease (CHD), is estimated to occur in approximately 7.3 per 10,000 live births [1]. CTD, comprising the tetralogy of Fallot (TOF), transposition of the great arteries (TGA), double outlet of right ventricle (DORV), persistent truncus arteriosus (PTA), pulmonary atresia with ventricular septal defect (PA/VSD), and interrupted aortic arch (IAA), is a complex form of CHD that requires surgical repair once diagnosed [2]. Although the etiology of the majority of CTD remains obscure, several studies have implicated genetic factors in its pathogenesis [3, 4].
CTD stems from perturbation of outflow tract (OFT) morphogenesis. A recently identified second heart field (SHF), which gives rise to OFT, may provide insights into the mechanism underlying CTD [5]. Indeed, SHF is subject to delicate regulation by various transcription factors and cofactors such as GATA4, TBX5, and NKX2-5 [6, 7]. Consistent with this finding, multiple mutations in the genes encoding these factors have been identified in CTD patients. GATA4, a zinc finger transcription factor, plays a crucial role in embryonic heart development such as proliferation of cardiomyocytes, endocardial cushion formation, development of right ventricle, and septation of the outflow tract [8, 9]. GATA4 can form a complex with ZFPM2/FOG2, which is related to the OFT development. Accordingly, mice harboring a GATA4 mutation that disrupts its interaction with ZFPM2/FOG2 recapitulate the abnormal heart phenotype, including CTD [3]. Most of previous studies showed that missense and nonsense mutations of GATA4 cause atrial and ventricular septal defects, often in association with pulmonary stenosis, and endocardial cushion defects [10, 11]. Several studies also reported that GATA4 has been linked to complex cardiovascular defects involving CTD [12]. Nemer et al. [13] first sequenced GATA4 among 26 TOF patients and revealed one mutation. The heterozygous mutation results in an amino acid substitution in the first zinc finger of GATA4 that reduced its transcriptional activation of downstream target genes, without affecting GATA4 ability to bind DNA, nor its interaction with ZFPM2. Subsequent independent studies also identified several mutations, mainly in TOF, but did not further investigate their pathogenicity [14]. With the advent and application of next-generation sequencing, large-population sequencing projects, such as the Exome Sequencing Project (ESP) and ClinSeq, have revealed in normal participants many variants that were previously assumed to be deleterious.
Although GATA4 mutations have been found in CTD patients, their functional significance is not known and the study populations and mutation spectra have thus far been limited. To further explore the role of GATA4 in CTD, we therefore screened for mutations in a larger CTD cohort and assessed potentially deleterious mutants. Also, this is the first time in Chinese Han population to carry out such a large sample of screening.
Materials and Methods
The Review Board of the Xinhua Hospital of the Shanghai Jiaotong University has approved this study. All subjects have consented to this research.
Study Subjects
From May 2013 to March 2015, a cohort of 600 unrelated patients diagnosed with conotruncal defects were recruited from Xinhua Hospital (Shanghai, China) and Children’s Medical Center (Shanghai, China). A thorough clinical examination of patients was carried out, including clinical history, physical examination, and a two-dimensional transthoracic echocardiography. From clinical information, the diagnosis of CTD was made by experienced pediatric cardiologists. A total of 300 unrelated healthy individuals used as controls were also matched to the patients in sex and age.
DNA Extraction
Approximately 3–5 ml peripheral blood was collected from each participant. Genomic DNA was extracted using the QIAamp DNA Blood Midi Kit (Qiagen, Dusseldorf, Germany) following the manufacturer’s instructions.
Mutation Sequencing
For each of the recruited 900 samples, all exons and intron–exon flanking regions of GATA4 were tested by target sequence technique for a mutation screen. Then all candidate mutations identified were confirmed by Sanger sequencing. DNA sequencing (of both strands) was carried out by an ABI 3730 sequencer (Applied Biosystems, USA). The sequence traces were aligned with the reference sequence using the GenBank BLAST program (http://blast.ncbi.nlm.nih.gov/Blast.cgi).
Multiple Sequence Alignments and Function Predictions
The multiple GATA4 protein sequences across species were aligned using the online program CLUSTALW (http://www.genome.jp/tools/clustalw/). The PolyPhen 2 (polymorphism phenotyping, http://genetics.bwh.harvard.edu/pph2/) and SIFT (Sorting Intolerant from Tolerant, http://sift.bii.astar.edu.sg/) programs were used to predict the disease-causing potential of a GATA4 variant.
Plasmid Construction and Site Directed Mutagenesis
The recombinant ANF-luciferase (ANF-Luc) reporter plasmid, which contains the 700-bp 5′-flanking region of the ANF gene, was kindly offered by Dr. Nimo from University of Ottawa. The full-length human GATA4 cDNA, NKX2.5 cDNA, and TBX5 cDNA were constructed and cloned into the pcDNA3.1 mammalian expression vector, respectively. Then the identified mutation was introduced into the GATA4 wild-type expression plasmid using KOD polymerase enzyme (TOYOBO) with a pair of mutated complementary primers. The mutant was sequenced to confirm the desired mutation and to exclude any other sequence variations.
Cell Cultures
Cos7 cells and C2C12 cells were maintained in DMEM (Invitrogen, California, USA) supplemented with 10% FBS (Invitrogen, California, USA), incubating at 37 °C in a humid atmosphere with 5% CO2.
Luciferase Assay
To examine the transcriptional activation function of GATA4 variants, the ANF-Luc reporter plasmid and an internal control reporter plasmid pGL4 (hRluc/CMV, Promega) were used in transient transfection assays. COS-7 cells were transfected with 300 ng of wild-type or mutant pcDNA3.1-GATA4 expression vector, 100 ng of ANF-Luc reporter construct, and 2 ng of pGL4.7 control reporter vector using the FuGENE Transfection Reagent (Promega, Madison, Wisconsin, USA). For analysis of the interaction between GATA4 and other transcriptional factors, 300 ng of pcDNA3.1-GATA4 plasmid (wild-type or each mutant), alone or together with 300 ng of NKX2.5 or TBX5 expression vector, and 100 ng of ANF-Luc were used. Firefly luciferase and Renilla luciferase activities were measured with the Dual-Glo luciferase assay system (Promega, Madison, Wisconsin, USA) and the Centro XS3 LB 960 Microplate Luminometer (Berthold, Bad Wildbad, Germany) 48 h after transfection. The results are the means of three independent experiments, each done in triplicate.
Western Blot
For Western blot analysis, C2C12 cells were harvested 48 h after transfection, lysed, and fractionated into nuclear and cytoplasmic extracts using the NE-PER kit (Thermo Fisher Scientific). Western blot analysis using the primary monoclonal antibody, anti-GATA4 (abcam), and the secondary antibody conjugated with anti-Rabbit-HRP (Thermo Fisher Scientific), was used to detect the presence of GATA4 protein in the cell fractions. Revelation was done using the Western Lightening Chemiluminescence Kit (Applied Biosystems). The protein bands were visualized by autoradiography.
Statistics
Statistical analyses were performed with the SPSS v.21.0 statistical software (SPSS, Chicago, IL, USA), and P < 0.05 was considered significant.
Results
In the present study, we have analyzed a total of 600 CTD patients. The percentages of CTD patients belonging to different categories were as follows; TOF: 38.17%, DORV: 16.50%, PA + VSD: 16.33%, TGA: 15.33%, SA + SV: 7.67%, IAA: 2.17%, PA + IVS: 2.17%, PTA: 1.67%. Age of all CTD patients ranged from 6 days to 5 years. However, maximum number of CHD patients taking part in this study were of <2 years (Table 1).
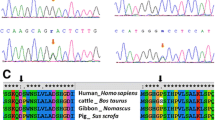
Human GATA4 located on chromosome 8p23.1-p22 consists of seven exons. GATA4 spans 50 kbp and is composed of 442 amino acids with two transactivation domains and two class IV zinc finger domains (N-terminal Zinc finger and a C-terminal zinc finger) along with a nuclear localization signal [15]. We investigated the genomic DNA for sequence variations in the entire coding regions, exon–intron boundaries, and untranslated regions of this gene for 600 patients with congenital heart diseases and 300 unaffected individuals. Eight heterozygous GATA4 missense changes (G64E, A66T, P163S, A167D, V267M, S377G, V380M, P407Q) were detected in 22 unrelated CTD patients (Table 2). One is a novel change (A167D) that involve amino acid located in conserved region of the transcriptional activation domain 2, at the N-terminus of GATA4 (Fig. 1). Sequencing of GATA4 in 300 healthy control subjects did not show the A167D change. We used the PolyPhen 2 and SIFT programs to predict the effect of the novel amino acid substitution and found that the A167D substitution was expected to influence protein function (Table 2). And as shown in Fig. 2, a cross-species alignment of multiple GATA4 protein sequences showed that the affected amino acids were highly conserved evolutionarily, indicating that the amino acids are functionally important. To investigate if the A167D change alters mammalian GATA4-mediated transactivation, we examined the ability of this mutation to activate the ANF promoter in transient transfection of COS7 cells in synergy with the transcriptional coregulators TBX5 and NKX2-5. We found that the mutant displayed normal activation ability when compared to wild-type GATA4, and the Western blot of GATA4 expression in transfected cells was also unchanged (Fig. 3).

Sequence traces showing the nonsynonymous nucleotide changes (C>A) causing amino acid replacements (A167D) in the GATA4 gene. The change is indicated with arrows

Multiple amino acid sequence alignment of different species shows the conservation of the mutated amino acid residue (amino acid marked) across species

Transactivation analysis of the A167D mutant protein. Inset shows Western blot of GATA4 expression in transfected cells with loading demonstrated by GAPDH levels
Four known changes, G64E, S377G, P163S, and A66T, were detected in 1 TOF, 2 DORV, 1 TGA, 3 SA + SV, and 1 PA + VSD patients (Table 2) of our cohort and in none of the 300 control subjects screened. This screen of all subjects also showed V380M, V267M, and P407Q nonsynonymous changes in both case group and healthy individuals. In our cohort, V380M and P407Q were all detected in four patients (4/600, 0.0067) and one control subjects (1/300, 0.0033). And V267M was showed in three patients (3/600, 0.005) and one healthy subject (1/300, 0.0033), respectively. There was no significant difference in the allele frequency of these three mutants between our CHD patients and control subjects.
Twelve additional variants of GATA4 gene were identified: six synonymous alterations, five intron mutants, and 1 splice site variant (Table 2). Six synonymous variants (A177, F110, C241, A442, A33, and Y244) are reported in the SNP database and have been previously described in CHD patients and control subjects.
Discussion
CHD is a common disease with a varied genetic background. Mutations in genes that encode factors involved in cardiac morphogenesis and development could lead to heart malformations [16]. GATA4 is an important transcriptional regulator of cardiogenesis. Its role has been verified in knockout mice that exhibit a variety of cardiac defects, including septal, valve, and outflow tract anomalies [17]. Small changes in the level of GATA4 protein expression can dramatically influence cardiac development and embryonic survival. In agreement with these observations, GATA4 variants have been reported in TOF and DORV patients, whereas they are absent in other types of CTD [18, 19]. Also several studies from different countries have established the association of GATA4 mutation with CTD, very few studies have been conducted in China. Therefore, we carried out this research to find the genetic link with GATA4 mutation from subjects with CTD.
In this study, one novel heterozygous GATA4 mutation of p.A167D was identified in one patient with DORV, which was located in the transactivation domain 2 (TAD2) required for GATA4 activity and is conserved between the different GATA factors. The missense mutation was absent in the 300 reference chromosomes from an ethnically matched control population. Alignment of the amino acid sequence from human, monkey, dog, cow, mouse, rabbit, frog, zebra fish, and sheep found that all of the nine species were much conserved in this site. The location and conservation together suggest that it may be disease-causing mutation, despite normal transactivation ability in our in vitro luciferase assay. Physical and comparative analysis suggested that the p.A167D substitution can be deleterious to protein function. In previous studies, only one missense mutation, p.P163S, was reported in TAD2 of GATA4 in patients with VSD and endocardial cushion defect [20]. Also, we found the same mutation in three patients with SA + SV, one patient with TOF, and one TGA.
Three changes, G64E, S377G, and A66T have been described previously in patients with septal defects or cardiac hypertrophy. And, in our cohort they were identified in five patients with CTDs, and in none of the control subjects. They both involve amino acids that are highly conserved between mammals, and PolyPhen analysis suggests a negative effect on protein function for G64E. The in vitro luciferase reporter assays showed that A66T significantly increased the ANF promoter activity [21]; Al-Azzouny et al. [22] applied three algorithms to predict the putative functional effect of S377G variant and predicted that S377G might lead to produce non-functional transcript, which would then lead to loss of phosphorylation legand in downstream aa 406. Another three mutations, V380M, V267M, and P407Q, were also found in control individuals and had been previously reported. Similarly, Schluterman et al. [18] reported the V380M in 3 out of 318 control subjects and Wang et al. [21] found the V267M in 4 out of 957 control individuals. On the other hand, the P407Q was firstly found in control group in this study.
In addition, six synonymous GATA4 sequence variants, p.A177A, p.F110F, p.C241C, p.A33A, p.Y244Y, and p.A442A, were found in 63 patients but not in control subjects. p.A177A, p.C241C, p.A33A, and p.A442A were found in various types of CTD, including TOF, TGA, DORV, PA + VSD, PA + IVS, and SA + SV. p.F110F and p.Y244Y were only found in two patients with IAA and TGA, respectively. The significance of these same sense mutations is unclear. However, “silent mutations” have been found to be associated with some genetic diseases [23]. Hence, these synonymous sequence variants may have influence at the posttranscriptional level as well as during posttranslational modification through exonic splicing enhancers or silencers. Many synonymous variants with GATA4 associated with CHD have been reported [24]. The present study showed that these synonymous variants may contribute to the pathogenesis of CHD.
The reasons for the distinct clinical phenotypes of GATA4 mutations are unknown, but may be related to complicated gene–gene and gene–environment interactions. Our study is limited by exploring only one variant rather than all. The selection of variants was based on the in silico results of PolyPhen-2 and SIFT. Thus, our results may in fact be representative of CTD patients in general. The issue of whether missense variants found in CTD patients are causative remains unresolved; however, because we have not investigated all aspects of GATA4 function. To fully investigate these variants, it is necessary to identify the downstream genes involved in cardiac development and assess the effects of these variants on transcription of the downstream genes.
Conclusions
In conclusion, we expanded the GATA4 variant profile in CTD patients by discovering one new missense variant (p.A167D). The function assay of this new mutant revealed that the missense mutation most likely to cause structural damage had no mild effect, if any, on the interaction with NKX2.5 or TBX5. Thus, the effects of these mutations may involve other known or unknown mechanisms underlying CTD or, alternatively, the mutations identified may not cause CTD. Our findings are a reminder for physicians to carefully evaluate gene variants associated with certain diseases, especially those lacking robust functional evidence, to confirm their pathogenicity.
References
Eisenberg L, Zhang W, Shen L, Deng Z, Ding Y, Mo X, Xu Z, Gao Q, Yi L (2014) Novel missense variants of ZFPM2/FOG2 identified in conotruncal heart defect patients do not impair interaction with GATA4. PLoS ONE 9:e102379
Xu YJ, Chen S, Zhang J, Fang SH, Guo QQ, Wang J, Fu QH, Li F, Xu R, Sun K (2014) Novel TBX1 loss-of-function mutation causes isolated conotruncal heart defects in Chinese patients without 22q11.2 deletion. BMC Med Genet 15:78
De Luca A, Sarkozy A, Ferese R, Consoli F, Lepri F, Dentici ML, Vergara P, De Zorzi A, Versacci P, Digilio MC, Marino B, Dallapiccola B (2011) New mutations in ZFPM2/FOG2 gene in tetralogy of Fallot and double outlet right ventricle. Clin Genet 80:184–190
Nakajima Y (2010) Second lineage of heart forming region provides new understanding of conotruncal heart defects. Congenit Anom (Kyoto) 50:8–14
Restivo A, Piacentini G, Placidi S, Saffirio C, Marino B (2006) Cardiac outflow tract: a review of some embryogenetic aspects of the conotruncal region of the heart. Anat Rec A Discov Mol Cell Evol Biol 288:936–943
Warburton D, Ronemus M, Kline J, Jobanputra V, Williams I, Anyane-Yeboa K, Chung W, Yu L, Wong N, Awad D, Yu CY, Leotta A, Kendall J, Yamrom B, Lee YH, Wigler M, Levy D (2014) The contribution of de novo and rare inherited copy number changes to congenital heart disease in an unselected sample of children with conotruncal defects or hypoplastic left heart disease. Hum Genet 133:11–27
Osoegawa K, Iovannisci DM, Lin B, Parodi C, Schultz K, Shaw GM, Lammer EJ (2014) Identification of novel candidate gene loci and increased sex chromosome aneuploidy among infants with conotruncal heart defects. Am J Med Genet A 164A:397–406
Zhou P, He A, Pu WT (2012) Regulation of GATA4 transcriptional activity in cardiovascular development and disease. Curr Top Dev Biol 100:143–169
Li J, Liu WD, Yang ZL, Yuan F, Xu L, Li RG, Yang YQ (2014) Prevalence and spectrum of GATA4 mutations associated with sporadic dilated cardiomyopathy. Gene 548:174–181
Mattapally S, Nizamuddin S, Murthy KS, Thangaraj K, Banerjee SK (2015) c.620C>T mutation in GATA4 is associated with congenital heart disease in South India. BMC Med Genet 16:7
Yang YQ, Wang J, Liu XY, Chen XZ, Zhang W, Wang XZ (2013) Mutation spectrum of GATA4 associated with congenital atrial septal defects. Arch Med Sci 9:976–983
Gittenberger-de Groot AC, Calkoen EE, Poelmann RE, Bartelings MM, Jongbloed MR (2014) Morphogenesis and molecular considerations on congenital cardiac septal defects. Ann Med 46:640–652
Nemer G, Fadlalah F, Usta J, Nemer M, Dbaibo G, Obeid M, Bitar F (2006) A novel mutation in the GATA4 gene in patients with Tetralogy of Fallot. Hum Mutat 27:293–294
Tomita-Mitchell A, Maslen CL, Morris CD, Garg V, Goldmuntz E (2007) GATA4 sequence variants in patients with congenital heart disease. J Med Genet 44:779–783
Reamon-Buettner SM, Borlak J (2005) GATA4 zinc finger mutations as a molecular rationale for septation defects of the human heart. J Med Genet 42:e32
Hoffman JI, Kaplan S (2002) The incidence of congenital heart disease. J Am Coll Cardiol 39:1890–1900
Rajagopal SK, Ma Q, Obler D, Shen J, Manichaikul A, Tomita-Mitchell A, Boardman K, Briggs C, Garg V, Srivastava D, Goldmuntz E, Broman KW, Benson DW, Smoot LB, Pu WT (2007) Spectrum of heart disease associated with murine and human GATA4 mutation. J Mol Cell Cardiol 43:677–685
Schluterman MK, Krysiak AE, Kathiriya IS, Abate N, Chandalia M, Srivastava D, Garg V (2007) Screening and biochemical analysis of GATA4 sequence variations identified in patients with congenital heart disease. Am J Med Genet A 143A:817–823
Zhang W, Li X, Shen A, Jiao W, Guan X, Li Z (2008) GATA4 mutations in 486 Chinese patients with congenital heart disease. Eur J Med Genet 51:527–535
Peng T, Wang L, Zhou SF, Li X (2010) Mutations of the GATA4 and NKX2.5 genes in Chinese pediatric patients with non-familial congenital heart disease. Genetica 138:1231–1240
Wang ESS, Qiao B, Duan W (2013) Identification of functional mutations in GATA4 in patients with congenital heart disease. PLoS ONE 8:e62138
Al-Azzouny MAERM, Issa HA, Behiry EG, Elsayed NR, Fayez AG (2016) Detection and putative effect of GATA4 gene variants in patients with congenital cardiac septal defects. Cell Mol Biol 62:5
Reamon-Buettner SM, Sattlegger E, Ciribilli Y, Inga A, Wessel A, Borlak J (2013) Transcriptional defect of an inherited NKX2-5 haplotype comprising a SNP, a nonsynonymous and a synonymous mutation, associated with human congenital heart disease. PLoS ONE 8:e83295
Xiong F, Li Q, Zhang C, Chen Y, Li P, Wei X, Li Q, Zhou W, Li L, Shang X, Xu X (2013) Analyses of GATA4, NKX2.5, and TFAP2B genes in subjects from southern China with sporadic congenital heart disease. Cardiovasc Pathol 22:141–145
Acknowledgements
The project was funded by a Grant (81670285) from the National Natural Science Foundation of China and the Three Years’ Action of Shanghai Health Bureau (GWTV-23). Without their assistance, this paper would not have been possible.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Informed Consent
Informed consent was obtained from all individual participants included in the study.
Rights and permissions
About this article

Cite this article
Liu, Y., Li, B., Xu, Y. et al. Mutation Screening of Gata4 Gene in CTD Patients Within Chinese Han Population. Pediatr Cardiol 38, 506–512 (2017). https://doi.org/10.1007/s00246-016-1542-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00246-016-1542-0