
Abstract
A number of mutations in GATA4 and NKX2.5 have been identified to be causative for a subset of familial congenital heart defects (CHDs) and a small number of sporadic CHDs. In this study, we evaluated common GATA4 and NKX2.5 mutations in 135 Chinese pediatric patients with non-familial congenital heart defects. Two novel mutations in the coding region of GATA4 were identified, namely, 487C>T (Pro163Ser) in exon 1 in a child with tetralogy of Fallot and 1220C>A (Pro407Gln) in exon 6 in a pediatric patient with outlet membranous ventricular septal defect. We also found 848C>A (Pro283Gln) in exon 2 of the NKX2.5 gene in a pediatric patient with ventricular septal defect, patent ductus arteriosus and aortic isthmus stenosis. None of the mutations was detected in healthy control subjects (n = 114). This study suggests that GATA4 and NKX2.5 missense mutations may be associated with congenital heart defects in pediatric Chinese patients. Further clinical studies with large samples are warranted.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Congenital heart defects (CHDs) are the most common developmental anomaly (about 1% in newborns in western countries) and are the leading non-infectious cause of mortality in newborns (Hoffman and Kaplan 2002; Sadowski 2009). Mendelian and chromosomal syndromes account for less than 20% of all CHDs. The genetic mechanism underlying non-chromosomal or non-Mendelian ‘sporadic’ CHDs accounting for the remaining 80%, is poorly understood (Bentham and Bhattacharya 2008). Transgenic and knockout mouse models have demonstrated that a number of candidate genes (mainly encoding transcriptional factors and receptors) contribute significantly to the etiology of CHDs, and the number of genes that have been shown to be needed for murine heart development probably exceeds 1,700 (Bentham and Bhattacharya 2008). However, so far only a few genes (e.g., GATA4, TBX-5, MYH6, NOTCH1 and NKX2.5/CSX1) involved in the pathogenesis of human CHDs have been identified using linkage analysis and candidate-gene approaches (Bentham and Bhattacharya 2008; Clark et al. 2006). GATA4 has been mapped to chromosome 8p23.1 and encodes a 442-amino acid protein of the GATA family of zinc finger transcription factors (Huang et al. 1995; White et al. 1995). A number of preclinical studies have demonstrated that GATA4 can regulate genes involved in embryogenesis and in myocardial differentiation and function (Brown et al. 2004; Dai et al. 2002; Liang et al. 2001; Molkentin 2000; Pikkarainen et al. 2004; Pu et al. 2004; Rivera-Feliciano et al. 2006; Rojas et al. 2008; Zeisberg et al. 2005). Myocardial expression of GATA4 is required for proliferation of cardiomyocytes, formation of the endocardial cushions, development of the right ventricle, and septation of the outflow tract. GATA4 binds to the consensus sequence 5′-AGATAG-3′ and acts as a transcriptional activator of the atrialnatriuretic factor in cooperation with NKX2.5 (Durocher et al. 1997). It appears that small changes in the level of GATA4 protein expression can dramatically influence cardiac morphogenesis and embryonic survival (Pu et al. 2004). Mice lacking Gata4 suffer from defective ventral morphogenesis and heart tube formation (Kuo et al. 1997). GATA binding factor-4 (GATA4) is an important zinc finger-containing transcription factor and its mutations have been found to be causative for a subset of familial atrial and ventricular septal defects (ASD & VSD) and pulmonary stenosis (PS) in several unrelated extended pedigrees (Chen et al. 2010a, b; D’Amato et al. 2010; Garg et al. 2003; Hirayama-Yamada et al. 2005; Okubo et al. 2004; Posch et al. 2010; Posch et al. 2008; Sarkozy et al. 2005). On the other hand, the importance of GATA4 mutations may be confined to specific subgroups of CHD phenotypes in specific populations among CHD patients without family history (i.e., sporadic CHDs), such as Erdheim–Chester disease (ECD) in Caucasians and double-inlet left ventricle (DILV) in Libyan and tetralogy of Fallot (TOF) in Lebanese (Nemer et al. 2006; Rajagopal et al. 2007). However, previously reported GATA4 mutations in Chinese patients are mostly associated with cardiac septal defects and TOF (Tang et al. 2006; Zhang et al. 2008; Zhang et al. 2009b).
The NK2 transcription factor related, locus 5 gene (NKX2.5/CSX1) has been mapped to chromosome 5q34 with 2 exons, encoding a 324-amino acid protein. NKX2.5 belongs to the NK-2 family of homeodomain-containing transcription factors, which are conserved from Drosophila to human (Bartlett et al. 2010; Reamon-Buettner and Borlak 2010). NKX2.5 is the earliest known marker of myocardial progenitor cells in all species studied (Komuro and Izumo 1993; Reamon-Buettner and Borlak 2010; Tanaka et al. 1998; Turbay et al. 1996). Through the 60-amino-acid homeodomain, NKX2.5 interacts with DNA through a helix-turn-helix DNA-binding motif of three α-helices, with helix 3 providing binding specificity. Other conserved regions including the TAD and the NK2-specific domain are also important for its function. Mutations of the Drosophila NKX2.5 homolog, tinman, results in failure of heart formation (Azpiazu and Frasch 1993; Bodmer 1993), and homozygous inactivation of NKX2.5 in mammals results in impaired cardiac looping and embryonic lethality (Lyons et al. 1995; Tanaka et al. 1999). In mice, heterozygous loss-of-function Nkx2.5 mutations lead to a mild conduction delay and atrial septal dysmorphogenesis manifest as an increased frequency of patent foramen ovale, atrial septal aneurysm, and ASD (Biben et al. 2000). Mutations in NKX2.5 have been identified as an important factor responsible for various clinical forms of CHDs. Up to now, more than 126 single nucleotide polymorphisms (SNPs) have been detected in human GATA4 gene (see http://www.ncbi.nlm.nih.gov/snp), but lesser SNPs and other mutations in NKX2.5 (<50) have been reported. There are few reports about NKX2.5 mutations in sporadic Chinese patients (Ding et al. 2009; Liu et al. 2009a, b; Zhang et al. 2009a, b). To evaluate the GATA4 and NKX2.5 mutations in CHD patients from China, we have screened 135 sporadic Chinese individuals with different CHD phenotypes and compared to 114 healthy control individuals using denaturing high-performance liquid chromatography (DHPLC) and DNA sequencing approaches.
Results
Clinical phenotypes and frequencies of GATA4 and NKX2.5 mutations
GATA4 (accession no. NM_002052) and NKX2.5 (NM_004387) mutation analysis was performed using DHPLC in a total of 135 unrelated Chinese pediatric CHD patients and 114 healthy subjects. In the patient group, 82 (62.1%) individuals had VSD and 19 (14.4%) suffered from secundum atrial septal defect (ASD; Table 1). TOF was present in 12 pediatric patients and 5 had patent ductus arteriosus (PDA). Altogether, three new non-synonymous SNPs of GATA4 and NKX2.5 were detected in the CHD patients but not in healthy controls, with an overall detection frequency of 2.22% in the patient group. We also observed three synonymous SNPs in exons of GATA4 and NKX2.5, which were also detected in healthy subjects.
Phenotypes of GATA4 mutations
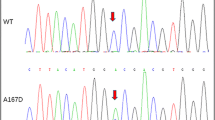
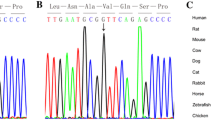
Two probands carried mutations in GATA4 (frequency = 1.48%). Our direct DNA sequencing analysis revealed the 487C>T (Pro163Ser) mutation in exon 1 and 1220C>A (Pro407Gln) in exon 6. The 1220C>A (Pro407Gln) mutation has not been previously reported in other ethnic groups with various CHDs. Notably, both Pro163 and Pro407 are conserved among several species including the human, mouse and rat. The affected individual in whom the Pro163Ser mutation was detected was a male child with TOF; the Pro407Gln mutation was found in a female patient with outlet membranous VSD (Table 2). However, the parents and siblings of these two patients were healthy. Their parents did not smoke and drink alcohol and had no history of exposure to toxic chemicals. Analysis of 114 healthy Chinese children did not found these two mutations. The odds ratio was 2.200, and 2.285 for 487C>T (Pro163Ser) and 1220C>A (Pro407Gln), respectively, and the P was 1.000 and 0.544, respectively (by Fisher’s exact test).
The synonymous mutation 99G>T (Ala333Al) in GATA4 was observed in 11 pediatric patients and 6 healthy subjects (P = 1.000; odds ratio = 1.583 by Fisher’s exact test). Two mutations in the introns, namely 50745A>T and 50946A>C, were also detected in 13 patients and 12 healthy subjects (P = 0.461 and 1.000, and odds ratio = 1.015 and 0.634, respectively, by Fisher’s exact test; Table 2).
Phenotypes of NKX2.5 mutations
In the 135 Chinese pediatric patients with CHDs, we only found one non-synonymous mutation in NKX2.5 exon 2 in a patient with VSD, PDA and ASD (Tables 3, 4). This gave a detection frequency of 0.74%, lower than those reported previously (1.4–4.8%) (Elliott et al. 2003; Esposito et al. 2009; McElhinney et al. 2003). The parents of this patient were non-smokers. Sequence analysis revealed a C to A transition at nucleotide 848 of NKX2.5 exon 2, which causes Pro283Gln (Table 3). This novel mutation has never been reported in other ethnic populations. This mutation was not found in any healthy control subjects. The odds ratio was 2.286 (P > 0.05).
Two known synonymous SNPs (rs2277923 and rs3729753) were detected in the NKX2.5 gene in both patients and healthy subjects (Table 3). In neither case did the allele frequency differ between the patients and healthy controls (P > 0.05 by Fisher’s exact test).
Discussion
A number of clinical studies have indicated that mutations of GATA4 are the cause of many familial and sporadic CHDs (Table 5; Chen et al. 2010a, b; D’Amato et al. 2010; Garg et al. 2003; Hirayama-Yamada et al. 2005; Okubo et al. 2004; Posch et al. 2008, 2010; Sarkozy et al. 2005). Defects in GATA4 are the cause of ASD type 2 (ASD2), which is a congenital heart malformation characterized by incomplete closure of the wall between the atria resulting in blood flow from the left to the right atria. In Chinese CHD patients, we revealed two GATA4 mutations, namely Pro163Ser mutation in patient with TOF and the other mutation Pro407Gln with a clinical phenotype of VSD. Rajagopal et al. (2007) have identified several mutations of GATA4, including Pro163Ser, two mutations in patients with ECD and another one in DILV patients and deduced GATA4 mutations as a cause of sporadic ECD in Caucasians and DILV in Libyan. However, our study together with other studies in Chinese patients (Tang et al. 2006; Zhang et al. 2008) have found that the mutations of GATA4 were associated with cardiac septal defects and TOF (Table 5). The reasons for the distinct clinical phenotypes of GATA4 mutations are unknown, but may be related to complicated gene–gene and gene–environment interactions.
GATA family members recognize the consensus motif (A/T)GATA(A/G) through a conserved multifunctional DNA-binding domain (Ko and Engel 1993). The DNA-binding domain of GATAs comprises two zinc fingers of the CX2CX17CX2C type and an adjacent basic domain. Like other GATA members, GATA4 is composed of two transactivation domains (TADs), two IV zinc fingers (N-terminal zinc finger, NZf; and C-terminal zinc finger, CZf), and a nuclear localization signal (Molkentin 2000). NZf is highly conserved that can interact with FOG2 and CZf may interact with other transcription factors such as NKX2.5, NF-At3, MEF2C, and HAND2 (Durocher et al. 1997; Lee et al. 1998; Molkentin et al. 1998; Morin et al. 2000). Until now, only one missense mutation, Pro163Ser, was reported in TADs of GATA4. The mutation 487C>T (Pro163Ser) is located in TAD2 of GATA4 (Rajagopal et al. 2007). Hirayama-Yamada et al. (2005) have found a mutation 155C>T in GATA4 exon 1, leading to a missense mutation, S52F, in TAD1. The identification and functional characterization of two evolutionarily conserved TADs suggests that each of these domains modulates critical functions in the transcriptional regulatory programs encoded by GATA4 during vertebrate development (Morrisey et al. 1997).
Another mutation, Pro407Gln, is located in the C-terminal domain of the GATA4 protein. Okubo et al. (2004) and Garg et al. (2003) have separately reported another two frameshift mutations S358X (1074delC) and E359X (1075delG) in the C-terminal of GATA4 and these two frameshifts may inactivate transcription of downstream genes and thus result in haploinsufficiency. As GATA4 functionally interacts with TBX5 and NKX2.5, the missense mutations may interfere with the coordinated interaction between GATA4 and other transcription factors in cardiogenesis (Durocher et al. 1997; Garg et al. 2003; Sepulveda et al. 2002).
To date, more than 42 mutations have been detected in NKX2.5, including 33 SNPs, 6 deletions and 2 insertions. A number of missense and nonsense mutations in NKX2.5 have been found in families with inherited autosomal-dominant ASD and atrioventricular conduction block (Table 6; Akcaboy et al. 2008; Benson et al. 1999; Ding et al. 2009; Elliott et al. 2003; Esposito et al. 2009; Gioli-Pereira et al. 2010; Goldmuntz et al. 2001; Gutierrez-Roelens et al. 2002; Hobbs et al. 2005; Hosoda et al. 1999; Ikeda et al. 2002; Liu et al. 2009a, b; McElhinney et al. 2003; Schott et al. 1998; Stallmeyer et al. 2010; Watanabe et al. 2002; Zhang et al. 2009a, b). Other congenital heart abnormalities have been observed at low penetrance in these families, including VSD, Ebstein’s anomaly, TOF, subvalvular aortic stenosis, and tricuspid valve abnormality. Many of these mutations are also found in sporadic CHDs. Studies in Chinese CHD patients have indicated NKX2.5 mutations are associated with CHDs, but are very rare in Chinese (Ding et al. 2009; Liu et al. 2009a, b; Zhang et al. 2009a, b). For Chinese patients, NKX2.5 mutation investigation should be limited within a number of familial and special phenotype of CHDs.
A novel mutation in the coding region of NKX2.5 (848C>A) was found in the C-terminal portion in this study. Experimental studies have shown that the C-terminal portion of NKX2.5 is important for its function distinct from the role of the homeodomain (Kasahara et al. 2000; Kasahara et al. 2001). Previous functional studies of NKX2.5 mutations have demonstrated decreased binding of mutant NKX2.5 proteins with mutations outside the homeodomain to dimeric DNA binding sites despite normal binding to monomeric sites (Kasahara et al. 2000). Kasahara et al. (2000) have suggested that this portion of the gene is critical for the cooperative homodimerization and heterodimerization (with other transcription factors such as GATA4) of the NKX2.5 protein on dimeric DNA binding sites. Similarly, the mutation Pro283Gln (848C>A) may also affect NKX2.5-DNA binding.
The genetic architecture of non-familial CHDs probably includes accumulation of rare nonsynonymous variants in cardiac developmental genes leading to mutational loading of cardiac developmental networks, copy number variation in cardiac developmental genes, and common variants that may not be obviously associated with cardiac development but may modulate genetic buffering pathways (e.g., folate and arachidonic acid metabolism) (Bentham and Bhattacharya 2008). GATA4 can interact with NKX2.5 and several other proteins which may directly and indirectly modify cardiac development and morphogenesis. For example, the cytochrome P450s CYP2C9 and 2C19 are regulated by GATA4 (Mwinyi et al. 2010a, b), and both CYP2C9 and 2C19 are involved in arachidonic acid metabolism, resulting in active metabolites that may modulate the activity and proliferation of cardiac endothelial cells.
In conclusion, this study has shown that genomic GATA4 and NKX2.5 missense mutations may be associated with non-familial CHDs with diverse clinical phenotypes in Chinese pediatric patients. However, the small sample size of this study does not allow us to establish such an association. A larger sample should be analyzed to determine how common GATA4 mutations are among patients with sporadic CHDs and the functional impact.
Materials and methods
Subjects
From September 2005 to April 2006, we recruited prospectively 135 cases diagnosed with CHD. All the subjects were assessed with regard to clinical and family history. All these patients had no family history of CHDs. Physical examination and echocardiogram were performed in Children’s Hospital of Fudan University (Shanghai, China). The clinical diagnosis of these patients included VSD, ASD, PDA, pulmonary artery stenosis (PAS) and others (Table 1). In addition, 114 unrelated healthy individuals served as the control. After informed consent was obtained, 5 mL blood sample was collected from each participant.
Genomic DNA extraction
Genomic DNA was isolated from peripheral blood leukocytes of participants using the DNA isolation Kit from Genebase (Vancouver, Canada), according to the manufacture’s protocol.
Polymerase chain reaction
The exons of GATA4 (accession no. NM_002052) and NKX2.5 (NM_004387) were amplified using Polymerase chain reaction (PCR). PCR was performed for exons 1–6 of GATA4, and exons 1 and 2 of NKX2.5. Primers were designed by Primer premier 5.1 or as described by previous studies (Tang et al. 2006; Zhang et al. 2009a). Primer sequences are presented in Supplementary Table 1. We divided GATA4 exon 1 and NKX2.5 exon 2 into two amplicons, as these exons were large, and used 2× GC buffer (Takara, Otsu, Japan) instead of 10× GC buffer for exon 1 of GATA4.
The PCR was performed in 25 μl standard PCR buffer, containing 50 ng genomic DNA, 0.2 mol/l forward and reverse primers, 1.5 mmol/l MgCl2, 0.2 mmol/l of each dNTP, and 1 U Taq DNA polymerase. The amplification program was one cycle of an initial denaturation step at 95°C for 5 min, followed by 35 cycles at 94°C for 30 s; 51–65°C for 30 s (different anneal temperatures with different primers); 72°C for 30 min, and a final extension at 72°C for 7 min. Reactions were performed on ABI9700 PCR machine (Applied Biosystems Inc., Carlsbad, CA).
For the analysis of various SNPs in GATA4 and NKX2.5 genes, the total number of chromosomes examined was slightly different in both patient and healthy subject groups. For the 99G>T SNP in GATA4, 264 and 228 chromosomes were analyzed in the patient and healthy subject groups, respectively. For 50745A>T, 50946A>C, and 1220C>A mutations in GATA4, 268 and 204 chromosomes were checked in the patient and healthy subject groups, respectively. For 63G>A, 606C>G, and 848C>A mutations in NKX2.5, 252, 268 and 270 chromosomes were tested in the patient group.
DHPLC analysis
Polymerase chain reaction products were denatured for 10 min at 95°C and subject to denaturing HPLC analysis system (Transgenomic Inc., San Jose, CA). The fragments were eluted at temperatures calculated by the DHPLC melting program for the successful resolution of heteroduplexes. Samples with double or triple-peaked DHPLC chromatograms were selected to run repeated PCR and DHPLC analysis. If there was a similarity between the two chromatograms, the samples were defined as “mutants”.
DNA sequence analysis
For samples with melting profiles different from the wild-type, PCR fragments were reamplified. PCR products with bidirectional extraction and DNA sequencing were performed using the BigDye Teminator Cycle Sequencing v3.1 Kit and an ABI PRISM 3730 Genetic Analyzer (Applied Biosystems Inc., Carlsbad, CA).
Statistical analysis
Comparison of allele frequencies between two groups was conducted by Fisher’s exact test. A P < 0.05 was considered statistically significant.
References
Akcaboy MI, Cengiz FB, Inceoglu B, Ucar T, Atalay S, Tutar E, Tekin M (2008) The effect of p.Arg25Cys alteration in NKX2–5 on conotruncal heart anomalies: mutation or polymorphism? Pediatr Cardiol 29:126–129
Azpiazu N, Frasch M (1993) Tinman and bagpipe: two homeo box genes that determine cell fates in the dorsal mesoderm of Drosophila. Genes Dev 7:1325–1340
Bartlett H, Veenstra GJ, Weeks DL (2010) Examining the cardiac NK-2 genes in early heart development. Pediatr Cardiol 31:335–341
Benson DW, Silberbach GM, Kavanaugh-McHugh A, Cottrill C, Zhang Y, Riggs S, Smalls O, Johnson MC, Watson MS, Seidman JG, Seidman CE, Plowden J, Kugler JD (1999) Mutations in the cardiac transcription factor NKX2.5 affect diverse cardiac developmental pathways. J Clin Invest 104:1567–1573
Bentham J, Bhattacharya S (2008) Genetic mechanisms controlling cardiovascular development. Ann N Y Acad Sci 1123:10–19
Biben C, Weber R, Kesteven S, Stanley E, McDonald L, Elliott DA, Barnett L, Koentgen F, Robb L, Feneley M, Harvey RP (2000) Cardiac septal and valvular dysmorphogenesis in mice heterozygous for mutations in the homeobox gene Nkx2-5. Circ Res 87:888–895
Bodmer R (1993) The gene tinman is required for specification of the heart and visceral muscles in Drosophila. Development 118:719–729
Brown CO 3rd, Chi X, Garcia-Gras E, Shirai M, Feng XH, Schwartz RJ (2004) The cardiac determination factor, Nkx2-5, is activated by mutual cofactors GATA-4 and Smad1/4 via a novel upstream enhancer. J Biol Chem 279:10659–10669
Chen MW, Pang YS, Guo Y, Pan JH, Liu BL, Shen J, Liu TW (2010a) GATA4 mutations in Chinese patients with congenital cardiac septal defects. Pediatr Cardiol 31:85–89
Chen Y, Mao J, Sun Y, Zhang Q, Cheng HB, Yan WH, Choy KW, Li H (2010b) A novel mutation of GATA4 in a familial atrial septal defect. Clin Chim Acta 411:1741–1745
Clark KL, Yutzey KE, Benson DW (2006) Transcription factors and congenital heart defects. Annu Rev Physiol 68:97–121
D’Amato E, Giacopelli F, Giannattasio A, D’Annunzio G, Bocciardi R, Musso M, Lorini R, Ravazzolo R (2010) Genetic investigation in an Italian child with an unusual association of atrial septal defect, attributable to a new familial GATA4 gene mutation, and neonatal diabetes due to pancreatic agenesis. Diabet Med 27:1195–1200
Dai YS, Cserjesi P, Markham BE, Molkentin JD (2002) The transcription factors GATA4 and dHAND physically interact to synergistically activate cardiac gene expression through a p300-dependent mechanism. J Biol Chem 277:24390–24398
Ding JD, Li KR, Zhang XL, Yao YY, Reng LQ, Tao SY, Fang X, Ma GS (2009) Preliminary exploration of transcription factor Nkx2.5 mutations and congenital heart diseases. Zhonghua Yi Xue Za Zhi 89:1114–1116
Durocher D, Charron F, Warren R, Schwartz RJ, Nemer M (1997) The cardiac transcription factors Nkx2-5 and GATA-4 are mutual cofactors. EMBO J 16:5687–5696
Elliott DA, Kirk EP, Yeoh T, Chandar S, McKenzie F, Taylor P, Grossfeld P, Fatkin D, Jones O, Hayes P, Feneley M, Harvey RP (2003) Cardiac homeobox gene NKX2-5 mutations and congenital heart disease: associations with atrial septal defect and hypoplastic left heart syndrome. J Am Coll Cardiol 41:2072–2076
Esposito G, Grutter G, Drago F, Costa MW, De Santis A, Bosco G, Marino B, Bellacchio E, Lepri F, Harvey RP, Sarkozy A, Dallapiccola B (2009) Molecular analysis of PRKAG2, LAMP2, and NKX2-5 genes in a cohort of 125 patients with accessory atrioventricular connection. Am J Med Genet A 149A:1574–1577
Garg V, Kathiriya IS, Barnes R, Schluterman MK, King IN, Butler CA, Rothrock CR, Eapen RS, Hirayama-Yamada K, Joo K, Matsuoka R, Cohen JC, Srivastava D (2003) GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature 424:443–447
Gioli-Pereira L, Pereira AC, Mesquita SM, Xavier-Neto J, Lopes AA, Krieger JE (2010) NKX2.5 mutations in patients with non-syndromic congenital heart disease. Int J Cardiol 138:261–265
Goldmuntz E, Geiger E, Benson DW (2001) NKX2.5 mutations in patients with tetralogy of fallot. Circulation 104:2565–2568
Gutierrez-Roelens I, Sluysmans T, Gewillig M, Devriendt K, Vikkula M (2002) Progressive AV-block and anomalous venous return among cardiac anomalies associated with two novel missense mutations in the CSX/NKX2-5 gene. Hum Mutat 20:75–76
Hirayama-Yamada K, Kamisago M, Akimoto K, Aotsuka H, Nakamura Y, Tomita H, Furutani M, Imamura S, Takao A, Nakazawa M, Matsuoka R (2005) Phenotypes with GATA4 or NKX2.5 mutations in familial atrial septal defect. Am J Med Genet A 135:47–52
Hobbs CA, Cleves MA, Keith C, Ghaffar S, James SJ (2005) NKX2.5 and congenital heart defects: a population-based study. Am J Med Genet A 134A:223–225
Hoffman JI, Kaplan S (2002) The incidence of congenital heart disease. J Am Coll Cardiol 39:1890–1900
Hosoda T, Komuro I, Shiojima I, Hiroi Y, Harada M, Murakawa Y, Hirata Y, Yazaki Y (1999) Familial atrial septal defect and atrioventricular conduction disturbance associated with a point mutation in the cardiac homeobox gene CSX/NKX2-5 in a Japanese patient. Jpn Circ J 63:425–426
Huang WY, Cukerman E, Liew CC (1995) Identification of a GATA motif in the cardiac α-myosin heavy-chain-encoding gene and isolation of a human GATA-4 cDNA. Gene 155:219–223
Ikeda Y, Hiroi Y, Hosoda T, Utsunomiya T, Matsuo S, Ito T, Inoue J, Sumiyoshi T, Takano H, Nagai R, Komuro I (2002) Novel point mutation in the cardiac transcription factor CSX/NKX2.5 associated with congenital heart disease. Circ J 66:561–563
Kasahara H, Lee B, Schott JJ, Benson DW, Seidman JG, Seidman CE, Izumo S (2000) Loss of function and inhibitory effects of human CSX/NKX2.5 homeoprotein mutations associated with congenital heart disease. J Clin Invest 106:299–308
Kasahara H, Usheva A, Ueyama T, Aoki H, Horikoshi N, Izumo S (2001) Characterization of homo- and heterodimerization of cardiac Csx/Nkx2.5 homeoprotein. J Biol Chem 276:4570–4580
Ko LJ, Engel JD (1993) DNA-binding specificities of the GATA transcription factor family. Mol Cell Biol 13:4011–4022
Komuro I, Izumo S (1993) Csx: a murine homeobox-containing gene specifically expressed in the developing heart. Proc Natl Acad Sci USA 90:8145–8149
Kuo CT, Morrisey EE, Anandappa R, Sigrist K, Lu MM, Parmacek MS, Soudais C, Leiden JM (1997) GATA4 transcription factor is required for ventral morphogenesis and heart tube formation. Genes Dev 11:1048–1060
Lee Y, Shioi T, Kasahara H, Jobe SM, Wiese RJ, Markham BE, Izumo S (1998) The cardiac tissue-restricted homeobox protein Csx/Nkx2.5 physically associates with the zinc finger protein GATA4 and cooperatively activates atrial natriuretic factor gene expression. Mol Cell Biol 18:3120–3129
Liang Q, Wiese RJ, Bueno OF, Dai YS, Markham BE, Molkentin JD (2001) The transcription factor GATA4 is activated by extracellular signal-regulated kinase 1- and 2-mediated phosphorylation of serine 105 in cardiomyocytes. Mol Cell Biol 21:7460–7469
Liu XY, Yang YQ, Yang Y, Lin XP, Chen YH (2009a) Mutation of NKX2-5 gene in patients with atrial septal defect. Zhonghua Er Ke Za Zhi 47:696–700
Liu XY, Yang YQ, Yang Y, Lin XP, Chen YH (2009b) Novel NKX2-5 mutations identified in patients with congenital ventricular septal defects. Zhonghua Yi Xue Za Zhi 89:2395–2399
Lyons I, Parsons LM, Hartley L, Li R, Andrews JE, Robb L, Harvey RP (1995) Myogenic and morphogenetic defects in the heart tubes of murine embryos lacking the homeo box gene Nkx2-5. Genes Dev 9:1654–1666
McElhinney DB, Geiger E, Blinder J, Benson DW, Goldmuntz E (2003) NKX2.5 mutations in patients with congenital heart disease. J Am Coll Cardiol 42:1650–1655
Molkentin JD (2000) The zinc finger-containing transcription factors GATA-4, -5, and -6. Ubiquitously expressed regulators of tissue-specific gene expression. J Biol Chem 275:38949–38952
Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, Robbins J, Grant SR, Olson EN (1998) A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell 93:215–228
Morin S, Charron F, Robitaille L, Nemer M (2000) GATA-dependent recruitment of MEF2 proteins to target promoters. EMBO J 19:2046–2055
Morrisey EE, Ip HS, Tang Z, Parmacek MS (1997) GATA-4 activates transcription via two novel domains that are conserved within the GATA-4/5/6 subfamily. J Biol Chem 272:8515–8524
Mwinyi J, Hofmann Y, Pedersen RS, Nekvindova J, Cavaco I, Mkrtchian S, Ingelman-Sundberg M (2010a) The transcription factor GATA-4 regulates cytochrome P4502C19 gene expression. Life Sci 86:699–706
Mwinyi J, Nekvindova J, Cavaco I, Hofmann Y, Pedersen RS, Landman E, Mkrtchian S, Ingelman-Sundberg M (2010b) New insights into the regulation of CYP2C9 gene expression: the role of the transcription factor GATA-4. Drug Metab Dispos 38:415–421
Nemer G, Fadlalah F, Usta J, Nemer M, Dbaibo G, Obeid M, Bitar F (2006) A novel mutation in the GATA4 gene in patients with Tetralogy of Fallot. Hum Mutat 27:293–294
Okubo A, Miyoshi O, Baba K, Takagi M, Tsukamoto K, Kinoshita A, Yoshiura K, Kishino T, Ohta T, Niikawa N, Matsumoto N (2004) A novel GATA4 mutation completely segregated with atrial septal defect in a large Japanese family. J Med Genet 41:e97
Pikkarainen S, Tokola H, Kerkela R, Ruskoaho H (2004) GATA transcription factors in the developing and adult heart. Cardiovasc Res 63:196–207
Poirier O, Nicaud V, McDonagh T, Dargie HJ, Desnos M, Dorent R, Roizes G, Schwartz K, Tiret L, Komajda M, Cambien F (2003) Polymorphisms of genes of the cardiac calcineurin pathway and cardiac hypertrophy. Eur J Hum Genet 11:659–664
Posch MG, Perrot A, Schmitt K, Mittelhaus S, Esenwein EM, Stiller B, Geier C, Dietz R, Gessner R, Ozcelik C, Berger F (2008) Mutations in GATA4, NKX2.5, CRELD1, and BMP4 are infrequently found in patients with congenital cardiac septal defects. Am J Med Genet A 146A:251–253
Posch MG, Boldt LH, Polotzki M, Richter S, Rolf S, Perrot A, Dietz R, Ozcelik C, Haverkamp W (2010) Mutations in the cardiac transcription factor GATA4 in patients with lone atrial fibrillation. Eur J Med Genet 53:201–203
Pu WT, Ishiwata T, Juraszek AL, Ma Q, Izumo S (2004) GATA4 is a dosage-sensitive regulator of cardiac morphogenesis. Dev Biol 275:235–244
Rajagopal SK, Ma Q, Obler D, Shen J, Manichaikul A, Tomita-Mitchell A, Boardman K, Briggs C, Garg V, Srivastava D, Goldmuntz E, Broman KW, Benson DW, Smoot LB, Pu WT (2007) Spectrum of heart disease associated with murine and human GATA4 mutation. J Mol Cell Cardiol 43:677–685
Reamon-Buettner SM, Borlak J (2005) GATA4 zinc finger mutations as a molecular rationale for septation defects of the human heart. J Med Genet 42:e32
Reamon-Buettner SM, Borlak J (2010) NKX2-5: an update on this hypermutable homeodomain protein and its role in human congenital heart disease (CHD). Hum Mutat
Reamon-Buettner SM, Cho SH, Borlak J (2007) Mutations in the 3′-untranslated region of GATA4 as molecular hotspots for congenital heart disease (CHD). BMC Med Genet 8:38
Rivera-Feliciano J, Lee KH, Kong SW, Rajagopal S, Ma Q, Springer Z, Izumo S, Tabin CJ, Pu WT (2006) Development of heart valves requires Gata4 expression in endothelial-derived cells. Development 133:3607–3618
Rojas A, Kong SW, Agarwal P, Gilliss B, Pu WT, Black BL (2008) GATA4 is a direct transcriptional activator of cyclin D2 and Cdk4 and is required for cardiomyocyte proliferation in anterior heart field-derived myocardium. Mol Cell Biol 28:5420–5431
Sadowski SL (2009) Congenital cardiac disease in the newborn infant: past, present, and future. Crit Care Nurs Clin North Am 21:37–48, vi
Sarkozy A, Conti E, Neri C, D’Agostino R, Digilio MC, Esposito G, Toscano A, Marino B, Pizzuti A, Dallapiccola B (2005) Spectrum of atrial septal defects associated with mutations of NKX2.5 and GATA4 transcription factors. J Med Genet 42:e16
Schott JJ, Benson DW, Basson CT, Pease W, Silberbach GM, Moak JP, Maron BJ, Seidman CE, Seidman JG (1998) Congenital heart disease caused by mutations in the transcription factor NKX2-5. Science 281:108–111
Sepulveda JL, Vlahopoulos S, Iyer D, Belaguli N, Schwartz RJ (2002) Combinatorial expression of GATA4, Nkx2-5, and serum response factor directs early cardiac gene activity. J Biol Chem 277:25775–25782
Stallmeyer B, Fenge H, Nowak-Gottl U, Schulze-Bahr E (2010) Mutational spectrum in the cardiac transcription factor gene NKX2.5 (CSX) associated with congenital heart disease. Clin Genet 78:533–540
Tanaka M, Kasahara H, Bartunkova S, Schinke M, Komuro I, Inagaki H, Lee Y, Lyons GE, Izumo S (1998) Vertebrate homologs of tinman and bagpipe: roles of the homeobox genes in cardiovascular development. Dev Genet 22:239–249
Tanaka M, Chen Z, Bartunkova S, Yamasaki N, Izumo S (1999) The cardiac homeobox gene Csx/Nkx2.5 lies genetically upstream of multiple genes essential for heart development. Development 126:1269–1280
Tang ZH, Xia L, Chang W, Li H, Shen F, Liu JY, Wang Q, Liu MG (2006) Two novel missense mutations of GATA4 gene in Chinese patients with sporadic congenital heart defects. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 23:134–137
Tomita-Mitchell A, Maslen CL, Morris CD, Garg V, Goldmuntz E (2007) GATA4 sequence variants in patients with congenital heart disease. J Med Genet 44:779–783
Turbay D, Wechsler SB, Blanchard KM, Izumo S (1996) Molecular cloning, chromosomal mapping, and characterization of the human cardiac-specific homeobox gene hCsx. Mol Med 2:86–96
Watanabe Y, Benson DW, Yano S, Akagi T, Yoshino M, Murray JC (2002) Two novel frameshift mutations in NKX2.5 result in novel features including visceral inversus and sinus venosus type ASD. J Med Genet 39:807–811
White RA, Dowler LL, Pasztor LM, Gatson LL, Adkison LR, Angeloni SV, Wilson DB (1995) Assignment of the transcription factor GATA4 gene to human chromosome 8 and mouse chromosome 14: Gata4 is a candidate gene for Ds (disorganization). Genomics 27:20–26
Zeisberg EM, Ma Q, Juraszek AL, Moses K, Schwartz RJ, Izumo S, Pu WT (2005) Morphogenesis of the right ventricle requires myocardial expression of Gata4. J Clin Invest 115:1522–1531
Zhang W, Li X, Shen A, Jiao W, Guan X, Li Z (2008) GATA4 mutations in 486 Chinese patients with congenital heart disease. Eur J Med Genet 51:527–535
Zhang W, Li X, Shen A, Jiao W, Guan X, Li Z (2009a) Screening NKX2.5 mutation in a sample of 230 Han Chinese children with congenital heart diseases. Genet Test Mol Biomarkers 13:159–162
Zhang WM, Li XF, Ma ZY, Zhang J, Zhou SH, Li T, Shi L, Li ZZ (2009b) GATA4 and NKX2.5 gene analysis in Chinese Uygur patients with congenital heart disease. Chin Med J (Engl) 122:416–419
Acknowledgments
The authors appreciate the support by National Leading Basic Research & Development Programs (973) entitled “Basic research on the pathogenesis and intervention of congenital heart diseases. Project 4: Mechanistic studies on the pathogenesis of conotruncal defects” (No. 2010CB529504).
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Peng, T., Wang, L., Zhou, SF. et al. Mutations of the GATA4 and NKX2.5 genes in Chinese pediatric patients with non-familial congenital heart disease. Genetica 138, 1231–1240 (2010). https://doi.org/10.1007/s10709-010-9522-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10709-010-9522-4