
Abstract
Congenital heart disease (CHD) is the most common congenital malformation, with evidence of a strong genetic component. We analyzed data from 223 consecutively ascertained families, each consisting of at least one child affected by a conotruncal defect (CNT) or hypoplastic left heart disease (HLHS) and both parents. The NimbleGen HD2-2.1 comparative genomic hybridization platform was used to identify de novo and rare inherited copy number variants (CNVs). Excluding 10 cases with 22q11.2 DiGeorge deletions, we validated de novo CNVs in 8 % of 148 probands with CNTs, 12.7 % of 71 probands with HLHS and none in 4 probands with both. Only 2 % of control families showed a de novo CNV. We also identified a group of ultra-rare inherited CNVs that occurred de novo in our sample, contained a candidate gene for CHD, recurred in our sample or were present in an affected sibling. We confirmed the contribution to CHD of copy number changes in genes such as GATA4 and NODAL and identified several genes in novel recurrent CNVs that may point to novel CHD candidate loci. We also found CNVs previously associated with highly variable phenotypes and reduced penetrance, such as dup 1q21.1, dup 16p13.11, dup 15q11.2-13, dup 22q11.2, and del 2q23.1. We found that the presence of extra-cardiac anomalies was not related to the frequency of CNVs, and that there was no significant difference in CNV frequency or specificity between the probands with CNT and HLHS. In agreement with other series, we identified likely causal CNVs in 5.6 % of our total sample, half of which were de novo.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Congenital heart disease (CHD) is the most common human congenital malformation, occurring in approximately 1 in 100 livebirths. Evidence for a genetic contribution to etiology is based on a sib recurrence risk 2–4 times higher than the overall incidence (Loffredo et al. 2004), the common occurrence of CHD in children with complete or partial aneuploidy (van Karnebeek and Hennekam 1999), the existence of several micro-deletion syndromes that include CHD (e.g., DiGeorge [MIM 188400], Williams [MIM 194050] and Alagille [MIM 118540]) and the documentation in CHD patients of point mutations in several genes known to be involved in heart development (Goldmuntz et al. 2001; Yang et al. 2012). Recent studies have also shown that rare de novo and inherited copy number variants (CNVs) occur in 5–10 % of probands with CHD, classified in a variety of ways (Breckpot et al. 2010, 2011; Erdogan et al. 2008; Greenway et al. 2009; Hitz et al. 2012; Lalani et al. 2013; Silversides et al. 2012; Soemedi et al. 2012b). This evidence points to great heterogeneity among the genetic factors implicated in CHD. So far the only single common cause identified is the 1–3 Mb 22q11.2 deletion that leads to the DiGeorge-velocardio-facial syndrome.
Our study was designed to provide an unbiased estimate of the contribution of de novo and rare inherited CNVs to two types of CHD, conotruncal anomalies (CNT) and hypoplastic left heart syndrome (HLHS). We selected these two because they are commonly considered to arise by discrete developmental mechanisms (Restivo et al. 2006; Grossfeld 2002). We hypothesized that different genetic pathways might be revealed by identifying CNVs. For both types of anomaly our sample is unique in analyzing only complete trios and including complete ascertainment of newborns irrespective of family history or presence of other malformations.
CNTs arise during the formation of the outflow vessels from the heart and include six defects, not mutually exclusive: (1) double outlet right ventricle (DORV), where both the aorta and the pulmonary artery arise from the right ventricle; (2) tetralogy of Fallot (TOF), where an anterior malalignment ventricular septal defect results in an overriding aorta, pulmonary stenosis and right ventricular hypertrophy (3) interrupted aortic arch (IAA), where the aorta is subdivided and cannot deliver blood efficiently to the body; (4) transposition of the great arteries (TGA), where the pulmonary artery and the aorta arise from the inappropriate ventricle; (5) truncus arteriosus (TA), where a single great vessel leaves the heart and gives rise to the aorta and pulmonary artery; and (6) conoventricular septal defect (CVSD) where a defect in the conoventricular portion of the interventricular septum results in malalignment. Several studies have studied CNVs in TOF only, further defined by family history or extra-cardiac defects, but only one (Soemedi et al. 2012b) has studied TOF without any restrictions. No study has considered CNT as a single group.
HLHS is characterized by severe stenosis or atresia of the mitral and/or aortic valves, a small left ventricle and aortic arch hypoplasia. Restricted flow into or out of the left ventricle leads to diminished ventricular blood and thereafter poor growth and development of the left ventricle and outflow tract. Because of its rarity (~1 % of CHD) only one study has reported CNVs in a substantial number of HLHS trios (Hitz et al. 2012), but that study focused on cases with a positive family history for CHD.
To detect CNVs we performed comparative genomic hybridization (CGH) on family trios that comprised a proband and both parents. We also studied affected sibs when available. Our primary analysis consisted of comparison of CGH microarray ratio data in probands and parents to detect de novo CNVs. Following validation of candidate de novo events, we assessed the genes involved for known or potential function in heart development. We also examined ultra-rare inherited variants to identify those that overlapped de novo events, recurred in our sample or included candidate genes. Secondary analyses compared the frequency and nature of CNVs between probands with CNT and HLHS, and between male and female probands. Finally, we assessed the genes found in CNVs for a role in CHD using both the literature and Ingenuity Pathway analysis.
Methods
Recruitment and protocol
We identified all infants <1 month with CNT and all children <5 years with HLHS seen in the Division of Cardiology of the Morgan Stanley Children’s Hospital of New York. The majority were inpatients. The difference in age range for the two diagnostic classes was necessary to achieve our sample size goals because HLHS is less common than CNT. We also recruited families with prenatal echocardiographic diagnoses of a CNT or HLHS, although only those infants delivered live at our hospital were eligible for the study. We identified cases from the end of November of 2006 to the end of May 2010. Initial cardiac classification was based on the diagnosis in the medical record. Our team cardiologist (IW) reviewed all echocardiograms and determined the research diagnosis. If diagnoses were inconsistent, the data were reviewed with a second cardiologist to reach a consensus.
This study was approved by the Institutional Review Boards of Columbia University Medical Center and Cold Spring Harbor Laboratory (CSHL). To be eligible for the study, we required that both biological parents be available and consent. Participation involved permission to review the medical records of parents and child, to interview both parents about demographic, medical, reproductive and family history, to obtain a blood sample from the child and parents, to carry out a genetic examination of the child for dysmorphic features and to perform standardized research echocardiograms on both parents to assess for previously undetected cardiac anomalies such as right aortic arch or bicuspid aortic valve. Information on developmental status at age ≥2 years was available on some patients via medical records, parental discussion or the genetic exam for HLHS patients ascertained at age two or older.
Sample collection and cytogenetics
Two blood samples were drawn from the proband and each parent: one in EDTA for DNA extraction and one in sodium heparin for chromosome preparations. Three-day PHA-stimulated cultures were set up and the fixed cells were stored at −20 °C for possible future metaphase preparations or fluorescence in situ hybridization (FISH). On rare occasions we used saliva samples from parents.
Karyotype analysis and FISH to detect the 22q11.2 DiGeorge deletion were carried out on most CNT patients and some HLHS patients as part of routine patient care in the Cytogenetics Laboratory at New York Presbyterian Hospital. On occasion, when a child’s blood sample was difficult to obtain or low in volume, we used a discarded blood sample from the clinical laboratory to set up a PHA-stimulated culture and isolate DNA. When no karyotype information was available from laboratory or hospital records, we used the saved fixed cells to prepare Giemsa-banded karyotypes and carry out FISH with the TUPLE or N25 (Abbot Molecular USA, LSI TUPLE: Vysis D22S75 LSI N25) probe to determine DiGeorge status.
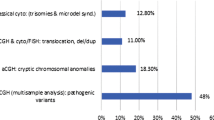
The disposition of the original 400 families ascertained with CNT or HLHS is shown in Table 1. Families in which there was an affected sibling (including twins) are counted only once. Of the 400 cases ascertained, 56 were ineligible for study. We list reasons for ineligibility, which includes complete aneuploidy, in the footnote of Table 1. Cases with unbalanced rearrangements were considered eligible since partial aneuploidies detectable by karyotype would still be informative. Cases with the 22q11.2 DiGeorge deletion detected by FISH were included so we could confirm that the microarray detected all known cases and to permit a detailed phenotype–genotype analysis, if desired. Of the 344 eligible cases, we obtained DNA samples from the proband, mother and father (a trio) in 238 (69.2 %): microarray analysis was successful in 223. Reasons for non-participation or microarray failure are given in the footnotes of Table 1.
Table 1 also shows the distribution of heart defects, sex ratio, and the frequency of the DiGeorge deletion among the 223 families with successful microarrays. CNT probands comprise 66.4 % of the sample. The overall male: female ratio is 2.0 and similar in both defect classes, in agreement with other reports (Loffredo 2000). Known DiGeorge syndrome cases made up 4.5 % of the sample (all had CNT). The distribution of heart defects, sex ratio and DiGeorge patients did not differ between the 121 eligible trios who did not complete the study and the 223 trios who had complete microarrays.
Microarray testing and data analysis
Blood or saliva samples were kept refrigerated and processed within 3 days. Genomic DNAs were isolated using Qiagen Flexigene kits (Qiagen Sciences, Germantown, MD, USA) and aliquots were sent to CSHL. Each sample was labeled with a unique identifier different from the family number and the laboratory was blind to all data except cardiac diagnosis, sex, ethnicity, and year of birth. We used CGH (Iafrate et al. 2004; Sebat et al. 2004) to analyze copy number variation. As control trios unaffected with CHD we used 750 trios from the Simons Simplex Collection (SSC), each consisting of a father, mother and unaffected child (Levy et al. 2011). These families were ascertained through an autistic child.
All samples were hybridized on the NimbleGen HD2 2.1-million probe microarray platform (http://www.nimblegen.com/products/cgh/wgt/human/2.1m/index.html) with oligonucleotides optimized for both hybridization performance and uniform genome coverage. Genomic DNAs were sent to NimbleGen’s Icelandic facility and hybridized against a male reference genome using a two-color protocol. CHD samples were labeled with Cy3 and the reference was labeled with Cy5.
Hybridization data underwent extensive processing before determining segments of altered copy number (Lee et al. 2012). We extracted signal and noise parameters from each hybridization, and used these for quality control and to model integer copy number states. To detect non-biological families we computed a relatedness measure for each pair of hybridizations, according to the same protocol as Levy et al. (2011), where the scoring methods are described in depth. For partitioning the genome into intervals of constant copy number, we used KS segmentation, minimization of variance and Kolmogorov–Smirnov statistics to establish significance (Grubor et al. 2009). We also employed a trio-based hidden Markov Model (HMM) to build databases of high-confidence events and transmissions. High-confidence events from 1500 control parents from the SSC (Levy et al. 2011) were utilized to determine the frequency of copy number variation for all regions represented on the HD2 microarray. We restricted calls to autosomal probes that did not have known extra mappings to the human genome (hg18 build) outside the event region, as well as probes that were rarely polymorphic (occurred in no more than 5 parents) in the parental database. We then relaxed these probe restrictions to consider lower quality trios, probes on the X-chromosome, and probes with higher frequencies of polymorphism in the controls (but not >20/1,500 parents), and de novo events of lower significance (p value <10−7). We then curated the resulting list by manual inspection of the graphics. The reasons for not using microarray data are shown in the footnote of Table 1.
Validation of CGH detected changes
We used FISH and real-time quantitative PCR (qPCR) to confirm de novo CNVs detected by microarray analysis. FISH was performed according to our previously published protocol (Jobanputra et al. 2005) using home-labeled probes obtained from BAC PAC Resources, CA. For qPCR we used a relative quantitation method (Kindich et al. 2005), which uses SYBR Green I to compare DNA copy number in the research sample relative to a reference sample.
Statistical and gene function analyses
Differences in the frequencies of CNVs by diagnosis or gender were tested by Chi square analysis. We imported the list of genes present in de novo and rare inherited CNVs into the Ingenuity pathway analysis (IPA, Ingenuity Systems, http://www.ingenuity.com) Web server. We chose human as the species option. We used the right-tailed Fisher’s exact test, corrected for multiple hypothesis testing, to calculate alpha levels for the genes that enriched in biological functions, canonical pathways or networks. A maximum of 35 molecules by default was set in each network and all evidence of experimentally observed, predicted high or moderate confidence was used. Gene networks were algorithmically generated based on their connectivity to the Ingenuity knowledge base.
Results
Karyotype analysis
Of the 223 probands with completed CGH, only one sample did not have a completed G-banded karyotype (this case had no detectable de novo CNVs). There were three cases with a non-exclusionary abnormal karyotype. One had a de novo unbalanced translocation, 46,XY,add(13)(p11.2), which the CGH and FISH data showed was an 18-Mb duplication of chromosome 1q42.2-q44. Another had an inherited balanced translocation, 46,XY,t(3;14)(p21;q22)mat, which had no CNVs at the breakpoints of the translocation (or elsewhere) and likely represents a truly balanced event. The third had a non-mosaic small marker, 47,XY,+M but no detectable CNVs indicating that the marker contained only repetitive DNA or a region heavily filtered in the CGH data analysis.
DiGeorge syndrome
Nine of the 223 probands had a de novo >2 Mb deletion of 22q11.2 in the DiGeorge region, as detected by both FISH and CGH: six of these patients had TOF, two had TA and one had IAA. A tenth case, a member of a set of monozygotic twins with a maternally inherited deletion, had TA; the twin had TOF. All cases previously diagnosed by FISH were confirmed on microarray. DiGeorge syndrome cases made up 4.5 % of the total sample and 6.8 % of all CNT cases. This rate is somewhat lower than that found in previous series (Goldmuntz et al. 1998), possibly because our ascertainment was only through CHD.
Parental echocardiograms
We obtained parental echocardiograms because the literature suggested that clinically undetected heart defects were increased in parents of children with CHD. In particular, right-sided aortic arch has been reported among parents of children with CNT and bicuspid aortic valve has been reported among parents with HLHS (Loffredo et al. 2004). Among the 223 trios, we carried out echocardiograms in 416 parents (208 mothers, 208 fathers). No parent had a right-sided aortic arch. Three fathers had a bicuspid aortic valve. Two of their offspring had DORV and the other TGA.
De novo CNVs detected by microarray analysis
Our first analysis examined trios to find de novo deletions or duplications. We classified 33 changes as probable de novo rare events. Only one of these changes, the 16p13.11 duplication, was seen once in a control SSC trio. De novo CNVs other than 22q11.21 deletions ranged in size from 6 to 18 Mb. All 12 CNVs ≥100 kb were confirmed by FISH with BAC probes. All but one CNV ≤100 kb were confirmed by qPCR using primers specific to the region. The 32 remaining de novo CNVs are described in Table 2. Representative array diagrams are shown in Figure 1S (Supplement).
Mosaicism
Validation studies identified three cases of mosaicism that had not been detected in the CGH analysis. In one case (ID 5105), FISH with two BACs present in the deleted region showed that about 10 % (8/82) of cells scored in the father had the same 12q24.31 deletion found in 100 % of cells from his son (Fig. 1). In another case (ID 5159) with a 7-kb duplication in 7q22.1, qPCR gave a value in the father that was intermediate between the son and the mother, suggesting that the 7-kb duplication present in the son with CHD was inherited from a mosaic father. Because of the small size, this event could not be confirmed by FISH. Although these lesions must have been inherited from the father, we have still scored them as de novo because in comparable studies without FISH (such as the control SSC sample), parental mosaicism at this level would not be identified. In a third case (ID 5144), we also detected that the 49-kb deletion in 2q23.1 found by CGH was present by FISH in only about 50 % of cells in the proband. An artifact of the FISH probes causing variable signal is ruled out because we ran FISH on each member of the trio, providing a positive and negative control for each probe (see Fig. 1).

FISH analysis on case 5105 using BAC probe RP11-4681113 to identify the deleted region in 12q24.31. Signal is seen on only one chromosome in the child (with a deletion) and on both chromosomes 12 in the mother. In the father, 11 % of 100 cells were missing the probe from one chromosome and 89 % of cells had two signals. This indices that the deletion was a somatic event in the father and was inherited rather than de novo as inferred from the array
FISH allowed us to see that the largest duplication was a >18-Mb segment of terminal 1q visible in the karyotype as additional non-heterochromatic material on chromosome 13p. In retrospect the 8.6-Mb terminal deletion in chromosome 1q was also just visible on the karyotype, but had been missed. The smallest verified lesion detected by CGH was 7.4 kb.
Table 2 lists the 32 de novo CNVs, including those with DiGeorge syndrome. Four de novo lesions contained no genes. Six contained genes or regions previously associated with pathology (see “Discussion” and Table 7)]. Altogether we detected 12 de novo deletions and 11 de novo duplications in 213 cases, excluding those with DiGeorge syndrome. Three cases had two de novo deletions each, giving a total of 20 (9.4 %) probands with at least one de novo lesion. This is significantly higher than the rate of 2 % found in the SSC trios. Details on the de novo events in the SSC families can be found in Levy et al. (2011), supplementary materials.
Table 3 summarizes the frequency of de novo CNVs by type of CHD and gender. The frequency of de novo CNVs did not differ significantly between probands with CNT (8 %) and probands with HLHS (12.7 %), nor between male (10.6 %) and female (7 %) probands. For CNTs, we show findings for probands with and without TOF because TOF was analyzed separately in other reports (Greenway et al. 2009; Soemedi et al. 2012a).
Ultra-rare inherited CNVs
We classified 224 CNVs in 163 trios, as ultra-rare inherited because they were not seen in more than one instance in 1500 control SSC parents. The numbers of events per family ranged from 0 to 5, with 122/224 (55 %) maternally inherited. There were eight cases where an inherited lesion was similar or identical to one occurring de novo. To reduce these data to those most likely to be meaningful, we focused on ultra-rare inherited CNVs that (1) overlapped de novo events, (2) were identical in an affected sib, (3) contained candidate genes for CHD, or (4) occurred in more than one trio. “Recurrence” was defined as overlapping that involved at least one gene. Deletions and duplications of the same region were considered different CNVs, but 19 different CNVs met these criteria. One was an inherited case of the standard DiGeorge deletion, which will not be considered in further analyses. Omitting the DiGeorge case, 40 instances of ultra-rare inherited CNVs occurred among 30 different probands. Twenty-four were inherited from the mother and 16 from the father (a non-significant difference). Table 4 lists probands with rare inherited CNVs meeting our criteria, their frequency in CHD and SSC trios, the genes involved and their ethnicity.
All CNVs in Table 4 contain genes except for a deletion in 9p21.1 that was seen in one de novo and two inherited cases. Two candidate genes were identified within lesions, CFC1 [MIM 605194] in 2q21.1 and NODAL [MIM 601265] in 10q22.1 found in an affected sibling and normal parent (see Figure 2S, Supplement). There was also an inherited duplication in the distal DiGeorge region, overlapping the region deleted in a de novo CNV.
Four CNVs were seen in at least three probands, either de novo or inherited: a duplication in 1q21.1, a duplication within the Prader–Willi region [MIM 176270], a deletion in 7q21.11 and a duplication in 8p23.1. The last two contain only one gene each (CD36 and BLK) that are not obvious CHD candidate genes.
Sample diversity
Our CHD sample is ethnically diverse: 40 % of probands were White non-Hispanic, 11 % were Black non-Hispanic, 33 % were at least half Hispanic and 5 % were Asian. Our control sample of SSC trios is 77 % white, 4 % black and 4 % Asian; data on Hispanic status is unavailable. Ethnicity cannot confound identification of de novo CNVs because the parents serve as ethnicity-matched controls. However, comparison of the rates or types of ultra-rare inherited CNVs between our sample and the SSC may be confounded by ethnic differences in the frequency of variants. One would expect a higher frequency of ultra-rare CNVs among the racial groups underrepresented in the SSC control sample. Table 5 shows that the proportions of probands with an ultra-rare inherited CNV did not differ significantly with ethnicity (p = 0.08) although the data suggest the rate may be higher among blacks.
Relationship between CNVs and extra-cardiac phenotypes
Based on the medical records and examination by a clinical geneticist with expertise in dysmorphology, 27 of the 213 cases without DiGeorge syndrome (13.4 %) had a major extra-cardiac abnormality. We classified children as having (1) another major malformation, (2) another minor malformation, (3) dysmorphology only, or (4) normal. The rates of de novo and ultra-rare inherited CNVs did not vary with the presence of non-cardiac features (Table 6).
Apart from the cases with DiGeorge syndrome, none of the cases with similar CNVs presented with similar extra-cardiac features. The most common dysmorphic features were microtia or other ear anomalies and micrognathia. There were also two with features of heterotaxy which did not have genes known to be involved in this defect.
Pathway analyses
Supplementary Table S1 shows the top five ranking functional categories, along with their P values, identified in an Ingenuity Pathway analysis of 107 genes from Tables 2 and 4. The most significant functional category is cardiovascular system development and function. There are 15 genes from identified CNV regions involved in this category. The top diseases and disorders are cardiovascular and developmental diseases. This analysis reaffirms that the genes identified by CNV analysis are mostly relevant to CHD. A similar but not identical pathway analysis (NETBAG) of the de novo CNVs found in the SSC (Gilman et al. 2011) identified a network of genes primarily related to neuron functioning and not to cardiovascular disease.
Discussion
CNVs likely related to CHD in proband
The substantially higher rate of de novo CNVs in probands with CHD than in control SSC families (9 vs. 2 %) indicates that many of these lesions are likely to be involved in the pathogenesis of CHD. Among the de novo or rare inherited CNVs we detected, there are 12 where we consider that the CNV is likely to be causally related to CHD. Table 7 lists these cases including the gene content of the CNV, any known clinical information about the child, and the evidence that this CNV may be related to the CHD. Three of the implicated genes, NODAL, CFC1, and NOMO3, are interactive in the nodal pathway, and suggest a major role for this pathway in CHD. Other CNVs, like the duplication in 1q21.1, have been reported in most other series.
CNVs with disease associations other than CHD
We detected three CNVs (Tables 2, 4) that have been reported in association with non-cardiac congenital anomalies, but not CHD. These could be either unrelated to the CHD or represent an enlarged spectrum of defects for these CNVs.
-
1.
A proband with HLHS had a de novo deletion of MBD5 [MIM 611472] in 2q23.1. This deletion has been reported in association with a variety of congenital anomalies (Noh and Graham 2012; Williams et al. 2010) including microcephaly, seizures and severe cognitive delay. Our proband had no extra-cardiac lesions and was developing normally at 2 years. The mosaicism in this patient complicates expectations for the phenotype.
-
2.
Two inherited and one de novo duplication included part of the Prader–Willi region of chromosome 15. Duplications in this region are associated with autism and other developmental disorders (Stewart et al. 2011) [MIM 608636]. One of our inherited cases had hemifacial microsomy, deafness and delayed development (classified clinically as Goldenhar syndrome). The other two cases had no extra-cardiac anomalies or developmental problems. We did not detect any cases with the deletion in the Prader–Willi region reported by Soemedi et al. (2012b) in association with CHD, especially HLHS.
-
3.
In a patient with TOF, a deletion in 17p13.3 included part of the proximal Miller–Dieker (MDS) region [MIM247200] containing YWHAE and CRK, but not LIS1 (the gene where deletion causes lissencephaly). The proband had no extra-cardiac malformations and by parental report was developing normally at 3 years. CHD was not reported among the features of 14 patients with deletions of 17p13.3 containing TUSC5, YWHAE and CRK reported by (Bruno et al. 2010).
Comparison with the literature and new findings indicated by this study
When we began this study there were almost no reports of CNVs in CHD. Our goal was to test whether CNVs were more common in cases than external controls and to examine whether specific CNVs were associated with either CNT or HLHS. Over the course of our study, it became apparent that CNVs contribute significantly to the etiology of CHD (Breckpot et al. 2010, 2011; Erdogan et al. 2008; Greenway et al. 2009; Hitz et al. 2012; Lalani et al. 2013; Silversides et al. 2012); Soemedi et al. 2012b). These same reports identified CNVs that contained genes involved in cardiac development. Reports vary widely in the type and sensitivity of the microarray platform used, the size used to define CNVs, the types of CHD included, the methods of ascertainment and selection for or against cases with extra-cardiac anomalies or positive family history. Our study and that of Soemedi et al. (2012b) are the only two to systematically ascertain all cases meeting with CHD diagnostic classes unselected for the presence or absence of extra-cardiac abnormalities or a positive family history. Use of unselected samples permits an unbiased estimate of the frequency of de novo CNVs within diagnostic classes as well as comparisons of cases with and without extra-cardiac abnormalities. Unlike other series parental studies were done in all cases, allowing an estimate of the frequency of de novo vs. inherited CNVs in each defect. Below we elaborate on the findings from our study, which provide new information on the relationship of CNVs to two types of CHD, CNT and HLHS.
Specificity of CNVs for CNT or HLHS and extra-cardiac anomalies
We hypothesized that HLHS and CNT differed sufficiently in pathogenesis such that the genes involved and possibly the frequency of CNVs would be different. Our data do not support this hypothesis. For example, the duplication in 1q21.1 was found in three cases with CNT and one with HLHS. The same inherited duplication in 8p23.1 was found in two cases with HLHS and two cases with CNT. The literature on CNVs in CHD also tends to report many of the same CNVs regardless of the diagnostic criteria for selection. Within our sample, the frequency of de novo CNVs did not differ significantly between probands with HLHS (12.3 %) and probands with CNT (7.4 %). For TOF alone (omitting cases of DiGeorge syndrome), de novo CNVs occurred among 6.1 %, a rate comparable to the rates of 4.6 % (Soemedi et al. 2012b) and 8 % Greenway et al. (2009) in other series. We know of no other systematic study of HLHS trios with which to compare our data. One possible limitation of our sample is that 35 % of probands with HLHS were ascertained at >1–60 months, potentially selecting for less lethal forms of the disorder. It is possible, therefore, that some of the CNVs we detected are associated with survival, rather than with the occurrence of HLHS. It is also possible that the HLHS cases with the highest mortality are caused more often or by different kinds of CNVs that increase the chance of death, e.g., by affecting multiple systems.
Although we expected that cases with extra-cardiac malformations would show a higher rate of de novo CNVs, this was not supported by our data (Table 6). This finding is contrary to some reports in the literature (e.g., Breckpot et al. 2011). We also found no recurrent extra-cardiac anomalies with the same CNV.
The lack of specificity of CNVs for HLHS, CNT and extra-cardiac malformations suggests that the specificity of the heart or other malformations must often lie in pathways downstream from the genes identified in recurrent CNVs.
Parental echocardiograms
We carried out parental echocardiograms, with special attention to right aortic arch and bicuspid aortic valve, to detect inherited lesions in apparently normal parents. The frequency of defects in our sample was lower (3/416 parents) than we expected based on previous studies and not related to the type of defect in the proband. This finding may reflect the unselected nature of our sample. The three defects did not occur among parents carrying inherited rare CNVs. We conclude that parental echocardiograms, which are difficult to obtain, are likely not needed in future studies of inherited lesions in CHD.
Mosaicism
We detected low-level parental mosaicism in 10 % (2/20) of probands with de novo CNVs. This observation shows the limitations of CGH and the value of confirmation by FISH or qPCR. Parental mosaicism has important implications for subsequent pregnancies. We therefore recommend examining at least 20 cells when confirming de novo status of CNVs by FISH. FISH also detected that one of the de novo deletions was 50 % mosaic in the proband, which, in retrospect, might have been suspected from the array ratios.
Significance of CNVs in the etiology of CHD
We conclude that 5.6 % (12/213) of probands had CNVs that are likely to be causally related to the CHD; half are de novo and half inherited from clinically normal parents.
Several CNVs recur in multiple studies. The most frequent appears to be the 1q21.1 duplication, which varies somewhat in size and coverage and where no particular gene can yet be designated as causal. This CNV occurs in at least 1 % of reported CHD cases. The largest study (Soemedi et al. 2012a) concluded that the 1q21.1 duplication was associated specifically with TOF and that the GJA5 gene was involved in all CHD cases. Neither our data nor those of Hitz et al. (2012) support these conclusions. None of our four cases with the duplication had TOF; only one of our 1q21.1 duplications (in a proband with HLHS) contained GJA5. Soemedi et al. (2012a) also concluded that a deletion of the 15q11.2 region was associated with HLHS but no study has replicated this result. It remains for further studies of carefully characterized patients to establish the phenotypic spectrum of even the most common CNVs.
Since the frequency of de novo CNVs among controls is approximately 2 %, one-fifth or more of de novo CNVs identified are expected to be chance occurrences unrelated to the CHD. Recurrence of a CNV strengthens the likelihood of a true association. Three types of CNVs can be distinguished among those found in CHD: (1) CNVs associated with well-described micro-deletion syndromes that include CHD (e.g., DiGeorge and William’s syndromes) or partial aneuploidy due to chromosomal rearrangements; (2) CNVs that include genes known or likely to be involved in heart development (e.g., GATA4 and three NODAL-related genes in our sample); and (3) CNVs associated with a wide variety of other phenotypes such as autism or schizophrenia, which often show reduced penetrance or inheritance from an unaffected parent.
Only CNVs not falling into the three classes described above contain genes that could be candidates for previously unrecognized pathways in heart development. Validation would need functional studies, replication in other series, or detection of gene mutations in other patients by direct sequencing or exome sequencing. Every study so far has identified a new set of candidates with very little overlap. We detected 12 CNVs that were recurrent either as de novo or ultra-rare inherited event (see Table 4), but contain no obvious candidate genes for CHD. With increasing knowledge this may change. For example, a rare inherited duplication in 8p23.1 containing only the gene BLK was present in four CHD trios but only one SSC family. BLK is a tyrosine kinase expressed chiefly in B-lymphocytes. However, it is only 144 kb distal to GATA4 and copy changes in the region might affect gene regulation.
Perhaps the most intriguing set of CNVs established as associated with CHD are those that are also associated with other phenotypes, most often autism or developmental disorders, such as dup1q21.1, dup16p13.11, dup15q11.2-13, and dup22q11.2. Characteristic of these CNVs is reduced penetrance demonstrated by frequent inheritance from a normal parent. Both deletions and duplications are commonly pathogenic. These chromosomal regions tend to be very complex, with multiple small and large repeats both on the same and different chromosomes (Mefford et al. 2008). Dosage changes in these regions may modify regulation of multiple pathways, leading to widespread and variable effects in development. Search for a single gene responsible for these variable developmental disorders may not be fruitful. Other modifying gene changes may needed (e.g. Jiang et al. 2011), or the relevant dosage changes may be in non-genic regions.
References
Ben-Shachar S, Ou Z, Shaw CA, Belmont JW, Patel MS, Hummel M, Amato S, Tartaglia N, Berg J, Sutton VR, Lalani SR, Chinault AC, Cheung SW, Lupski JR, Patel A (2008) 22q11.2 distal deletion: a recurrent genomic disorder distinct from DiGeorge syndrome and velocardiofacial syndrome. Am J Hum Genet 82:214–221. doi:10.1016/j.ajhg.2007.09.014
Breckpot J, Thienpont B, Peeters H, de Ravel T, Singer A, Rayyan M, Allegaert K, Vanhole C, Eyskens B, Vermeesch JR, Gewillig M, Devriendt K (2010) Array comparative genomic hybridization as a diagnostic tool for syndromic heart defects. J Pediatr 156:810–7, 817 e1–817 e4. doi:10.1016/j.jpeds.2009.11.049
Breckpot J, Thienpont B, Arens Y, Tranchevent LC, Vermeesch JR, Moreau Y, Gewillig M, Devriendt K (2011) Challenges of interpreting copy number variation in syndromic and non-syndromic congenital heart defects. Cytogenet Genome Res 135:251–259. doi:10.1159/000331272
Bruno DL, Anderlid BM, Lindstrand A, van Ravenswaaij-Arts C, Ganesamoorthy D, Lundin J, Martin CL, Douglas J, Nowak C, Adam MP, Kooy RF, Van der Aa N, Reyniers E, Vandeweyer G, Stolte-Dijkstra I, Dijkhuizen T, Yeung A, Delatycki M, Borgstrom B, Thelin L, Cardoso C, van Bon B, Pfundt R, de Vries BB, Wallin A, Amor DJ, James PA, Slater HR, Schoumans J (2010) Further molecular and clinical delineation of co-locating 17p13.3 microdeletions and microduplications that show distinctive phenotypes. J Med Genet 47:299–311. doi:10.1136/jmg.2009.069906
Cooper GM, Coe BP, Girirajan S, Rosenfeld JA, Vu TH, Baker C, Williams C, Stalker H, Hamid R, Hannig V, Abdel-Hamid H, Bader P, McCracken E, Niyazov D, Leppig K, Thiese H, Hummel M, Alexander N, Gorski J, Kussmann J, Shashi V, Johnson K, Rehder C, Ballif BC, Shaffer LG, Eichler EE (2011) A copy number variation morbidity map of developmental delay. Nat Genet 43:838–846. doi:10.1038/ng.909
Erdogan F, Larsen LA, Zhang L, Tumer Z, Tommerup N, Chen W, Jacobsen JR, Schubert M, Jurkatis J, Tzschach A, Ropers HH, Ullmann R (2008) High frequency of submicroscopic genomic aberrations detected by tiling path array comparative genome hybridisation in patients with isolated congenital heart disease. J Med Genet 45:704–709. doi:10.1136/jmg.2008.058776
Gilman SR, Iossifov I, Levy D, Ronemus M, Wigler M, Vitkup D (2011) Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron 70:898–907
Goldmuntz E, Bamford R, Karkera JD, dela Cruz J, Roessler E, Muenke M (2002) CFC1 mutations in patients with transposition of the great arteries and double-outlet right ventricle. Am J Hum Genet 70:776–780. doi:10.1086/339079
Goldmuntz E, Clark BJ, Mitchell LE, Jawad AF, Cuneo BF, Reed L, McDonald-McGinn D, Chien P, Feuer J, Zackai EH, Emanuel BS, Driscoll DA (1998) Frequency of 22q11 deletions in patients with conotruncal defects. J Am Coll Cardiol 32:492–498
Goldmuntz E, Geiger E, Benson DW (2001) NKX2.5 mutations in patients with tetralogy of fallot. Circulation 104:2565–2568
Greenway SC, Pereira AC, Lin JC, DePalma SR, Israel SJ, Mesquita SM, Ergul E, Conta JH, Korn JM, McCarroll SA, Gorham JM, Gabriel S, Altshuler DM, Quintanilla-Dieck Mde L, Artunduaga MA, Eavey RD, Plenge RM, Shadick NA, Weinblatt ME, De Jager PL, Hafler DA, Breitbart RE, Seidman JG, Seidman CE (2009) De novo copy number variants identify new genes and loci in isolated sporadic tetralogy of Fallot. Nat Genet 41:931–935. doi:10.1038/ng.415
Grossfeld P (2002) The genetics of hypoplastic left heart syndrome. Futura Publishing Co., Inc., Armonk
Grubor V, Krasnitz A, Troge JE, Meth JL, Lakshmi B, Kendall JT, Yamrom B, Alex G, Pai D, Navin N, Hufnagel LA, Lee YH, Cook K, Allen SL, Rai KR, Damle RN, Calissano C, Chiorazzi N, Wigler M, Esposito D (2009) Novel genomic alterations and clonal evolution in chronic lymphocytic leukemia revealed by representational oligonucleotide microarray analysis (ROMA). Blood 113:1294–1303. doi:10.1182/blood-2008-05-158865
Gu H, Smith FC, Taffet SM, Delmar M (2003) High incidence of cardiac malformations in connexin40-deficient mice. Circ Res 93:201–206. doi:10.1161/01.RES.0000084852.65396.70
Hitz MP, Lemieux-Perreault LP, Marshall C, Feroz-Zada Y, Davies R, Yang SW, Lionel AC, D’Amours G, Lemyre E, Cullum R, Bigras JL, Thibeault M, Chetaille P, Montpetit A, Khairy P, Overduin B, Klaassen S, Hoodless P, Nemer M, Stewart AF, Boerkoel C, Scherer SW, Richter A, Dube MP, Andelfinger G (2012) Rare copy number variants contribute to congenital left-sided heart disease. PLoS Genet 8:e1002903. doi:10.1371/journal.pgen.1002903
Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, Scherer SW, Lee C (2004) Detection of large-scale variation in the human genome. Nat Genet 36:949–951. doi:10.1038/ng1416
Jiang Q, Ho YY, Hao L, Nichols Berrios C, Chakravarti A (2011) Copy number variants in candidate genes are genetic modifiers of Hirschsprung disease. PLoS ONE 6:e21219. doi:10.1371/journal.pone.0021219
Jobanputra V, Sebat J, Troge J, Chung W, Anyane-Yeboa K, Wigler M, Warburton D (2005) Application of ROMA (representational oligonucleotide microarray analysis) to patients with cytogenetic rearrangements. Genet Med 7:111–118. doi:10.109701.GIM.0000153661.11110.FB
Kaminsky EB, Kaul V, Paschall J, Church DM, Bunke B, Kunig D, Moreno-De-Luca D, Moreno-De-Luca A, Mulle JG, Warren ST, Richard G, Compton JG, Fuller AE, Gliem TJ, Huang S, Collinson MN, Beal SJ, Ackley T, Pickering DL, Golden DM, Aston E, Whitby H, Shetty S, Rossi MR, Rudd MK, South ST, Brothman AR, Sanger WG, Iyer RK, Crolla JA, Thorland EC, Aradhya S, Ledbetter DH, Martin CL (2011) An evidence-based approach to establish the functional and clinical significance of copy number variants in intellectual and developmental disabilities. Genet Med 13:777–784. doi:10.1097/GIM.0b013e31822c79f9
Kindich R, Florl AR, Jung V, Engers R, Muller M, Schulz WA, Wullich B (2005) Application of a modified real-time PCR technique for relative gene copy number quantification to the determination of the relationship between NKX3.1 loss and MYC gain in prostate cancer. Clin Chem 51:649–652. doi:10.1373/clinchem.2004.045013
Kodo K, Nishizawa T, Furutani M, Arai S, Ishihara K, Oda M, Makino S, Fukuda K, Takahashi T, Matsuoka R, Nakanishi T, Yamagishi H (2012) Genetic analysis of essential cardiac transcription factors in 256 patients with non-syndromic congenital heart defects. Circ J 76:1703–1711
Kuang SQ, Guo DC, Prakash SK, McDonald ML, Johnson RJ, Wang M, Regalado ES, Russell L, Cao JM, Kwartler C, Fraivillig K, Coselli JS, Safi HJ, Estrera AL, Leal SM, Lemaire SA, Belmont JW, Milewicz DM, Gen TACI (2011) Recurrent chromosome 16p13.1 duplications are a risk factor for aortic dissections. PLoS Genet 7:e1002118. doi:10.1371/journal.pgen.1002118
Lalani SR, Shaw C, Wang X, Patel A, Patterson LW, Kolodziejska K, Szafranski P, Ou Z, Tian Q, Kang SH, Jinnah A, Ali S, Malik A, Hixson P, Potocki L, Lupski JR, Stankiewicz P, Bacino CA, Dawson B, Beaudet AL, Boricha FM, Whittaker R, Li C, Ware SM, Cheung SW, Penny DJ, Jefferies JL, Belmont JW (2013) Rare DNA copy number variants in cardiovascular malformations with extracardiac abnormalities. Eur J Hum Genet 21:173–181. doi:10.1038/ejhg.2012.155
Lee YH, Ronemus M, Kendall J, Lakshmi B, Leotta A, Levy D, Esposito D, Grubor V, Ye K, Wigler M, Yamrom B (2012) Reducing system noise in copy number data using principal components of self–self hybridizations. Proc Natl Acad Sci USA 109:E103–E110. doi:10.1073/pnas.1106233109
Levy D, Ronemus M, Yamrom B, Lee YH, Leotta A, Kendall J, Marks S, Lakshmi B, Pai D, Ye K, Buja A, Krieger A, Yoon S, Troge J, Rodgers L, Iossifov I, Wigler M (2011) Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron 70:886–897. doi:10.1016/j.neuron.2011.05.015
Loffredo CA (2000) Epidemiology of cardiovascular malformations: prevalence and risk factors. Am J Med Genet 97:319–325
Loffredo CA, Chokkalingam A, Sill AM, Boughman JA, Clark EB, Scheel J, Brenner JI (2004) Prevalence of congenital cardiovascular malformations among relatives of infants with hypoplastic left heart, coarctation of the aorta, and d-transposition of the great arteries. Am J Med Genet A 124A:225–230. doi:10.1002/ajmg.a.20366
Mefford HC, Sharp AJ, Baker C, Itsara A, Jiang Z, Buysse K, Huang S, Maloney VK, Crolla JA, Baralle D, Collins A, Mercer C, Norga K, de Ravel T, Devriendt K, Bongers EM, de Leeuw N, Reardon W, Gimelli S, Bena F, Hennekam RC, Male A, Gaunt L, Clayton-Smith J, Simonic I, Park SM, Mehta SG, Nik-Zainal S, Woods CG, Firth HV, Parkin G, Fichera M, Reitano S, Lo Giudice M, Li KE, Casuga I, Broomer A, Conrad B, Schwerzmann M, Raber L, Gallati S, Striano P, Coppola A, Tolmie JL, Tobias ES, Lilley C, Armengol L, Spysschaert Y, Verloo P, De Coene A, Goossens L, Mortier G, Speleman F, van Binsbergen E, Nelen MR, Hochstenbach R, Poot M, Gallagher L, Gill M, McClellan J, King MC, Regan R, Skinner C, Stevenson RE, Antonarakis SE, Chen C, Estivill X, Menten B, Gimelli G, Gribble S, Schwartz S, Sutcliffe JS, Walsh T, Knight SJ, Sebat J, Romano C, Schwartz CE, Veltman JA, de Vries BB, Vermeesch JR, Barber JC, Willatt L, Tassabehji M, Eichler EE (2008) Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N Engl J Med 359:1685–1699. doi:10.1056/NEJMoa0805384
Mohapatra B, Casey B, Li H, Ho-Dawson T, Smith L, Fernbach SD, Molinari L, Niesh SR, Jefferies JL, Craigen WJ, Towbin JA, Belmont JW, Ware SM (2009) Identification and functional characterization of NODAL rare variants in heterotaxy and isolated cardiovascular malformations. Hum Mol Genet 18:861–871. doi:10.1093/hmg/ddn411
Nagamani SC, Erez A, Bader P, Lalani SR, Scott DA, Scaglia F, Plon SE, Tsai CH, Reimschisel T, Roeder E, Malphrus AD, Eng PA, Hixson PM, Kang SH, Stankiewicz P, Patel A, Cheung SW (2011) Phenotypic manifestations of copy number variation in chromosome 16p13.11. Eur J Hum Genet 19:280–286. doi:10.1038/ejhg.2010.184
Noh GJ, Graham JM Jr (2012) 2q23.1 microdeletion of the MBD5 gene in a female with seizures, developmental delay and distinct dysmorphic features. Eur J Med Genet 55:59–62. doi:10.1016/j.ejmg.2011.10.001
Ou Z, Berg JS, Yonath H, Enciso VB, Miller DT, Picker J, Lenzi T, Keegan CE, Sutton VR, Belmont J, Chinault AC, Lupski JR, Cheung SW, Roeder E, Patel A (2008) Microduplications of 22q11.2 are frequently inherited and are associated with variable phenotypes. Genet Med 10:267−277. doi:10.1097/GIM.0b013e31816b64c2
Restivo A, Piacentini G, Placidi S, Saffirio C, Marino B (2006) Cardiac outflow tract: a review of some embryogenetic aspects of the conotruncal region of the heart. Anat Rec A Discov Mol Cell Evol Biol 288:936–943. doi:10.1002/ar.a.20367
Schinzel A (2001) Catalogue of unbalanced chromosome aberrations in man, 2nd edn. Walter de Gruyter, New York, pp 49–72
Sebat J, Lakshmi B, Troge J, Alexander J, Young J, Lundin P, Maner S, Massa H, Walker M, Chi M, Navin N, Lucito R, Healy J, Hicks J, Ye K, Reiner A, Gilliam TC, Trask B, Patterson N, Zetterberg A, Wigler M (2004) Large-scale copy number polymorphism in the human genome. Science 305:525–528. doi:10.1126/science.1098918
Selamet Tierney ES, Marans Z, Rutkin MB, Chung WK (2007) Variants of the CFC1 gene in patients with laterality defects associated with congenital cardiac disease. Cardiol Young 17:268–274. doi:10.1017/S1047951107000455
Silversides CK, Lionel AC, Costain G, Merico D, Migita O, Liu B, Yuen T, Rickaby J, Thiruvahindrapuram B, Marshall CR, Scherer SW, Bassett AS (2012) Rare copy number variations in adults with tetralogy of Fallot implicate novel risk gene pathways. PLoS Genet 8:e1002843. doi:10.1371/journal.pgen.1002843
Soemedi R, Topf A, Wilson IJ, Darlay R, Rahman T, Glen E, Hall D, Huang N, Bentham J, Bhattacharya S, Cosgrove C, Brook JD, Granados-Riveron J, Setchfield K, Bu’lock F, Thornborough C, Devriendt K, Breckpot J, Hofbeck M, Lathrop M, Rauch A, Blue GM, Winlaw DS, Hurles M, Santibanez-Koref M, Cordell HJ, Goodship JA, Keavney BD (2012a) Phenotype-specific effect of chromosome 1q21.1 rearrangements and GJA5 duplications in 2436 congenital heart disease patients and 6760 controls. Hum Mol Genet 21:1513–1520. doi:10.1093/hmg/ddr589
Soemedi R, Wilson IJ, Bentham J, Darlay R, Topf A, Zelenika D, Cosgrove C, Setchfield K, Thornborough C, Granados-Riveron J, Blue GM, Breckpot J, Hellens S, Zwolinkski S, Glen E, Mamasoula C, Rahman TJ, Hall D, Rauch A, Devriendt K, Gewillig M, O’Sullivan J, Winlaw DS, Bu’Lock F, Brook JD, Bhattacharya S, Lathrop M, Santibanez-Koref M, Cordell HJ, Goodship JA, Keavney BD (2012b) Contribution of global rare copy-number variants to the risk of sporadic congenital heart disease. Am J Hum Genet 91:489–501. doi:10.1016/j.ajhg.2012.08.003
Stewart LR, Hall AL, Kang SH, Shaw CA, Beaudet AL (2011) High frequency of known copy number abnormalities and maternal duplication 15q11-q13 in patients with combined schizophrenia and epilepsy. BMC Med Genet 12:154. doi:10.1186/1471-2350-12-154
Tomita-Mitchell A, Maslen CL, Morris CD, Garg V, Goldmuntz E (2007) GATA4 sequence variants in patients with congenital heart disease. J Med Genet 44:779–783. doi:10.1136/jmg.2007.052183
van Karnebeek CD, Hennekam RC (1999) Associations between chromosomal anomalies and congenital heart defects: a database search. Am J Med Genet 84:158–166
Williams SR, Mullegama SV, Rosenfeld JA, Dagli AI, Hatchwell E, Allen WP, Williams CA, Elsea SH (2010) Haploinsufficiency of MBD5 associated with a syndrome involving microcephaly, intellectual disabilities, severe speech impairment, and seizures. Eur J Hum Genet 18:436–441. doi:10.1038/ejhg.2009.199
Yang YQ, Li L, Wang J, Liu XY, Chen XZ, Zhang W, Wang XZ, Jiang JQ, Liu X, Fang WY (2012) A novel GATA4 loss-of-function mutation associated with congenital ventricular septal defect. Pediatr Cardiol 33:539–546. doi:10.1007/s00246-011-0146-y
Acknowledgments
We thank the families and their doctors whose cooperation made this study possible. We thank Alma Cruz, Renee Davenport, Alyssa Lanz and Michele Waste who carried out the fieldwork. We thank Drs. Stephanie Levasseur and the late Charles Kleinman, who assisted in diagnostic reviews. We thank Ann Kinney who carried out the statistical programming. This research was supported by NIH grant R01HL-080146 from NHLBI to DW and MW. The research was also supported in part by the National Center for Advancing Translational Sciences, National Institutes of Health, through Grant Number UL1 TR000040, formerly the National Center for Research Resources, Grant Number UL1 RR024156.
Author information
Authors and Affiliations
Corresponding author
Additional information
D. Warburton, M. Ronemus, and J. Kline contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
{kind=link}
{kind=link}
Rights and permissions
About this article
Cite this article
Warburton, D., Ronemus, M., Kline, J. et al. The contribution of de novo and rare inherited copy number changes to congenital heart disease in an unselected sample of children with conotruncal defects or hypoplastic left heart disease. Hum Genet 133, 11–27 (2014). https://doi.org/10.1007/s00439-013-1353-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-013-1353-9