
Abstract
Sorghum (Sorghum bicolor (L.) Moench) is a multipurpose crop grown for food, fodder, and bioenergy production. Its cultivated varieties, along with their wild counterparts, contribute to the core genetic pool. Despite the availability of several re-sequenced sorghum genomes, a variable portion of sorghum genomes is not reported during reference genome assembly and annotation. The present analysis used 223 publicly available RNA-seq datasets from seven sweet sorghum cultivars to construct superTranscriptome. This approach yielded 45,864 Representative Transcript Assemblies (RTAs) that showcased intriguing Presence/Absence Variation (PAV) across 15 published sorghum genomes. We found 301 superTranscripts were exclusive to sweet sorghum, including 58 de novo genes encoded core and linker histones, zinc finger domains, glucosyl transferases, cellulose synthase, etc. The superTranscriptome added 2,802 new protein-coding genes to the Sweet Sorghum Reference Genome (SSRG), of which 559 code for different transcription factors (TFs). Our analysis revealed that MULE-like transposases were abundant in the sweet sorghum genome and could play a hidden role in the evolution of sweet sorghum. We observed large deletions in the D locus and terminal deletions in four other NAC encoding loci in the SSRG compared to its wild progenitor (353) suggesting non-functional NAC genes contributed to trait development in sweet sorghum. Moreover, superTranscript-based methods for Differential Exon Usage (DEU) and Differential Gene Expression (DGE) analyses were more accurate than those based on the SSRG. This study demonstrates that the superTranscriptome can enhance our understanding of fundamental sorghum mechanisms, improve genome annotations, and potentially even replace the reference genome.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sweet sorghum (Sorghum bicolor (L.) Moench) is an ideal crop for future food, feed, and fuel security because of its high cellular biomass, stalk sugar content, and average grain yield. The crop is further characterized by high photosynthetic, water, and nutrient use efficiency; it helps to withstand changing environmental conditions (Rao et al. 2019). Therefore, it is a potential alternative to fossil fuels to achieve future bioenergy needs. The current breeding schemes need an overhaul to adapt to changing environmental conditions. Instead of relying solely on recombination within one population, we should consider exploring variability across sorghum species and sub-types. Its diverse primary races and intermediate varieties offer substantial natural variation (Venkateswaran et al. 2018). However, intensive selection and breeding have led to a loss of genetic diversity in modern germplasm, especially in specific agroecological zones (Smith et al. 2019).
Studies from the past suggest that natural selection played a big role in the evolution of sweet sorghum. Midrib color and stem juiciness are controlled by non-functional alleles for genes related to secondary cell wall development (Zhang et al. 2018). Interestingly, the midrib color strongly correlates with traits such as sugar yield, juice volume, and moisture content (Burks et al. 2015). Both sweet and grain sorghum show remarkable differences at the phenotypic level and are supposed to carry genome-level changes due to accumulating mutation load during crop domestication (Jiang et al. 2013; Smith et al. 2019). The primary gene pool of sweet sorghum constitutes 35,467 genes along with several Structural Variations (SV). Past studies reported deletions were more frequent in sweet sorghum than duplications, marking an important aspect of sweet sorghum evolution (Cooper et al. 2019). Comparative genomics of sweet and grain sorghum genomes identified distinct genes that were associated with variations which helped to distinguish between these sub-types at the genome level (Zheng et al. 2011).
Most genome-wide studies emphasize the number of genes annotated in the reference genome; however, some part of the genome is only shared by a subset of individuals within the species, termed as dispensable genome (Yao et al. 2015). In addition, dispensable genes were associated with complex genomic regions affected by SV, and are likely to be missed during the reference genome assembly (Gerdol et al. 2020). The comparative studies using reference genomes from a single organism or sub-types are unreliable, as the population shows considerable variation in intraspecies genomes (Bhatti et al. 2020). The third revolution in sequencing technologies leads to a decreased cost of sequencing (Jiao and Schneeberger 2017), allowing researchers to sequence more individuals to trap a major portion of dispensable genes among the population. RNA-seq offers a cost-effective alternative to genome sequencing for identifying functional genes and regulatory elements in plants with complex genomic architecture (Jin et al. 2016).
Representative Transcript Assemblies (RTAs) are a group of genes in the cluster that work as a superstructure and are a great way to characterize core, dispensable, and private genes in a population using RNA-seq datasets (Hirsch et al. 2014). In the present investigation, we have constructed RTAs or superTranscripts; these superTranscripts facilitated population-level identification of genes and non-coding RNAs and helped improve existing sorghum genome annotations. The superTranscript-based gene presence/absence on 15 sorghum genomes helped to provide a better understanding of the evolution of domesticated sorghum.
Material and Methods
Sorghum Genomes/Transcriptomes and Pre-Processing
The Sweet Sorghum Reference Genome version 2.1 (SSRG), along with 13 cultivated sorghum genomes belonging to diverse racial types were retrieved from Phytozome [https://phytozome-next.jgi.doe.gov] and SorghumBase [https://www.sorghumbase.org] from past studies (McCormick et al. 2018; Varoquaux et al. 2019; Cooper et al. 2019; Voelker et al. 2023) (Supplemental Table S1). This includes 9 parent genotypes from the Carbon-Partitioning Nested Association Mapping (CP-NAM) population representing diverse bioenergy sorghum sub-types and 4 cultivated sorghum reference genomes (Boatwright et al. 2021) and one elite sweet sorghum cultivar Wray (Broadhead et al. 1981). To compare the number of annotated genes and PAVs between cultivated sorghum (Sorghum bicolor subsp. bicolor) and its wild progenitor, one wild sorghum genome (Sorghum bicolor subsp. Verticilliflorum, accession 353) was included from sorghum pan-genome study (Tao et al. 2021). The pan-genome representing 390 diverse bioenergy sorghum accessions was downloaded from the ICRISAT repository [http://dataverse.icrisat.org] (Ruperao et al. 2021). The 223 sweet sorghum RNA-seq accessions for seven diverse sweet sorghum genotypes namely Rio (Li et al. 2019b; Cooper et al. 2019), Keller, SIL05 (Mizuno et al. 2016), Della, Dochna (Zhou et al. 2022), Roma (Sui et al. 2015), and M-81E (Sui et al. 2015) from past studies were retrieved from NCBI Sequence Read Archive (SRA) database using IBM Aspera Connect data transfer protocol and quality trimmed with FastP (Chen et al. 2018) to remove poor quality reads and adapter contamination for assembly (Supplemental Data S1).
superTranscriptome Construction
We used the Necklace (Davidson and Oshlack 2018) pipeline to build the superTranscriptome with a few improvements. It includes individual implementation of genome-guided, de novo transcriptome assembly, clustering, superTranscript construction, and gene expression counts steps for large datasets to avoid time loss due to errors (Fig. 1a; Supplemental Figure S1). The RNA-seq reads from sweet sorghum were aligned to SSRG using HISAT2 (Kim et al. 2019) with default parameters and known splice site information. The Sequence Alignment Map (SAM) to Binary Alignment Map (BAM) conversion and BAM sorting was done using SAMtools (Li et al. 2009). Genome-guided transcript assemblies for individual samples were obtained using StringTie (Pertea et al. 2015) with default parameters and merged into a single *.gtf file with the stringtie –-merge option. Further, merged *.gtf was flattened with gtf2flatgtff.pl to extend the gene boundaries for getting longer-length, genome-guided transcripts. Finally, genome-guided transcripts were obtained with GffRead (Pertea and Pertea 2020). The RNA-seq reads further assembled into de novo transcripts by using Trinity (Grabherr et al. 2011) with –max_memory 100 G. Protein-coding ORFs for de novo transcripts were obtained with grain sorghum (BTX623, BTX642, and RTX430) proteomes using BLAT (Kent 2002) with parameters -t = dnax -q = dnax -minScore = 200 and chimera_braker tool came along with necklace pipeline. Genome-guided and de novo transcripts were clustered into groups with parameters -minScore = 200 -minIdentity = 90 using blat. Finally, a superTranscript representing each cluster/group was constructed using Lace (Davidson et al. 2017) with cluster and sequence information. The entire workflow for superTranscriptome construction was divided into three steps and automated with Bpipe (Sadedin et al. 2012).

a The necklace pipeline (Davidson and Oshlack 2018) used in the present analysis integrates reference-guided transcriptome assembly and de novo transcriptome assembly to get a complete protein-coding orthologs. b Sweet sorghum superTranscriptome constructed by using necklace pipeline identified with three types of sequences: (1) Annotated on sweet sorghum reference genome (2) Annotated on grain sorghum (3) Unannotated
superTranscriptome Quality
The superTranscriptome assembly quality was analyzed with TransRate (Smith-Unna et al. 2016) using sequence information to obtain N50 values. Additionally, superTranscriptome constructed with 98%, 90%, 80%, and 70% sequence identities were checked with TransRate to see whether clustering influences contiguity. The superTranscriptome and six sorghum reference genomes were further searched for complete, partial, and missing gene orthologs within the Poaceae family with the Poales database using BUSCO (Simão et al. 2015). Further, RNA-seq samples from Rio were aligned to superTranscriptome and six published sorghum genomes using HISAT2 (Kim et al. 2019) with default parameters to obtain percentage read coverage by each assembly.
Functional Annotation and Characterization of Unannotated Genes
The genome-guided assembly reported 13,815 new genomic regions on SSRG, which were first annotated with SwissProt (Boutet et al. 2016) for protein-coding genes using the Trinotate (Ghaffari et al. 2014) pipeline. The superTranscripts missing annotations from Swiss-Prot were searched for homology with RNAcentral (Sweeney et al. 2020) using BLASTN with parameters pid = 90 & qcov = 90 to report various mi-RNAs and sno-RNAs encoding genes responsible for alternative splicing and silencing. The superTranscripts that missed annotations from the above three approaches were evaluated for coding potentials using CPC2 (Kang et al. 2017) and characterized into putative protein-coding and non-coding categories. Finally, lncRNAs from putative non-coding transcripts were annotated with the PLncDB database using BLASTN (parameters pid = 90 and qcov = 90).
superTranscript-Based Genome Annotation and Gene Densities
The superTranscripts were used as c-DNA/ESTs to re-annotate 15 diverse sorghum genomes using the PASA gene structure annotation pipeline (Haas et al. 2008) through the alignAssembly method with blat and gmap aligners. To report novel genes, alternative splice sites, and gene structure updates, a superTranscript-based annotation comparison of 14 sorghum genomes with existing ones was done using the PASA annotComapre method. Using PASA, alternative splicing updates over 15 sorghum genomes with superTranscriptome were also reported. This identified transcripts with retained intronic regions, spliced exons, and splice site donors-acceptors. The genome-wide annotations, gene structure, and splicing updates were visualized as a bar diagram with the ggplot R package. The numbers of genes per chromosome for SSRG and superTranscriptome annotated SSRG were obtained and a bar diagram showing chromosome-wise gene counts was plotted with a custom R script. The superTranscripts were annotated on SSRG using the PASA pipeline (Haas et al. 2008) produced *.gff3 annotation file and was used to obtain gene densities for tile size 100 kb with feature exon by using GFFex function of RIdeogram (Hao et al. 2020) R package. Similarly, gene densities for SSRG were obtained using past gene annotation *. gff3 file from Phytozome. Finally, an ideogram highlighting transcribed regions across ten sorghum chromosomes with the overlay method was prepared.
Orthogroups Identification, Synteny Analysis, and Identification of De Novo Genes
The superTranscript-based gene models over 15 genomes and singleton genes were predicted for protein sequences using TransDecoder [https://github.com/TransDecoder/TransDecoder] with a minimum sequence length of 100. This gave several protein sequences per gene. Therefore, the longest protein sequence per gene across all cultivars was selected for orthogroup identification. Finally, orthogroup identification was made using OrthoMCL (Li et al. 2003) with BLASTP e-value < 10−5 and inflation factor = 1.5 as previously mentioned for sorghum pan-genome (Tao et al. 2021). Gene duplication events are major contributors to speciation, we used OrthoFinder (Emms and Kelly 2019) to report duplicated orthogroups across assemblies. Syntenic gene families across assemblies were identified using the MCScanX_h algorithm of MCScanX (Wang et al. 2012) with BLASTN e-value < 10−5 and -max_target_seqs = 5 for homology search. To report de novo genes in sweet sorghum we looked for syntenic matches with other organisms as described previously for rice (Zhang et al. 2019). Briefly, the synteny match of sweet sorghum with grain sorghum (McCormick et al. 2018), wild sorghum (353) (Tao et al. 2021), and several other C3 -C4 grasses such as Saccharum spontaneum, Panicum hallii, Setaria viridis, Zea mays, Setaria italica, Aegilops tauschii, Eragrostis curvula, Triticum aestivum, Oryza sativa, Brachypodium distachyon, Avena sativa, Hordeum vulgare, and model plant species Arabidopsis thaliana from the Gramene database [https://www.gramene.org/] (Gupta et al. 2016) were obtained using MCScanX. Syntenic genes with the mentioned organisms were marked as 1 and nonsyntenic as 0. This provided a binary matrix (0/1) used for pairwise distance calculations and phylogeny construction with the Unweighted Pair-Group Method with Arithmetic mean (UPGMA) clustering using Phylip (Mansour 2009). The genes with missing synteny match with the above organisms were noted as de novo genes. These de novo genes were further looked for ORFs using TransDecoder with the default setting. The transcriptional evidence for de novo genes was obtained based on DGE-assigned p-values using RNA-seq. The de novo genes were further investigated to determine if they originated from transposases, based on internal sequence matches of MULE, TNT, and RE transposases with them using BLASTN (e-value < 10–37) as described previously for rice (Juretic et al. 2005).
Identification of Core, Dispensable, and Private Genes in the Population
superTranscripts locations were annotated on 15 sorghum genomes using the PASA pipeline (F. et al. 2006; Haas et al. 2008). The superTranscripts shared locations on any genomes using the PASA pipeline were marked as 1 else 0. The core, dispensable, and cloud genes were characterized based on their presence/absence on several genomes, a similar approach that was previously described (Jobson and Roberts 2022). Briefly, superTranscripts were categorized as core if they shared locations across all 15 genomes, dispensable if they shared locations across 4–14 genomes, and cloud if they shared locations across 1–3 genomes. Private superTranscripts were those that were exclusive to a single genome. The proportion of core, dispensable, and cloud genes per genome was obtained and visualized as a bar diagram, as mentioned in the past investigation on the sorghum pan-genome (Tao et al. 2021). The superTranscripts that did not share any locations on the above-mentioned 15 sorghum genomes were marked as orphan genes because they lacked homologs or had partial homologs on those genomes (Yao et al. 2017).
Origin of Sweet Sorghum-Specific Genes
We assigned ages to the genes exclusive to sweet sorghum genomes namely Rio, Chinese amber, Leoti and Wray. The pairwise sequence alignments of these genes with c-DNA sequences of grain sorghum (McCormick et al. 2018), wild sorghum (353) (Tao et al. 2021), several other C3-C4 plant outgroups, and Arabidopsis was performed using blat (pid = 60 and minscore = 100). In pairwise sequence alignment, genes were classified based on their hits with target species: those showed hits with Arabidopsis sequences were categorized as belonging to core angiosperm or Mesangiospermae; hits with Hordeum, Oryza, or any other C3 grasses were classified as Poaceae origin; hits with Saccharum, Panicum, or any other C4 grasses were identified as Andropogoneae origin; sequences aligning with grain or wild sorghum were categorized as sorghum-specific or Sorghinae origins; and If there were no hits then the gene was designated as sweet sorghum-specific. These given hits matrix (0/1) for all organisms which is further used for pairwise distance calculations and UPGMA phylogeny construction using phylip (Mansour 2009).
Differential Exon Usage (DEU) Analysis
The DEU comparison was performed as per the methodology described (Davidson et al. 2017). The RNA-seq reads of sweet sorghum Rio from past studies (Cooper et al. 2019) were aligned to SSRG and superTranscriptome using HISAT2. Novel splice sites were extracted for superTranscriptome to understand blocking within the superTranscripts. The *.gtf file for superTranscript blocks was prepared with the make_block tool in the necklace pipeline. The exon bin or blockwise expression count was obtained using FeatureCouts, (Liao et al. 2014). Statistical significance for exon usage was tested with DEXSeq, (Anders et al. 2012) R package. Per gene q-values for differential exon usage were obtained for both approaches. Once, per gene q-values were obtained, the true positives and true negatives were selected based on q-value cutoffs. The superTranscripts with a q-value below 0.05 were considered true positive, and superTranscripts with a q-value of 0.9 and above were considered true negative. The Sci-Kit-learn Python module was used to train the datasets with a logistic regression method. This helped to report the differences between actual and predicted labels for the above two approaches using machine learning. The ROC curve showing DEU performance was plotted with a custom R script. Similar to this, the KNeighborsClassifier method of the Sci-Kit-learn module was used to train datasets and prepare the confusion matrix for both. A DEU analysis of Rio and PR22 internode was also done to report differentially spliced transcripts between these two genotypes during internode growth.
Differential Gene Expression Analysis
To report changes in differential gene expression with two different references, i.e. SSRG, and superTranscriptome, the RNA-seq reads from leaf, meristem, and internode tissues from six-time points of sweet sorghum Rio were aligned to both, followed by expression counts using FeatureCounts (Liao et al. 2014) with a transcript count method. Finally, time point transcript count normalization and log2fold changes were estimated with the Likelihood Ratio Test (LRT) method using the DESeq2 (Love et al. 2014) R package. The volcano plots representing DGE for SSRG and superTranscriptome were plotted using the EnhancedVolcano (Blighe K et al. 2022) R package. The top 50 highly expressed genes with a P value < 0.05 from the above two methods were visualized as a heat map with the geom_tile function of the ggplot2 (Gómez-Rubio 2017) R package.
Similarly, differential gene expression analysis of Rio and PR22 during internode growth was done using superTranscriptome as a reference. The top 50 highly expressed genes between these two genotypes during internode growth were visualized as a heatmap. The gene ontology enrichment analysis was performed to report significantly enriched GO terms with AnnotationForge (Pagès et al. 2022) based custom database using superTranscriptome annotations. Significantly enriched GO terms (P value < 0.05) between Rio and PR22 during internode growth were reported using the clusterProfiler (Wu et al. 2021) R package.
Identification of Agronomically Essential Genes and Phylogenetic Analysis
The agronomically essential genes involved in DNA packaging, transcription, sugar transport, sugar metabolism, and stress responses were selected for MSA. These include genes encoding for histones, cytochrome b5, hexokinases, zinc finger domain, MULE-like transposase, NAC, MYB, SWEET and SUT transporters, Invertase (INV), expansin, Universal Stress Protein (USP), etc. The phylogenetic analysis was performed to report sequence level changes across these genes in sweet sorghum in comparison with grain sorghum, other C4-C3 outgroups, and Arabidopsis. The shared ortholog sequences for these genes with grain sorghum, Saccharum sp., maize, barley, and Arabidopsis were obtained from Phytozome [https://phytozome-next.jgi.doe.gov/]. The multiple sequence alignment of shared orthologs was done using MUSCLE (Edgar 2004). To refine alignment, the alignment trimming was performed using trimAl (Capella-Gutiérrez et al. 2009) with parameters (-gt 0.5 -resoverlap 0.5 -seqoverlap 50). Finally, a maximum-likelihood phylogenetic analysis was performed using IQ-tree (Nguyen et al. 2015) with standard substitution model selection (-m TEST) for 1000 bootstrap repetitions. The phylogenetic tree for these genes was visualized and annotated using iTOL (Letunic and Bork 2021).
Genomic Landscape of Sweet Sorghum Evolution
Through the utilization of ScanPAV (Giordano et al. 2018), we conducted a comparison of the genomes of grain (BTX623) and wild sorghum (353) to SSRG. Our findings revealed significant deletions (> 1 kb) that hold evolutionary significance. ScanPAV uses a sliding window (SW) approach for aligning sequences to reference using BWA (Li and Durbin 2010), reports missing blocks in reference i.e. > 1 kb, and writes missing blocks as well as their genomic coordinates in *.fasta and *.vcf file format. To characterize, genic and intergenic deletions in grain and wild sorghum, missing blocks with SSRG were looked for protein-coding gene overlap using BEDTools (Quinlan and Hall 2010). Similarly, missing regions on cultivated sorghum i.e. Rio and BTX623 genomes compared to wild sorghum were identified. This reported several large deletions that occurred in cultivated sorghum compared to wild sorghum throughout sorghum domestication.
Results
superTranscriptome Construction
Collectively, 223 RNA-seq accessions of seven diverse sweet sorghum genotypes from the public repository formed 45,049 genome-guided transcripts with SSRG and 886,115 de novo transcripts, constituting 45,864 gene clusters. The representative sequence of each cluster was prepared using Lace (Davidson et al. 2017). The superTranscriptome reported 45,864 genes with three types of sequences: genome-guided sequences, which were already annotated on SSRG; unannotated or novel transcribed genomic regions, which require further functional annotation; and de novo spliced isoforms or novel genes, which showed less than 90% similarity with SSRG genome-guided contigs but they were annotated on grain sorghum cultivars genomes such as BTX623, BTX642, and RTX430. The third sequence types were Open Reading Frames (ORFs) from transcripts using protein sequences retrieved from Phytozome [https://phytozome-next.jgi.doe.gov/] (Fig. 1b). This approach yielded 886,115 de novo transcripts but only 163,651 were reported with ORFs. Of these 163,651; 161,712 successfully incorporated known protein-coding superTranscripts from SSRG to retrieve complete protein-coding sequence ortholog. The remaining 1,939 de novo transcripts did not show significant similarity with any genome-guided transcript formed 815 separate clusters. This suggests there were some genes in sweet sorghum from grain sorghum background exclusively. The superTranscripts number may vary when we change the clustering parameters; it leads to the loss of some annotated genes from the reference genome. However, no significant change in contiguity was reported when we changed the clustering parameters (Supplemental Table S2).
superTranscriptome Quality
Contiguity analysis for superTranscriptome was done using the TransRate tool. This reported the N50 value of 3,551 bp for superTranscriptome. We found 30,697 (66.93%) and 572 (1.24%) superTranscripts were above 1 kb and 10 kb sequence lengths respectively. Only 184 (0.4%) superTranscripts were below 200 bp in sequence length (Table 1; Supplemental Data S2) and the rest 14,411 (31.42%) sequences showed lengths ranging from 200–800 bp. The result suggested that most sequences were contiguous, could be complete gene orthologs, and highlighted the diversity of superTranscript length distribution (Supplemental Figure S2a). BUSCO assembly completeness analysis suggested that 94.19% of superTranscripts shared complete orthologs with Poales and a 6% duplication rate. Therefore, duplication could be the reason for reporting more orthologs for some essential genes (Fig. 2). The superTranscriptome also reported incomplete/partial genes in comparison with Rio, SC187, BTX642, RTX430, and BTX623 sorghum genomes, suggesting that there were specific genes in the population with missing start/stop codons or both codons that play an essential role in trait development. To check the quality of the assembly, 48 RNA-seq accessions of sweet sorghum Rio were aligned to superTranscriptome along with six published sorghum genomes using HISAT2 (Kim et al. 2019). We reported that SSRG showed the highest average read alignment (84.06%) among the six genomes, followed by grain sorghum genomes BTX623 (82.29%), BTX642 (82.07%), SC187 (82.06%), RTX430 (81.87%), and superTranscriptome (79.18%) in that particular order. Results suggest that an average of 5% read coverage was reduced in superTranscriptome over SSRG (Supplemental Figure S2b).

BUSCO assembly completeness analysis with Poacecae database by using superTranscriptome and six published sorghum reference genomes showing complete (single copy/duplicated), fragmented and missing genes
Functional Annotation and Characterization of Unannotated Genes
The sweet sorghum superTranscriptome comprises 45,864 genes. Among these, 31,234 were annotated with SSRG, 815 with grain sorghum, and 13,815 remain unannotated. Within this unannotated group, the Trinotate pipeline identified and annotated 2,802 new protein-coding genes/superTranscripts with SwissProt. Among these newly annotated protein-coding genes, 559 encodes for various transcription factors (TFs) including NAC, MYB, and chromo-domain proteins as determined by their DNA-binding domains using PLantTFcat (Dai et al. 2013), PLantTFDB (Guo et al. 2008) and iTAK (Zheng et al. 2016) online server databases (Supplemental Data S3, sheet2). The rest of the 11,013 sequences, 572 genes/superTranscripts, were annotated with diverse roles in intron splicing, gene silencing, and ribosomal assembly when searched for non-coding RNA annotation against the RNAcentral database. The remaining 10,441 superTranscripts were checked for coding potential with CPC2 and reported 954 protein-coding and 9,487 non-coding regions. Putative 9,487 non-coding genes/superTranscripts were potential sources of lncRNAs, of which 6,516 were annotated with PLncDB (Jin et al. 2021) using BLASTN (Fig. 3; Supplemental Figure S3, Supplemental Data S3). The results suggest that about 21.93% of sweet sorghum genomes transcribe non-coding RNAs involved in diverse functions, including gene silencing, intron splicing, and gene regulation. Long non-coding RNAs occupied 14% of total expressed sequences; however, this proportion may increase when an independent study is performed.

Donut plot showing functional annotations of superTranscriptome with proportion of annotated sequences with different databases
superTranscript-Based Genome Annotation and Gene Densities
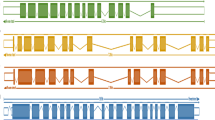
Of a total of 45,864 genes/superTranscripts, 41,169 (89.76%) were annotated on 15 diverse sorghum genomes using the PASA gene structure annotation tool (Haas et al. 2008) and 4,695 (10.24%) remains singleton. These genes were selectively transcribed across sorghum genotypes and contributed to variability. In comparison to other genomes, the SSRG recorded the highest number of annotated genes 40,901(89.17%). This was much higher in contrast to the previous study on sweet sorghum genome annotation (Cooper et al. 2019). Additionally, chromosome-wise gene counts and gene density in SSRG were reported higher when annotated with superTranscriptome using PASA (Supplemental Figure S4a, b); Supplemental Data S4). The superTranscripts showed several gene structure updates on 14 diverse sorghum genomes (excluding wild progenitor 353). The updates include new gene additions, single gene model updates, and alt-splice site additions. Additionally, the superTranscript-based approach reported extensive alternative splicing on these 15 cultivar genomes. The results demonstrate that superTranscriptome improved genome annotations, gene structures, and alternative splicing (Fig. 4; Tables 2, 3).

Bar diagram showing various types of gene structure updates on 15 published sorghum genomes when annotated with superTranscriptome using PASA pipeline
Orthogroups Identification, Synteny Analysis, and Identification of De Novo Genes
A total of 454,772 protein-coding gene models across 15 genome assemblies and 4,695 singletons genes were used for orthogroups identification. Of the total 459,467 genes, 448,293 (97.57%) were assigned with orthologroups and the rest of the 11,174 (2.43%) were unassigned groups. The present analysis reported a total of 44,222 orthogroups in the sweet sorghum (Supplemental Data S5 and Supplemental Data S5(a)). Similar results for orthogroups identifications were obtained previously for sorghum pan-genome analysis (Tao et al. 2021). Our OrthoFinder analysis reported several orthogroups that showed duplications on 15 cultivar genomes. This includes the top three namely RTX430 (3,001), BTX642 (1471), and Rio (792) with the highest number of duplicated orthogroups (Supplemental Figure S5a). This infers gene duplication is frequent in sorghum and cultivars RTX430, BTX642, and Rio were selected with duplicated genes during domestication. Of 540,286 superTranscript-based gene models across 15 genome assemblies, 516,825 (95.66%) were syntenic genes, forming 20,419 syntenic gene families. The results demonstrate that more than 90% of genes across 15 sorghum genome assemblies were collinear with few syntenic re-arrangements. This reported 1,422,576 syntenic gene pairs were located on the same chromosome or scaffold over 15 genome assemblies. However, 7,885 were reported with syntenic re-arrangements. We compared the synteny of sweet sorghum (SSRG) with wild progenitor (353) and grain sorghum (BTX623). This reported 108,856 syntenic gene pairs on the same chromosome/scaffold and 1,528 syntenic re-arrangements between these genomes. We have taken subsets of these syntenic genes and visualized them as ideograms (Supplemental Figure S5b); Supplemental Data S5(b)). The tandem duplications during synteny analysis were also reported these include several tandem duplicates across each assembly. Rio reported the highest number of tandem duplicates 2,154 followed by RTX430 (2,101), Wray (613), PI510757 (342), PI329311 (332), etc. (Supplemental Table S3). Most of the tandem duplications in Rio genes were related to the Gene Ontologies (GOs) such as protein binding, DNA binding, protein kinase activity, nucleic acid binding, catalytic activity, nucleotide binding, DNA-binding transcription factor activity, and metabolic processes (Supplemental Data S5, Sheet 3 and 4). This infers that duplication among these genes was frequent in sweet sorghum and could significantly contribute to its evolution.
To report de novo genes, 37,003 genes on SSRG were used for synteny comparison. We found that 34,288 were syntenic with grain sorghum, wild sorghum, other C3-C4 grasses, and Arabidopsis (Fig. 5). The remaining 2,715 were de novo genes because they lacked synteny with the above organisms. These 2,715 genes were evaluated for de novo ORFs and transcriptional evidence using RNA-seq. These reported 1,669 with complete/partial ORFs and 1,543 with transcriptional evidence in the leaf, meristem, and internode tissues of Rio. The minimum and maximum ORF lengths for de novo genes were 91 and 8,346 amino acids (aa) respectively. Of 1,669 de novo ORFs only 895 were transcribed in Rio and 774 were non-transcribed. A total of 1,046 de novo genes lack ORFs of which 648 were transcribed in Rio. This infers that further mutations may trigger their transnational activity because they lack a start codon for transnational activity. The rest of the 398 de novo genes were non-transcribed non-ORF types and could be pseudogenes (Supplemental Table S4). Most of the de novo genes encoded elements such as TNT-1, RE-1, RE-2, DUF domain, MYB/SANT-like domain, reverse transcriptase, zinc finger domain, core and linker histones, ribonuclease H, etc. (Supplemental Data S5(c)). A total of 173 de novo genes were found to retain internal sequences from MULE, TNT, and RE transposases when the internal sequences of these transposases were queried using BLASTN (e-value < 10–37) (Supplemental Data S5(c), sheet 3). This highlights the role of these transposases in de novo gene formation.

Synteny-based phylogenetic grouping of organisms using UPGMA clustering
Identification of Core, Dispensable, and Private Genes in the Sweet Sorghum
The Pan-genome analysis classified the gene families into core, dispensable, and cloud categories for many plants such as sorghum (Ruperao et al. 2021; Tao et al. 2021; Wang et al. 2021), rice (Sun et al. 2017; Zhao et al. 2018; Qin et al. 2021), maize (Hirsch et al. 2014), and pea (Yang et al. 2022). The superTranscripts annotated 15 diverse sorghum genomes based on genomic presence/absence and were classified into core (20,743), dispensable (18,915), and cloud (1,511) genes (Fig. 6a, b). The remaining 4,695 singleton superTranscripts were marked as orphan genes because they either lack homologs on the above 15 genomes or have only partial homologs. Collectively, 24,625 (53.69%) superTranscripts were reported with remarkable presence/absence variation on 15 sorghum genomes (excluding single genome superTranscripts and core genes), suggesting that these genes contributed to variability in sorghum (Supplemental Data S6, Sheet 2). The GO enrichment analysis of these genes suggests that they encoded various Transposable Elements (TEs), proteolytic enzymes, and regulatory elements of biological processes. This suggests that proteolysis, transposition, and gene regulation of several biological processes were significant contributors to the observed variability in sorghum (Supplemental Figure S6a). Additionally, some superTranscripts were exclusively located on specific cultivar genomes (Fig. 6c), suggesting that sweet sorghum carries some genes from cellulosic, grain, and forage backgrounds; however, the function of these genes in sweet sorghum was not known and might carry some modifications. Phylogenetic analysis based on gene presence/absence across 15 cultivar genomes was performed using the UPGMA clustering previously described for genome-wide SNPs and PAVs (Tao et al. 2021). This reported the PAV-based grouping of 15 genotypes according to their geographical origin. We found that US-origin (Rio and Wray) and Asia-Europe origin (Chinese amber and Leoti) sweet sorghum formed a group; suggesting that the gene set carried by sweet sorghum varies with geographical origin (Fig. 6d). Further, 301 superTranscripts were exclusive to sweet sorghum genomes (Rio, Wray, Leoti, and Chinese amber). These include genes encoding hexokinases, core and linker histones i.e. H2A, H3, H4, and H1, cytochromes, MULE-like transposase, glucosyl transferases, chitinases, cell wall-associated receptor kinases, MYB-TF, and some known/novel lncRNAs. Gene ontology studies of these genes showed that most of them were connected to the transport of different kinds of molecules across membranes, like electrons, metal ions, organic and inorganic ions, and more. They were also linked to catalytic enzymes, cell signaling, gene regulation, chromosome assembly, and DNA packaging. The results showed that sweet sorghum has extra alleles for maintaining cellular homeostasis, managing energy and carbohydrates, controlling chromatin, and DNA packaging (Supplemental Figure S6b; Supplemental Data S6, Sheet 3). These genes were exclusive to sweet sorghum genomes and probably evolved later. The orphan genes contribute 9.64% of superTranscriptome for which KEGG Orthology (KO) analysis was performed using KAAS (Moriya et al. 2007). The majority of orphan genes were coded for TFs, signaling molecules, transporters, and catalytic enzymes that play a significant role in cellular physiology, cell wall development, abiotic and biotic stress resistance and probably help in the development of sweet sorghum-specific traits (Supplemental Figures S6c, d).

a Pie chart showing number of core, dispensable and cloud genes identified on 15 published sorghum genomes. b Bar diagram showing proportion of core, dispensable and cloud genes across each sorghum genome when annotated with superTranscriptome using PASA. c Flower plot showing distribution of core, shell and private genes across 15 diverse sorghum genomes; helps to report sweet sorghum specific genes and genes from various cultivar background in sweet sorghum. d Unweighted Pair-Group Method with Arithmetic mean (UPGMA) based phylogenetic grouping of 15 cultivars based on gene presence/absence reveals that Asia-Europe and the US-origin sweet sorghum carry distinct sets of genes
Origin of Sweet Sorghum-Specific Genes
Of 301 sweet sorghum-specific genes, only 25 were reported hits with Arabidopsis; infer that these sequences have Mesangiospermae origin. A total of 185 sequences reported hits with C3 grasses of which 160 were added over Arabidopsis, suggesting that those sequences were exclusively from the Poaceae family. Next, 238 sequences showed hits with C4 grasses of which 53 were added over Poales, indicating that those 53 sequences have Andropogoneae origin. A total of 270 sequences were reported hits with wild sorghum (353) and 283 with grain sorghum (BTX623). These include 32 and 13 added sequences to wild and grain sorghum respectively. This demonstrates that those sequences were sorghum-specific or Sorghinae origin. The remaining 18 were specific to sweet sorghum, did not display any hits, and encoded histone H1/H5, unknown proteins and non-coding genes. This infers that these 18 genes were selected in sweet sorghum (Figs. 7a, b); Supplemental Data S7a). Interestingly, out of these 301 sweet sorghum-specific genes, only 58 were identified as de novo genes and assigned gene ages (Supplemental Figure S7; Supplemental Data S7b). Based on the taxonomic age of the target organism or family, the ages of all 301 genes were determined using the TimeTree5 database (Supplemental Table S5) (Kumar et al. 2022). We found that a total of 270 (89.70%) genes have ages > 9 Million Years (MYA), while the rest of the 31 (10.30%) have ages < 3.55 MYA evidenced that they were evolved in sorghum.

aThe 301 genes were exclusive to sweet sorghum genomes, assigned gene ages based on their sequence similarity hits with c-DNA sequences of several C3-C4 grasses and Arabidopsis. b Sequence similarity hits (0/1) of 301 sweet sorghum-specific genes with several other C3- C4 grasses and Arabidopsis facilitated phylogenetic grouping based on shared genes using UPGMA clustering
Differential Exon Usage (DEU) Analysis
The traditional genome-based approach with a standard blocking scheme reported only 148,847 exons on SSRG. superTranscripts followed a dynamic blocking scheme when aligned with RNA-seq reads which reported 461,355 exon bins, suggesting that exon splicing was extensive in superTranscripts. For example, the standard blocking scheme employed for DEU analysis with SSRG yielded five exons for the NLP2 TF coding gene which was located on chromosome 6 (SbRio.06G148100), of which three were differentially used for spliced transcript formation under six developmental stages. Where superTranscriptome followed a dynamic blocking scheme for the same gene (NLP2 TF, gene id: SbRio.06G148100), it identified 19 different exon bins, of which eight were differentially used for transcript formation (Supplemental Figures S8a, b); Supplemental Data S8). This suggested that superTranscript-based dynamic blocking was more informative for the same gene than the SSRG-based standard blocking. Additionally, this increased the probability of finding more alternatively spliced transcripts; those may be involved in adaptation, stress responses, and trait development. The DEU testing using two references i.e. SSRG and superTranscriptome, reported 20,942 and 22,554 genes with differential exon usage. The true positives (with q-value < 0.05) and true negatives (with q-value > 0.9) reported with SSRG and superTranscriptome were 12,793 (61.08%), 4,583 (21.88%) and 13,556 (60.10%), 5,749 (25.48%) respectively. The superTranscriptome-based approach found a better classifier for true labels than the SSRG when trained datasets using the logistic regression method (Fig. 8a). Additionally, the confusion matrix prepared for the above two approaches using the KNeighborsClassifier method reported more true labels in superTranscritome than the SSRG-based standard approach (Fig. 8b, c)). The results established that the genes identified by the superTranscriptome were real transcripts, as shown by their ability to give more accurate estimates of DEU when tested with supervised machine learning and differential splicing. The DEU analysis of Rio and PR22 during internode growth reported that 8,787 and 5,769 genes were differentially spliced (q-value < 0.05) in Rio and PR22 (Supplemental Figures S8c, d). This clearly showed that the differential splicing of the genes plays a crucial role in phenotype development.

a ROC curve for trained datasets with logistic regression method showing superTranscritptome with dynamic blocking giving better estimates of DEU than SSRG-based standard blocking approach. b Confusion matrix prepared for DEU with SSRG showing true and predicted labels when trained datasets using KNeighborsClassifier method. c Confusion matrix prepared for DEU with superTranscriptme reported more true labels than SSRG-based approach when trained datasets using KNeighborsClassifier method
Differential Gene Expression (DGE) Analysis
Gene-level abundance estimates and DGE analysis using SSRG and superTranscriptome as a reference reported a total of 20,940 and 22,036 genes respectively, with significant (P value < 0.05) changes in gene expression across the leaf, meristem, and internode tissues over six-time points (Figs. 9a, b); Table 4; Supplemental Figure S9a). Among 50 highly expressed genes in the leaf, meristem, and internode tissues, 83% were the same for the above two references, and the rest of the 17% genes showed slightly altered gene expression (Supplemental Figures S9b, c). Past studies reported minor differences in gene expression when performed de novo and reference-based DGE analysis (Davidson and Oshlack 2014); suggesting that the choice of references for DGE analysis has little influence on gene expression. The ROC curve based on trained datasets using the logistic regression, showed that superTranscriptome gave better DGE estimates than SSRG (Supplemental Figure S9d). This infers that the superTranscript-based approach identifies more truly expressed genes than SSRG and highlights its suitability for DGE analysis. The list of the top 50 highly expressed genes during internode growth differed between Rio and PR22. Rio highlighted several Light Harvesting Complexes (LHCs) but did not show expression of metallothionein-II within the top 50 genes. Conversely, PR22 showed a lower number of LHCs but expressed metallothionein-II. This suggested that the internode phenotype is likely influenced by active photosynthesis and metal ion transport (Fig. 9c). Further, the growing meristem of Rio reported expression of metallothionein-II indicated that this gene appears to be essential during the active growth of the meristem but not necessarily required for the sugar-accumulating internode. Differential gene expression showed that GO terms related to moving metal ions, secondary metabolites, cells, and other things were more common in PR22 during internode growth (Supplemental Figure S9e). Therefore, active transport of metal ions, secondary metabolites, and organic compounds contributes to dry or pithy stems in sorghum. Further, both genotypes reported 9,028 common genes that showed differential (P value < 0.05) expression during internode growth; however, the expression of 6,092 and 2,840 genes were exclusive to Rio and PR22 respectively. (Supplemental Figure S9f). Interestingly, 487 and 348 lncRNAs were also reported to be differentially expressed during the internode growth of Rio and PR22 respectively. It is inferred that the lncRNAs could be hidden players that control internode development and confer sugary internode (Supplemental Figures S9g, h)). Further, the top 50 highly expressed lncRNAs and TFs were listed and their expression patterns were correlated with percentage (%) brix readings; which were taken previously during internode growth for both genotypes (Cooper et al. 2019). This reported several lncsRNAs especially novel lnc SBRIO_LNC019054 along with NAC-2–1 and NAC-60 showed elevated expression with sugar accumulation in Rio; suggesting that they were positive regulators of sucrose-related genes. However, during the internode growth of PR22, it was reported that FHA-SMAD-21, Tify-13, and conserved SBIC_LNC004344 showed higher expression with decreasing sucrose levels. This implies that they may function as negative regulators of sucrose (Supplemental Figures S9i, j).

a Volcano plot showing differentially (p-value < 0.05) expressed genes across Leaf, Internodes and Meristems tissues when used SSRG as a reference for DGE analysis. b Volcano plot reported more differentially (p-value < 0.05) expressed genes across Leaf, Internodes and Meristems tissues when superTranscriptome used as a reference for DGE analysis. c Top 50 highly expressed genes in Rio and PR22 during internode growth shows that Rio reported comparatively more numbers of expressed LHCs along with missing expression of Metallothionein II
Identification of Agronomically Essential Genes and Phylogenetic Analysis
The agronomically important genes such as histones, MULE-like transposase, zinc finger domain, hexokinases, cytochrome b5, SWEET, SUT, invertase, expansin, USP, NAC, and MYB transcription factor reported more in number in sweet sorghum than grain sorghum and some of them were exclusive to sweet sorghum genomes. We found the majority of novel histone genes either formed separate clades or clades with Hordeum sp. (Supplemental Figures S10a–d). Further, the chromosomal locations of these novel histone genes on SSRG showed that they were located at the lower arm of chromosome 8; suggesting that this region of chromosome 8 carries duplications of histone genes in sweet sorghum (Supplemental Figure S10e). The hexokinases, cytochrome b5, and MYB transcription factor genes from sweet sorghum were found to be related to sorghum and Saccharum sp. This suggests that these genes may be ancestral copies of genes from sorghum and sugarcane (Supplemental Figures S10f–h). MULE-like transposases were abundant in the sweet sorghum genome, of which SwSb − MULE − 21** was reported to be exclusive to sweet sorghum genomes and found similar to existing ones in sweet sorghum (Supplemental Figure S10i). This inferred that MULE genes might have duplicated on sweet sorghum genomes. The zinc finger domain is a major transcription factor family associated with abiotic stress responses in plants (Han et al. 2020). Of several, five zinc finger loci namely SwSb-zf-2**, SwSb-zf-3**, SwSb-zf-4**, SwSb-zf-5**, and SwSb-zf-6** were exclusive to sweet sorghum and showed similarity with existing sweet and grain sorghum genes (Supplemental Figure S10j). This demonstrated that some zinc finger genes were also duplicated in sweet sorghum. The SUT4, one of the six SUT genes was identified in sweet sorghum. This gene was reported to be deleted on SSRG due to large deletions (Cooper et al. 2019). The present analysis reported the presence of a SUT4 ortholog named SwSb − SUT − 6* through de novo transcriptome assembly (Supplemental Figure S10k). The Sugars Will Eventually Exported Types of Transporters (SWEET) is a gene family involved in phloem loading and unloading. The superTranscriptomic approach reported 24 SWEET genes with one additional SWEET locus SwSb − SWEET − 24* located on chromosome 8 with only one exon (Supplemental Figure S10l). The plant-specific NAC transcription factor qualitatively controls dry biomass, sugar production, and grain yield (Xia et al. 2018). superTranscriptome identified 130 NAC-TF encoding loci, of which six were newly reported on SSRG. The new NAC loci were more similar to ancestral types i.e. sorghum, Saccharum sp., and maize than other outgroup species (Supplemental Figure S10m). Further, the rest of the newly annotated genes which include invertase, expansin, and USP formed separate clades in phylogeny; suggesting that these sequences carry some evolutionary modifications in sweet sorghum (Supplemental Figures S10n–p).
The numbers and chromosomal locations of agronomically important genes on SSRG and grain sorghum reference genomes (BTX623) reported that these loci were more abundant in sweet sorghum than grain sorghum (Figs. 10a, b). This suggests that sweet sorghum carries additional loci for agronomically important genes. We find more and fuller gene orthologs for agriculturally important genes by combining de novo transcriptome assembly and genome-guided assembly methods. Nevertheless, we were unable to validate the functions of newly discovered genes in sweet sorghum. Therefore, further GWAS analysis may provide more information about these genes.


a Genomic locations agronomically important QTLs such as histone, cytochrome, hexokinases, MULE-like transposase and MYB-TF on grain sorghum genome. b Genomic locations agronomically important QTLs such as novel histones, cytochrome, hexokinases, MULE-like transposase and MYB-TF on sweet sorghum genome
Genomic Landscape of Sweet Sorghum Evolution
Compared to sweet sorghum (SSRG), ScanPAV reported 14,293 (31.6 MB) and 20,869 (47.99 MB) large deletions (> 1 kb) in grain (BTX623) and wild (353) sorghum respectively. These include the loss of 2,974 genes in grain sorghum and 5,028 genes in wild sorghum. This indicated that wild sorghum reported with greater number of deletions in both gene and intergenic regions than grain sorghum. Interestingly, 58 and 51 sweet sorghum-specific or private genes were reported with deletions in wild and grain sorghum respectively (Table 5ii). The results validated that sweet sorghum-specific genes showed deletions in grain and wild sorghum; suggesting that insertions for these genes were selected in sweet sorghum. Sequence loss in cultivated sorghum i.e. grain and sweet sorghum compared to wild sorghum accounted for 20,495 (47.94 MB) and 20,501 (48.82 MB) large deletions in grain sorghum and sweet sorghum respectively (Table 5i). This included deletions of 2,394 and 2,177 genes in grain and sweet sorghum respectively (Table 5iii). This infers that a comparatively higher number of gene deletions (2,394) were found in grain sorghum than sweet sorghum (2,177); suggesting that more genic deletions were selected in grain sorghum. Whereas, sweet sorghum showed fewer genic (2,177) and a comparatively higher number of intergenic deletions (18,324) than grain sorghum (18,101); inferred that more intergenic deletions were selected in sweet sorghum. The results suggest that both deletion and insertions were frequent in cultivated sorghum and selections of genes with SVs lead to subtype development.
Dry locus or D locus (Sobic.006G147400) encodes plant-specific NAC transcription factor which qualitatively controls midrib color and stem composition in sorghum (Xia et al. 2018; Zhang et al. 2018). The present analysis reported large deletions in the D locus of sweet sorghum compared with grain and wild type. This includes 2.05 kb and 2.29 kb deletions in the D locus of sweet sorghum compared to wild and grain sorghum respectively (Supplemental Figures S11a–c). In comparison with wild sorghum, a total of 3 and 5 NAC encoding loci (including the D locus) were deleted in grain and sweet sorghum respectively (Table 5iii; Supplemental Data S11). This includes an additional four NAC encoding loci other than the D locus located on chromosomes 1, 2, 4, and 6 reported with deletions in sweet sorghum (Supplemental Figures S11d–h). We found that these four NAC encoding loci were affected by terminal deletions and could be non-functional genes due to the loss of upstream/downstream promoter regions. Results demonstrated that sweet sorghum NAC encoding loci were affected by deletions compared with wild progenitor (353). This confirms that deletions in NAC encoding genes could be one of the reasons for trait development in sweet sorghum.
Discussion
superTranscriptome identified a total of 45,864 genes, which was higher than the previously reported in sorghum (McCormick et al. 2018), sweet sorghum (Cooper et al. 2019), sorghum pan-genome (Ruperao et al. 2021; Tao et al. 2021; Wang et al. 2021), rice pan-genome (Zhao et al. 2018; Qin et al. 2021), barley (Jayakodi et al. 2020), and much lesser than maize pan-genome (Hufford et al. 2021). The results validated that the superTranscriptome approach identified more expressed genes and suggested that a single reference genome cannot report all genes expressed in sweet sorghum. Since superTranscripts are probabilistic gene models for sweet sorghum, their functions may vary from genome to genome depending on the gene structure. The N50 value for superTranscriptome was 3551 bp, much higher than the N50 value previously reported for pan-transcriptome assemblies of maize (Hirsch et al. 2014; Jin et al. 2016), alfalfa (Medina et al. 2021), potato (Petek et al. 2020), and barley (Ma et al. 2019) and showed the increased contiguity in superTranscriptome assembly. BUSCO assembly completeness studies reported several gene duplications and missing/partial genes that could promote trait development in sorghum. Past studies on sorghum pan-genome reported BUSCO gene completeness ranging from 94.4 to 98% in the sorghum (Tao et al. 2021), supporting our findings. Past results showed partial genes were associated with the sugarcane aphid (SCA) resistance for the cultivated sorghum variety TX278 (Wang et al. 2021). Furthermore, a comparative analysis of maize genomes also reported that more incomplete genes and missing genes contribute to maize adaptation under diverse environments and crop improvement (Yang et al. 2017; Li et al. 2019a). The comparative analysis of sweet and grain sorghum genomes reported 2,175 genome re-arrangements, these include several deletions and duplications along with 98,723 base substitutions (missense: silent ratio = 1:1) in coding regions (Cooper et al. 2019). This infers that the sweet sorghum genome carries several evolutionary changes during its domestication. The superTranscriptome reported an average of 5% less read coverage over SSRG; demonstrating that we lost an average of 5% transcribed sequences over SSRG during superTranscripts construction.
The lncRNAs contribute 14% of superTranscriptome. However, past studies on lncRNAs suggest they are highly tissue-specific and condition-specific (Statello et al. 2021). In the present analysis, we have reported some lncRNAs with significant changes in expression during the internode development. There were between 8000 and 23,309 lncRNAs found in maize (Wang et al. 2015; Lv et al. 2019), sorghum (Sun et al. 2020), rice (Zhou et al. 2021), and barley (Unver and Tombuloglu 2020) which showed that lncRNAs make up most of the RNA. Recent studies on ten high-quality genome assemblies of diverse bioenergy sorghum genotypes reported similar results for lncRNAs (Voelker et al. 2023). The present analysis reported 44,222 orthogroups in sweet sorghum which was similar to previously reported for sorghum pan-genome analysis (Tao et al. 2021). The genes in the population are most likely affected by SVs and duplication events during speciation, which results in the loss or formation of new orthogroups (Brasó-Vives et al. 2022; Prabh and Rödelsperger 2022; Berdan et al. 2024). Our synteny analysis revealed that Rio had the highest number of tandem duplicates (2,154), which indicates duplications were frequent in Rio and could be associated with the activity of various transposases. We reported 2,715 de novo genes in sweet sorghum, of which 648 (23.68%) were transcribed without ORFs. This suggests that the absence of start codons rendered them non-functional, implying that any mutation could potentially affect their functions. Further, 398 (14.65%) de novo genes were non-transcribed non-ORFs, evidencing they could be pseudogenes. We identified 173 de novo genes that likely originated from the activity of MULE, TNT, and RE transposases. The origins of the remaining de novo genes remain undetermined. The dispensable genes, a major player contributing to variability that codes for various Transposable Elements (TEs), proteolytic enzymes, and regulators of biological processes; their proportion was about 53.69% in the superTranscriptome. However, previous studies reported 63.6% of dispensable genes in sorghum compared with rice, soybean, and Brachypodium sp., suggesting that sorghum is more genetically diverse than other crops (Xin et al. 2021). The past studies on sorghum reported that Transposable Elements (TEs) may play a role in the gene content variation (Voelker et al. 2023) and this work substantiated it but processes, such as proteolysis and gene regulation, were not reported. Our findings showed that proteolysis and gene regulation contribute to the observed sorghum variability. Phylogenetic analysis based on gene presence/absence reported separate grouping of US and Asia-Europe origin sweet sorghum, highlighting geography-based selection of genes within sweet sorghum. Similar results were reported for gene PAV and SNP-based phylogenetic analysis of ten sorghum genome assemblies (Voelker et al. 2023). The 301 genes are exclusive to sweet sorghum genomes, including hexokinases, core histones, cytochromes, etc. These 301 were assigned gene ages based on their sequence similarity hits with c-DNA sequences of several C3–C4 outgroups and Arabidopsis which reported 18 were sweet sorghum-specific ones. The synteny-based comparison revealed that out of these 301, only 58 were de novo genes for which gene ages were determined. These genes were newly evolved in sweet sorghum. A recent study reported that some histones and unique chromatin remodeling factors were newly evolved in sorghum (Hu et al. 2022).
Our DEU analysis reported more true positive genes in the superTranscriptome-based approach than SSRG when datasets were trained with the KNeighborsClassifier method and gave better estimates of DEU when trained with the logistic regression method. superTranscriptome reported more differentially spliced genes; therefore, they were true transcripts. Additionally, the count-based method we used for gene-level abundance estimates and statistical inferences was superior to traditional TPM-based methods (Soneson et al. 2016). DEU analysis of Rio and PR22 during internode development suggested that differential splicing is extensive in Rio and could be one of the reasons for the trait development in Rio.
Using two different sources, superTranscriptome, and SSRG, we have reported more differentially expressed genes (P value < 0.05) with superTranscriptome than with SSRG. superTranscriptome gives better estimates of DGE than SSRG when trained datasets with the logistic regression method. This demonstrated superTranscriptome reduces bias caused by using a single reference genome for genetic analysis such as DEU and DGE (Davidson et al. 2017; Davidson and Oshlack 2018; Wang et al. 2023). DGE analysis of Rio and PR22 during internode development reported more LHCs and missing expression of Metallothionein-II in Rio under the Top 50 category, suggesting that higher expression of LHCs and lower expression of Metallothioneins contributes to sugary internode. Metallothioneins move metal ions around, like copper (Cu2+), and a lack of them causes metal ions to build up in different parts of plants (R. Benatti et al. 2014). The past studies on Chenopodium murale (Llerena et al. 2021), Colobanthus quitensis (Contreras et al. 2018), and sugarcane (Agarwala et al. 1993) reported copper concentrations stimulate sugar levels in various plant parts, indicating the presence of the metal ions inside the cell positively regulates sugar accumulation in internode tissues.
SUT, SWEET, MYB, and NAC were important gene families controlling internode development in sorghum (Mizuno et al. 2016; Zhang et al. 2018; Hennet et al. 2020). Past studies reported six SUT genes with elevated gene expression in sweet sorghum stem internodes when compared with grain sorghum (Li et al. 2014; Babst et al. 2021). In previous studies, SUT4 was reported with putative deletions in Rio (Cooper et al. 2019); our de novo transcriptome assembly analysis with 223 RNA-seq accessions reported its presence, suggesting that SUT4 is present in the sweet sorghum population. Several SUT genes were reported in maize (4–7)(Leach et al. 2017), rice (5)(Aoki et al. 2003; Hirose et al. 2010), wheat (4)(Deol et al. 2013), and barley (5)(Radchuk et al. 2017) with a variety of functions in growth and development. To date, 23 SWEET genes were reported in the sweet sorghum (Mizuno et al. 2016); however, SSRG reported only 21 SWEET genes, along with two with putative deletions, namely SWEET3–3 and SWEET8–2; but we reported 24 orthologs using a superTranscriptome-based approach. Interestingly, maize and foxtail millet also have the same number (24) of SWEET orthologs (Liu et al. 2022), suggesting that there could be a history of introgression of SWEET genes between sorghum, maize, and foxtail millets. Genome introgression between wild relatives and crops within the same family was reported in other crops also (Ellstrand et al. 1999; Hufford et al. 2013; Ananda et al. 2020). In sorghum, plant-specific NAC-TFs control a variety of traits, including high cellular biomass (Xia et al. 2018), insect-pest resistance (Zhang et al. 2013), and drought/salinity tolerance (Sanjari et al. 2019; Punia et al. 2021), etc. In this study, we reported six new loci for NAC genes. One of them was involved in secondary cell wall development, i.e., NAC-73–2* with remarkable PAV on 15 genotypes, proposing that this gene contributes to variability in sweet sorghum. The number of NAC genes identified by the superTranscriptome approach was comparatively less than previously reported in the grain sorghum genome (Kadier et al. 2017) and the loss of NAC genes might be the reason for sweet phenotype development. Novel histone genes were located at the lower arm of chromosome 8 on SSRG, suggesting that this region carries duplications of histone genes. The comparative genomics of sorghum and rice reported that 68.6% of genes were collinear between these two crops with several duplicated regions on chromosome 5 and 8 in sorghum (Paterson et al. 2009) which supports our findings. Most of our sweet sorghum-specific loci that encoded genes such as hexokinases, cytochrome b5, MYB, MULE-like transposes, and Zinc finger domain were greater in sweet sorghum than grain sorghum and formed clades with sorghum and sugarcane in the phylogeny; suggesting that these genes might be ancestral copies. Past studies reported that most of the sorghum duplications were derived from ancestral gene families and a new transposon family was a major contributor to recent duplication in sorghum (Guo et al. 2019). Interestingly, we found that MULE-like transposase genes were abundant in sweet sorghum, implying that this transposase family may contribute to ancestral gene duplications in sweet sorghum. However, the functions of those duplicated genes in sweet sorghum remained unknown. MULE-mediated duplication of host genes usually results in pseudogenes rather than new functional protein-coding genes (Juretic et al. 2005).
Our comparative analysis of one wild and two cultivated sorghum genomes using ScanPAV reported that both deletions and insertions were frequent during sorghum evolution. The dry gene or the D locus on chromosome 6 regulates internode growth and midrib color qualitatively and natural mutants for this gene produce sugary internodes (Zhang et al. 2018). We reported major deletions ~ 2 kb in the D locus in sweet sorghum (Rio) in comparison with the grain sorghum (BTX623) and wild type (353); suggesting that the partial D locus was a major player that governs internode phenotype. These findings were consistent with the earlier sorghum report on juicy grain type sorghum (BTX623) that carries ~ 1.8 kb deletions in the D locus when compared to dry grain type (Ji2731) (Zhang et al. 2018). We also identified four additional NAC loci in sweet sorghum, beyond the previously known D locus, that have been affected by terminal deletions compared to its wild progenitor (353). This suggests that deletions in NAC loci might play a significant role in the evolution of sweet sorghum. Previous research on the genomes of sweet and grain sorghum indicated that deletions and duplications could be found in a single gene or groups of related genes at the same time (Cooper et al. 2019). This suggests that deletions or duplications occurring in specific genes or clusters of related genes could potentially contribute to the differences observed between wild and cultivated sorghum.
Conclusion
The superTranscriptome identified 44,222 orthogroups in sweet sorghum consisting of 45,864 genes, with 301 genes unique to sweet sorghum. These genes are involved in functions such as chromatin organization, gene regulation, sugar metabolism, and cell wall synthesis. A gene age analysis suggested that 18 of these genes might be newly evolved in sweet sorghum. superTranscript-based Differential Exon Usage (DEU) and Differential Gene Expression (DGE) analysis provided more accurate estimates than traditional methods and showed potential as replacements. The DGE analysis of Rio and PR22 showed that the top 50 highly expressed gene lists differed between these two genotypes during internode growth. In Rio, several Light Harvesting Complexes (LHCs) were prominent, while metallothionein-II was absent in the top 50 categories. This suggests that active photosynthesis and metal ion transport play key roles in sugary internode development in Rio. Additionally, expression patterns of various long non-coding RNAs (lncRNAs) and transcription factors (TFs) were linked to sugar accumulation. The superTranscriptome also identified more orthologs for agronomically important genes and several tandem duplications across 15 genome assemblies. Rio reported the highest number (2,154) of tandem duplicates across all genomes, possibly due to transposase activity. Significant deletions (> 2 kb) in the D locus and terminal deletions in the other four NAC encoding loci were observed in sweet sorghum compared to its wild type (353) which could be one of the contributors to sweet sorghum evolution.
Data Availability
The superTranscriptome assembly is publicly available at the European Nucleotide Archive (ENA) [https://www.ebi.ac.uk/ena/browser/home] with accession number GCA_963506585. Improved genome annotations (*. gff3 files) for 15 diverse sorghum genomes, which include new gene additions and splicing updates using superTranscriptome with PASA gene structure annotation pipeline have been provided with supplementary datasets. The R and Python codes used in the present analysis are publicly available on GitHub with repositories named Sorghum superTranscriptome [https://github.com/nikhilshinde0909/Sorghum-superTranscriptome] and Modified necklace pipeline [https://github.com/nikhilshinde0909/Modified-Necklace-Pipeline]. The supplemental data and tables have been submitted along with the manuscript.
Abbreviations
- PAV:
-
Presence/absence variation
- ePAV:
-
Expression presence/absence variation
- gPAV:
-
Genomic presence/absence variation
- SV:
-
Structural variations
- kb:
-
Kilobases
- RTAs:
-
Representative transcript assemblies
- CNV:
-
Copy number variation
- HISAT2:
-
Hierarchical indexing for spliced alignment of transcripts version 2
- BUSCO:
-
Benchmarking universal single-copy orthologs
- CPC2:
-
Coding potential calculator version 2
- GFF3:
-
General feature format type 3
- SAM:
-
Sequence alignment map
- BAM:
-
Binary alignment map
- LHCs:
-
Light harvesting complexes
- DGE:
-
Differential gene expression
- MB:
-
Mega base pair
- DEU:
-
Differential exon usage
- NCBI:
-
National center for biotechnology information
- SRA:
-
Sequence read archives
- aa:
-
Amino acid
- bp:
-
Base pairs
References
Agarwala SC, Chatterjee C, Nautiyal N, Jain R (1993) Sugar-cane response to copper in refined sand. TROPAGRIC 70. https://journals.sta.uwi.edu/ojs/index.php/ta/article/view/7125
Ananda GKS, Myrans H, Norton SL et al (2020) Wild sorghum as a promising resource for crop improvement. Front. Plant Sci. 11:1108. https://doi.org/10.3389/fpls.2020.01108
Anders S, Reyes A, Huber W (2012) Detecting differential usage of exons from RNA-seq data. Genome Res. https://doi.org/10.1101/gr.133744.111
Aoki N, Hirose T, Scofield GN et al (2003) The sucrose transporter gene family in rice. Plant Cell Physiol. https://doi.org/10.1093/pcp/pcg030
Babst BA, Karve A, Sementilli A et al (2021) Physiology and whole-plant carbon partitioning during stem sugar accumulation in sweet dwarf sorghum. Planta. https://doi.org/10.1007/s00425-021-03718-w
Benatti RM, Yookongkaew N, Meetam M et al (2014) Metallothionein deficiency impacts copper accumulation and redistribution in leaves and seeds of Arabidopsis. New Phytol. https://doi.org/10.1111/nph.12718
Berdan EL, Aubier TG, Cozzolino S et al (2024) Structural variants and speciation: multiple processes at Play. Cold Spring Harb Perspect Biol. https://doi.org/10.1101/cshperspect.a041446
Bhatti A, Shah FS, Azhar J et al (2020) Pan-transcriptomics and its applications. In: Pan-genomics: Applications challenges and future prospects. https://doi.org/10.1016/B978-0-12-817076-2.00018-4
Blighe K, Rana S, Lewis M (2022) Publication-ready volcano plots with enhanced colouring and labeling. R package version 1.0 (2019):10-18129
Boatwright JL, Brenton ZW, Boyles RE et al (2021) Genetic characterization of a Sorghum bicolor multiparent mapping population emphasizing carbon-partitioning dynamics. G3 Genes Genomes, Genet. https://doi.org/10.1093/g3journal/jkab060
Boutet E, Lieberherr D, Tognolli M et al (2016) Uniprotkb/swiss-prot, the manually annotated section of the uniprot knowledgebase: How to use the entry view. In: Methods in Molecular Biology. https://doi.org/10.1007/978-1-4939-3167-5_2
Brasó-Vives M, Marlétaz F, Echchiki A et al (2022) Parallel evolution of amphioxus and vertebrate small-scale gene duplications. Genome Biol. https://doi.org/10.1186/s13059-022-02808-6
Broadhead DM, Freeman KC, Zummo N (1981) Registration of wray sweet sorghum 1 (Reg. No. 119). Crop Sci. https://doi.org/10.2135/cropsci1981.0011183x002100060048x
Burks PS, Kaiser CM, Hawkins EM, Brown PJ (2015) Genomewide association for sugar yield in sweet sorghum. Crop Sci. https://doi.org/10.2135/cropsci2015.01.0057
Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T (2009) trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. https://doi.org/10.1093/bioinformatics/btp348
Chen S, Zhou Y, Chen Y, Gu J (2018) Fastp: An ultra-fast all-in-one FASTQ preprocessor. In: Bioinformatics. https://doi.org/10.1093/bioinformatics/bty560
Contreras RA, Pizarro M, Köhler H et al (2018) Copper stress induces antioxidant responses and accumulation of sugars and phytochelatins in Antarctic Colobanthus quitensis (Kunth) Bartl. Biol Res. https://doi.org/10.1186/s40659-018-0197-0
Cooper EA, Brenton ZW, Flinn BS et al (2019) A new reference genome for Sorghum bicolor reveals high levels of sequence similarity between sweet and grain genotypes: implications for the genetics of sugar metabolism. BMC Genomics. https://doi.org/10.1186/S12864-019-5734-X
Dai X, Sinharoy S, Udvardi M, Zhao PX (2013) PlantTFcat: an online plant transcription factor and transcriptional regulator categorization and analysis tool. BMC Bioinformatics. https://doi.org/10.1186/1471-2105-14-321
Davidson NM, Oshlack A (2014) Corset: Enabling differential gene expression analysis for de novo assembled transcriptomes. Genome Biol. https://doi.org/10.1186/s13059-014-0410-6
Davidson NM, Oshlack A (2018) Necklace: combining reference and assembled transcriptomes for more comprehensive RNA-Seq analysis. Gigascience. https://doi.org/10.1093/gigascience/giy045
Davidson NM, Hawkins ADK, Oshlack A (2017) SuperTranscripts: a data driven reference for analysis and visualisation of transcriptomes. Genome Biol. https://doi.org/10.1186/s13059-017-1284-1
Deol KK, Mukherjee S, Gao F et al (2013) Identification and characterization of the three homeologues of a new sucrose transporter in hexaploid wheat (Triticum aestivum L.). BMC Plant Biol. https://doi.org/10.1186/1471-2229-13-181
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. https://doi.org/10.1093/nar/gkh340
Ellstrand NC, Prentice HC, Hancock JF (1999) Gene flow and introgression from domesticated plants into their wild relatives. Annu Rev Ecol Syst. https://doi.org/10.1146/annurev.ecolsys.30.1.539
Emms DM, Kelly S (2019) OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. https://doi.org/10.1186/s13059-019-1832-y
F. C, B.J. H, S.M.D. G, et al (2006) Sequencing Medicago truncatula expressed sequenced tags using 454 Life Sciences technology. BMC Genomics
Gerdol M, Moreira R, Cruz F et al (2020) Massive gene presence-absence variation shapes an open pan-genome in the Mediterranean mussel. Genome Biol. https://doi.org/10.1186/s13059-020-02180-3
Ghaffari N, Sanchez-Flores A, Doan R et al (2014) Novel transcriptome assembly and improved annotation of the whiteleg shrimp (Litopenaeus vannamei), a dominant crustacean in global seafood mariculture. Sci Rep. https://doi.org/10.1038/srep07081
Giordano F, Stammnitz MR, Murchison EP, Ning Z (2018) ScanPAV: a pipeline for extracting presence-absence variations in genome pairs. Bioinformatics. https://doi.org/10.1093/bioinformatics/bty189
Gómez-Rubio V (2017) ggplot2—elegant graphics for data analysis 2nd Edition. J Stat Softw. https://doi.org/10.18637/jss.v077.b02
Grabherr MG, Haas BJ, Yassour M et al (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. https://doi.org/10.1038/nbt.1883
Guo AY, Chen X, Gao G et al (2008) PlantTFDB: a comprehensive plant transcription factor database. Nucleic Acids Res. https://doi.org/10.1093/nar/gkm841
Guo H, Jiao Y, Tan X et al (2019) Gene duplication and genetic innovation in cereal genomes. Genome Res. https://doi.org/10.1101/gr.237511.118
Gupta P, Naithani S, Tello-Ruiz MK et al (2016) Gramene database: navigating plant comparative genomics resources. Curr Plant Biol. https://doi.org/10.1016/j.cpb.2016.12.005
Haas BJ, Salzberg SL, Zhu W et al (2008) Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. https://doi.org/10.1186/gb-2008-9-1-r7
Han G, Lu C, Guo J et al (2020) C2H2 zinc finger proteins: master regulators of abiotic stress responses in plants. Front. Plant Sci. 11:115. https://doi.org/10.3389/fpls.2020.00115
Hao Z, Lv D, Ge Y et al (2020) RIdeogram: drawing SVG graphics to visualize and map genome-wide data on the idiograms. PeerJ Comput Sci. https://doi.org/10.7717/peerj-cs.251
Hennet L, Berger A, Trabanco N et al (2020) Transcriptional regulation of sorghum stem composition: key players identified through co-expression gene network and comparative genomics analyses. Front Plant Sci. https://doi.org/10.3389/fpls.2020.00224
Hirose T, Zhang Z, Miyao A et al (2010) Disruption of a gene for rice sucrose transporter, OsSUT1, impairs pollen function but pollen maturation is unaffected. J Exp Bot. https://doi.org/10.1093/jxb/erq175
Hirsch CN, Foerster JM, Johnson JM et al (2014) Insights into the maize pan-genome and pan-transcriptome. Plant Cell. https://doi.org/10.1105/tpc.113.119982
Hu Y, Chen X, Zhou C et al (2022) Genome-wide identification of chromatin regulators in Sorghum bicolor. 3 Biotech. https://doi.org/10.1007/s13205-022-03181-8
Hufford MB, Lubinksy P, Pyhäjärvi T et al (2013) The Genomic signature of crop-wild introgression in maize. PLoS Genet. https://doi.org/10.1371/journal.pgen.1003477
Hufford MB, Seetharam AS, Woodhouse MR et al (2021) De novo assembly, annotation, and comparative analysis of 26 diverse maize genomes. Science. https://doi.org/10.1126/science.abg5289
Jayakodi M, Padmarasu S, Haberer G et al (2020) The barley pan-genome reveals the hidden legacy of mutation breeding. Nature. https://doi.org/10.1038/s41586-020-2947-8
Jiang SY, Ma Z, Vanitha J, Ramachandran S (2013) Genetic variation and expression diversity between grain and sweet sorghum lines. BMC Genomics. https://doi.org/10.1186/1471-2164-14-18
Jiao WB, Schneeberger K (2017) The impact of third generation genomic technologies on plant genome assembly. Curr. Opin. Plant Biol. 36:64. https://doi.org/10.1016/j.pbi.2017.02.002
Jin M, Liu H, He C et al (2016) Maize pan-transcriptome provides novel insights into genome complexity and quantitative trait variation. Sci Rep. https://doi.org/10.1038/srep18936
Jin J, Lu P, Xu Y et al (2021) PLncDB V.20: a comprehensive encyclopedia of plant long noncoding RNAs. Nucleic Acids Res. https://doi.org/10.1093/nar/gkaa910
Jobson E, Roberts R (2022) Genomic structural variation in tomato and its role in plant immunity. Mol Hortic. https://doi.org/10.1186/s43897-022-00029-w
Juretic N, Hoen DR, Huynh ML et al (2005) The evolutionary fate of MULE-mediated duplications of host gene fragments in rice. Genome Res. https://doi.org/10.1101/gr.4064205
Kadier Y, Zu Y, yi, Dai Q min, et al (2017) Genome-wide identification, classification and expression analysis of NAC family of genes in sorghum [Sorghum bicolor (L.) Moench]. Plant Growth Regul. https://doi.org/10.1007/s10725-017-0295-y
Kang YJ, Yang DC, Kong L et al (2017) CPC2: a fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. https://doi.org/10.1093/nar/gkx428
Kent WJ (2002) BLAT —the BLAST -like alignment tool. Genome Res. https://doi.org/10.1101/gr.229202
Kim D, Paggi JM, Park C et al (2019) Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. https://doi.org/10.1038/s41587-019-0201-4
Kumar S, Suleski M, Craig JM et al (2022) Timetree 5: an expanded resource for species divergence times. Mol Biol Evol. https://doi.org/10.1093/molbev/msac174
Leach KA, Tran TM, Slewinski TL et al (2017) Sucrose transporter2 contributes to maize growth, development, and crop yield. J Integr Plant Biol. https://doi.org/10.1111/jipb.12527
Letunic I, Bork P (2021) Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. https://doi.org/10.1093/nar/gkab301
Li H, Durbin R (2010) Fast and accurate long-read alignment with burrows-wheeler transform. Bioinformatics. https://doi.org/10.1093/bioinformatics/btp698
Li L, Stoeckert CJ, Roos DS (2003) OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. https://doi.org/10.1101/gr.1224503
Li H, Handsaker B, Wysoker A et al (2009) The sequence alignment/map format and SAMtools. Bioinformatics. https://doi.org/10.1093/bioinformatics/btp352
Li X, Su M, Li X et al (2014) Molecular characterization and expression patterns of sucrose transport-related genes in sweet sorghum under defoliation. Acta Physiol Plant. https://doi.org/10.1007/s11738-014-1505-0
Li C, Song W, Luo Y et al (2019a) The HuangZaoSi maize genome provides insights into genomic variation and improvement history of maize. Mol Plant. https://doi.org/10.1016/j.molp.2019.02.009
Li Y, Wang W, Feng Y et al (2019b) Transcriptome and metabolome reveal distinct carbon allocation patterns during internode sugar accumulation in different sorghum genotypes. Plant Biotechnol J. https://doi.org/10.1111/pbi.12991
Liao Y, Smyth GK, Shi W (2014) FeatureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. https://doi.org/10.1093/bioinformatics/btt656
Liu Z, Fan H, Ma Z (2022) Comparison of SWEET gene family between maize and foxtail millet through genomic, transcriptomic, and proteomic analyses. Plant Genome. https://doi.org/10.1002/tpg2.20226
Llerena JPP, Coasaca RL, Rodriguez HOL et al (2021) Metallothionein production is a common tolerance mechanism in four species growing in polluted Cu mining areas in Peru. Ecotoxicol Environ Saf. https://doi.org/10.1016/j.ecoenv.2021.112009
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. https://doi.org/10.1186/s13059-014-0550-8
Lv Y, Hu F, Zhou Y et al (2019) Maize transposable elements contribute to long non-coding RNAs that are regulatory hubs for abiotic stress response. BMC Genomics. https://doi.org/10.1186/s12864-019-6245-5
Ma Y, Liu M, Stiller J, Liu C (2019) A pan-transcriptome analysis shows that disease resistance genes have undergone more selection pressure during barley domestication 06 biological sciences 0604 genetics. BMC Genomics. https://doi.org/10.1186/s12864-018-5357-7
Mansour A (2009) Phylip and phylogenetics. Focus Bioinforma Genes, Genomes Genomics 3:46
McCormick RF, Truong SK, Sreedasyam A et al (2018) The sorghum bicolor reference genome: improved assembly, gene annotations, a transcriptome atlas, and signatures of genome organization. Plant J. https://doi.org/10.1111/tpj.13781
Medina CA, Samac DA, Yu LX (2021) Pan-transcriptome identifying master genes and regulation network in response to drought and salt stresses in Alfalfa (Medicago sativa L.). Sci Rep. https://doi.org/10.1038/s41598-021-96712-x
Mizuno H, Kasuga S, Kawahigashi H (2016) The sorghum SWEET gene family: stem sucrose accumulation as revealed through transcriptome profiling. Biotechnol Biofuels. https://doi.org/10.1186/s13068-016-0546-6
Moriya Y, Itoh M, Okuda S et al (2007) KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. https://doi.org/10.1093/nar/gkm321
Nguyen LT, Schmidt HA, Von Haeseler A, Minh BQ (2015) IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. https://doi.org/10.1093/molbev/msu300
Pagès H, Carlson M, Falcon S, Li N (2022) AnnotationDbi: Manipulation of SQLite-based annotations in Bioconductor. R Packag version 1
Paterson AH, Bowers JE, Bruggmann R et al (2009) The Sorghum bicolor genome and the diversification of grasses. Nature. https://doi.org/10.1038/nature07723
Pertea M, Pertea GM, Antonescu CM et al (2015) StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. https://doi.org/10.1038/nbt.3122
Pertea G, Pertea M (2020) GFF Utilities: GffRead and GffCompare. F1000Research. https://doi.org/10.12688/f1000research.23297.2
Petek M, Zagorščak M, Ramšak Ž et al (2020) Cultivar-specific transcriptome and pan-transcriptome reconstruction of tetraploid potato. Sci Data. https://doi.org/10.1038/s41597-020-00581-4
Prabh N, Rödelsperger C (2022) Multiple Pristionchus pacificus genomes reveal distinct evolutionary dynamics between de novo candidates and duplicated genes. Genome Res. https://doi.org/10.1101/gr.276431.121
Punia H, Tokas J, Malik A et al (2021) Genome-wide transcriptome profiling, characterization, and functional identification of nac transcription factors in sorghum under salt stress. Antioxidants. https://doi.org/10.3390/antiox10101605
Qin P, Lu H, Du H et al (2021) Pan-genome analysis of 33 genetically diverse rice accessions reveals hidden genomic variations. Cell. https://doi.org/10.1016/j.cell.2021.04.046
Quinlan AR, Hall IM (2010) BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics. https://doi.org/10.1093/bioinformatics/btq033
Radchuk V, Riewe D, Peukert M et al (2017) Down-regulation of the sucrose transporters HvSUT1 and HvSUT2 affects sucrose homeostasis along its delivery path in barley grains. J Exp Bot. https://doi.org/10.1093/jxb/erx266
Rao PS, Vinutha KS, Kumar GSA, et al (2019) Sorghum: A multipurpose bioenergy crop. In: Sorghum: State of the Art and Future Perspectives. https://doi.org/10.2134/agronmonogr58.c18
Ruperao P, Thirunavukkarasu N, Gandham P et al (2021) Sorghum pan-genome explores the functional utility for genomic-assisted breeding to accelerate the genetic gain. Front Plant Sci. https://doi.org/10.3389/fpls.2021.666342
Sadedin SP, Pope B, Oshlack A (2012) Bpipe: a tool for running and managing bioinformatics pipelines. Bioinformatics. https://doi.org/10.1093/bioinformatics/bts167
Sanjari S, Shirzadian-Khorramabad R, Shobbar ZS, Shahbazi M (2019) Systematic analysis of NAC transcription factors’ gene family and identification of post-flowering drought stress responsive members in sorghum. Plant Cell Rep. https://doi.org/10.1007/s00299-019-02371-8
Simão FA, Waterhouse RM, Ioannidis P et al (2015) BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. https://doi.org/10.1093/bioinformatics/btv351
Smith O, Nicholson WV, Kistler L et al (2019) A domestication history of dynamic adaptation and genomic deterioration in Sorghum. Nat Plants. https://doi.org/10.1038/s41477-019-0397-9
Smith-Unna R, Boursnell C, Patro R et al (2016) TransRate: reference-free quality assessment of de novo transcriptome assemblies. Genome Res. https://doi.org/10.1101/gr.196469.115
Soneson C, Matthes KL, Nowicka M et al (2016) Isoform prefiltering improves performance of count-based methods for analysis of differential transcript usage. Genome Biol. https://doi.org/10.1186/s13059-015-0862-3
Statello L, Guo CJ, Chen LL, Huarte M (2021) Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 22:96. https://doi.org/10.1038/s41580-020-00315-9
Sui N, Yang Z, Liu M, Wang B (2015) Identification and transcriptomic profiling of genes involved in increasing sugar content during salt stress in sweet sorghum leaves. BMC Genomics. https://doi.org/10.1186/s12864-015-1760-5
Sun C, Hu Z, Zheng T et al (2017) RPAN: rice pan-genome browser for ∼3000 rice genomes. Nucleic Acids Res. https://doi.org/10.1093/nar/gkw958
Sun X, Zheng H, Li J et al (2020) Comparative transcriptome analysis reveals new lncRNAs responding to salt stress in sweet sorghum. Front Bioeng Biotechnol. https://doi.org/10.3389/fbioe.2020.00331
Sweeney BA, Tagmazian AA, Ribas CE et al (2020) exploring non-coding RNAs in RNAcentral. Curr Protoc Bioinforma. https://doi.org/10.1002/cpbi.104
Tao Y, Luo H, Xu J et al (2021) Extensive variation within the pan-genome of cultivated and wild sorghum. Nat Plants. https://doi.org/10.1038/s41477-021-00925-x
Unver T, Tombuloglu H (2020) Barley long non-coding RNAs (lncRNA) responsive to excess boron. Genomics. https://doi.org/10.1016/j.ygeno.2019.11.007
Varoquaux N, Cole B, Gao C et al (2019) Transcriptomic analysis of field-droughted sorghum from seedling to maturity reveals biotic and metabolic responses. Proc Natl Acad Sci U S A. https://doi.org/10.1073/pnas.1907500116
Venkateswaran K, Elangovan M, Sivaraj N (2018) Origin, domestication and diffusion of Sorghum bicolor. In: Breeding Sorghum for Diverse End Uses. https://doi.org/10.1016/B978-0-08-101879-8.00002-4
Voelker WG, Krishnan K, Chougule K et al (2023) Ten new high-quality genome assemblies for diverse bioenergy sorghum genotypes. Front Plant Sci. https://doi.org/10.3389/fpls.2022.1040909
Wang Y, Tang H, Debarry JD et al (2012) MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. https://doi.org/10.1093/nar/gkr1293
Wang H, Niu QW, Wu HW et al (2015) Analysis of non-coding transcriptome in rice and maize uncovers roles of conserved lncRNAs associated with agriculture traits. Plant J. https://doi.org/10.1111/tpj.13018
Wang J, Yang W, Zhang S et al (2023) A pangenome analysis pipeline provides insights into functional gene identification in rice. Genome Biol. https://doi.org/10.1186/s13059-023-02861-9
Wang B, Jiao Y, Chougule K et al (2021) Pan-genome analysis in sorghum highlights the extent of genomic variation and sugarcane aphid resistance genes. bioRxiv. https://doi.org/10.1101/2021.01.03.424980
Wu T, Hu E, Xu S et al (2021) clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innov. https://doi.org/10.1016/j.xinn.2021.100141
Xia J, Zhao Y, Burks P et al (2018) A sorghum NAC gene is associated with variation in biomass properties and yield potential. Plant Direct. https://doi.org/10.1002/pld3.70
Xin Z, Wang M, Cuevas HE et al (2021) Sorghum genetic, genomic, and breeding resources. Planta 254:114. https://doi.org/10.1007/s00425-021-03742-w
Yang N, Xu XW, Wang RR et al (2017) Contributions of zea mays subspecies mexicana haplotypes to modern maize. Nat Commun. https://doi.org/10.1038/s41467-017-02063-5
Yang T, Liu R, Luo Y et al (2022) Improved pea reference genome and pan-genome highlight genomic features and evolutionary characteristics. Nat Genet. https://doi.org/10.1038/s41588-022-01172-2
Yao W, Li G, Zhao H et al (2015) Exploring the rice dispensable genome using a metagenome-like assembly strategy. Genome Biol. https://doi.org/10.1186/s13059-015-0757-3
Yao C, Yan H, Zhang X, Wang R (2017) A database for orphan genes in poaceae. Exp Ther Med. https://doi.org/10.3892/etm.2017.4918
Zhang LM, Leng CY, Luo H et al (2018) Sweet sorghum originated through selection of dry, a plant-specific nac transcription factor gene[open]. Plant Cell. https://doi.org/10.1105/tpc.18.00313
Zhang L, Ren Y, Yang T et al (2019) Rapid evolution of protein diversity by de novo origination in Oryza. Nat Ecol Evol. https://doi.org/10.1038/s41559-019-0822-5
Zhang H, Huang Y, Zhang H, Huang Y (2013) Genome-wide survey and characterization of greenbug induced nac transcription factors in sorghum [Sorghum bicolor (L.) Moench]. Plant Anim Genome
Zhao Q, Feng Q, Lu H et al (2018) Pan-genome analysis highlights the extent of genomic variation in cultivated and wild rice. Nat Genet. https://doi.org/10.1038/s41588-018-0041-z
Zheng LY, Sen GX, He B et al (2011) Genome-wide patterns of genetic variation in sweet and grain sorghum (Sorghum bicolor). Genome Biol. https://doi.org/10.1186/gb-2011-12-11-r114
Zheng Y, Jiao C, Sun H et al (2016) iTAK : a program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinase Mol Plant 9:1667. https://doi.org/10.1016/j.molp.2016.09.014
Zhou R, Sanz-Jimenez P, Zhu XT et al (2021) Analysis of rice transcriptome reveals the LncRNA/CircRNA Regulation in tissue development. Rice. https://doi.org/10.1186/s12284-021-00455-2
Zhou W, Zhou Y, Wang Z et al (2022) Comparative transcriptome analysis in three sorghum (Sorghum bicolor) cultivars reveal genomic basis of differential seed quality. Plant Biosyst. https://doi.org/10.1080/11263504.2020.1851790
Acknowledgements
We thank the SRM Institute of Science and Technology in Kattankulathur, Chennai (India), for providing fellowships and the essential infrastructure for this research. We also acknowledge the SRM HPCC facility for offering computational support.
Funding
None.
Author information
Authors and Affiliations
Contributions
Conceptualization: SN, NSR. Methodology: SN, HSM. Investigation: SN, NSR. Visualization: SN, NSR, HSM. Supervision: NSR, HSM. Writing-original draft: SN, NSR. Writing-review & editing: SN, NSR.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Handling editor: Bojian Zhong.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Nikhil, S., Mohideen, H.S. & Sella, R.N. Unveiling the Genomic Symphony: Identification Cultivar-Specific Genes and Enhanced Insights on Sweet Sorghum Genomes Through Comprehensive superTranscriptomic Analysis. J Mol Evol (2024). https://doi.org/10.1007/s00239-024-10198-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00239-024-10198-5