
Abstract
We sequenced to completion the circular plastid genome of the red alga Gracilaria tenuistipitata var. liui. This is the first plastid genome sequence from the subclass Florideophycidae (Rhodophyta). The genome is composed of 183,883 bp and contains 238 predicted genes, including a single copy of the ribosomal RNA operon. Comparisons with the plastid genome of Porphyra pupurea reveal strong conservation of gene content and order, but we found major genomic rearrangements and the presence of coding regions that are specific to Gracilaria. Phylogenetic analysis of a data set of 41 concatenated proteins from 23 plastid and two cyanobacterial genomes support red algal plastid monophyly and a specific evolutionary relationship between the Florideophycidae and the Bangiales. Gracilaria maintains a surprisingly ancient gene content in its plastid genome and, together with other Rhodophyta, contains the most complete repertoire of plastid genes known in photosynthetic eukaryotes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Plastids are the photosynthetic organelles of eukaryotes and their genomes range in size from approximately 35 to 200 kb. Relatively few of these sequences have been determined (35 completed) in comparison to the mitochondrial genome (529 completed; see http://www.ncbi.nlm.nih.gov:80/genomes/static/euk_o.html). Furthermore, most of the sequenced plastid genomes (25 in total) are of chlorophyll b–containing algae and plants. The handful that have been sequenced from the diverse group of “nongreen” protists include representatives of the Alveolata (Eimeria tenella, Toxoplasma gondii), Cryptophyta (Guillardia theta), Euglenozoa (Euglena longa and Euglena gracilis), Glaucophyta (Cyanophora paradoxa), Stramenopiles (Odontella sinensis), and Rhodophyta (Cyanidium caldarium, Cyanidioschyzon merolae, and Porphyra purpurea) (see Table 1). Importantly, these nongreen taxa comprise the wealth of protistan diversity including the major phytoplankton in the oceans (e.g., chlorophyll c–containing forms such as diatoms and haptophytes) and many toxic species (e.g., dinoflagellates and brown algae). Here we present the complete plastid genome sequence of the florideophyte red alga, Gracilaria tenuistipitata var. liui, with the aim of further elucidating plastid evolution.
It is now generally accepted that plastids originated through endosymbiosis, whereby a single-celled protist engulfed and retained a foreign photosynthetic cell inside a vacuole (Gray 1992; Bhattacharya and Medlin 1995). Over time, the foreign cell was reduced to a plastid and transmitted to subsequent generations. Plastid origin occurred via primary and secondary endosymbiosis. The first involves the engulfment of a photosynthetic prokaryote (cyanobacterium) and is believed to have occurred once in evolution (but see Stiller and Hall 1997; Stiller et al. 2003), giving rise to the protoalga that is the ultimate root of all plastids. This primary plastid eventually became established in the first algae that split into two lineages. The first produced the glaucophyte algae, whereas the second gave rise to the highly successful red algae and their sister group the green algae and land plants (Moreira et al. 2000; Palmer 2003). Once the three primary algal lineages were established, the stage was set for secondary endosymbiosis, whereby a protist engulfed an existing alga. This type of eukaryotic–eukaryotic endosymbiosis accounts for the vast majority of algal diversity (Bhattacharya et al. 2004). Secondary plastids are found in the ecologically and economically important chlorophyll c–containing chromalveolates that define a broadly diverse group including the Chromista (cryptophyte, haptophyte, and stramenopile algae) and the Alveolata (parasitic apicomplexans, apparently plastidless ciliates, and dinoflagellate algae). The chromalveolate hypothesis is based primarily on the idea that all taxa containing a chromophytic plastid share a common origin (Cavalier-Smith 1986). The chromalveolate plastid is believed to have originated from a single red algal secondary endosymbiosis that occurred ca. 1.2 billion years ago in the ancestor of this group with the ensuing evolution of chlorophyll c2 (Yoon et al. 2002). This scenario, however, remains to be substantiated with analysis of nuclear genes from chromalveolates (Bhattacharya et al. 2004). The plastid was putatively lost in ciliates and parasitic/saprobic stramenopiles like oomycetes (e.g., the water mold Achlya), and the genome was reduced to a 35-kb DNA circle in the apicomplexans (McFadden 2001; Andersson and Roger 2002). The red algae have, therefore, contributed the photosynthetic organelle to a major branch of the protist tree of life. Secondary endosymbiosis also explains the origin of the green (i.e., chlorophyll b-containing) plastids of the Euglenozoa (e.g., Hannaert et al. 2003; Martin and Borst 2003) and Chlorarachniophyta (Gilson and McFadden 1995; Archibald et al. 2003). However, it is unclear whether these organelles arose independently or from a single endosymbiotic event (Bhattacharya et al. 2004).
Important challenges that remain in the field of endosymbiosis research are to document genome evolution in different plastids, to understand the timing and nature of gene transfer to the nuclear compartment, to use the multigene approach with complete plastid genome sequences to generate a robust plastid phytogeny, to definitively test the monophyly of red algal-derived secondary plastids, and to provide a framework for mapping gene transfer and the evolution of other characters associated with photosynthesis. We used comparative methods with the complete Gracilaria plastid genome sequence to address these issues. Our data also open up the possibility for genetic engineering of this economically important red seaweed (Bock 2001; Gewolb 2002).
Materials and Methods
Algal Cultures and Plastid DNA Purification
Gracilaria tenuistipitata var. liui Zhang et Xia was collected in Haikou, China (5 June 1990) by E.C. de Oliveira. Tetrasporophyte cultures were maintained at 25°C under a 14-h light period (150–170 μmol photons m−2 s−1) with constant aeration in sterilized filtered seawater (20 ppm) that was changed weekly and supplemented with Von Stosch nutrients (Oliveira et al. 1995). Purification of plastid DNA from an enriched rhodoplast fraction was done as described by Hagopian et al. (2002).
Genomic Library Construction and Sequencing
The complete plastid genome sequence was generated using a combination of shotgun libraries and PCR strategies. Shotgun libraries were constructed with fragments (1–3 kb) of nebulized, purified plastid DNA cloned into pUC18 (Pharmacia). Sequencing reactions were performed with BigDye terminators and analyzed on ABI Prism 377 and 3700 automated sequencers.
Genome Sequence Assembly
Assembly was accomplished with the phred-phrap-consed package (Gordon et al. 1998), which was used to screen for vector and common contaminant sequences. The initial assembly of random clone sequences generated three large plastid contigs that contained 89% of all screened reads. This value provided an accurate estimate of plastid DNA purity. Contigs were unified and overall coverage was enhanced by reverse sequencing of selected clones. A final gap of about 500 bp was amplified in multiple PCR reactions that were directly sequenced. The final sequence quality standard was achieved by sequencing select clones with 1 of 26 primers. Every base of the plastid genome has minimum quality with Phred value of at least 20 and was confirmed in both directions by a minimum of three reads. There are no unexplained high-quality discrepancies, and the overall error estimate is less than 0.001 in every 10,000 bases. A total of 4441 reads were used in the genome assembly, providing approximately nine-fold genome coverage. The genome sequence has been deposited in GenBank with the accession number AY673996.
Genome Annotation and Analysis
Open reading frames (ORFs) were identified using GLIMMER (Delcher et al. 1999) and alignment against the National Center for Biotechnology Information (NCBI) protein database using BLASTX (Altschul et al. 1997). BLASTX searches were carried out to find additional putative protein-coding genes at the intergenic spacers. All ORFs were inspected manually and checked against Cluster of Orthologous Groups of Proteins (COG), Protein Family Database (PFAM), and the Kyoto Encyclopedia of Genes and Genomes (KEGG). RNA species were identified using BLASTN (Altschul et al. 1997), secondary structure analysis, and tRNAscan-SE (Lowe and Eddy 1997). Identified genes were named as in Stoebe et al. (1998). For a full list of ORFs, gene map, and comparative tables, refer to the supplementary material at http://gracilaria.lbi.ic.unicamp.br/chloroplast Whole plastid genomes were compared at the nucleotide level using the program Cross-Match (Gordon et al. 1998).
Phylogenetic Analyses
We analyzed a concatenated protein data set from 23 completely sequenced plastid genomes (including Gracilaria) and two cyanobacteria (Nostoc sp. PCC 7120 [GenBank NC 003272] and Synechocystis sp. PCC 6830 [NC_000911]) as the outgroup (see http://www.ncbi.nlm.nih.gov ). The chosen taxa represent a broad sample of photosynthetic eukaryotes including Glaucophyta, Rhodophyta, Chromista, Chlorophyta, and Streptophyta (Martin et al. 1998, 2002). A total of 41 protein-coding genes shared by these evolutionarily divergent genomes were combined to create a data set of 8810 amino acids of unambiguously aligned sequence positions (alignment available at http://gracilaria.lbi.ic.unicamp.br/chloroplast). The proteins that we used in the phylogenetic analyses and the length of each aligned data set are as follows: atpA (451 aa), atpB (459 aa), atpE (116 aa), atpF (32 aa), atpH (80 aa), ccsA (156 aa), petA (263 aa), petB (214 aa), petD (159), petG (35 aa), psaA (743 aa), psaB (734 aa), psaC (79 aa), psaJ (36 aa), psbA (343 aa), psbB (504 aa), psbC (449 aa), psbD (338 aa), psbE (73 aa), psbF (34 aa), psbH (58 aa), psbL (36 aa), psbN (41 aa), psbT (30 aa), rp12 (236 aa), rpl14 (121 aa), rpl16 (128 aa), rp120 (80 aa), rpoA (176 aa), rpoB (667 aa), rpoC1 (406 aa), rpoC2 (475 aa), rps2 (217 aa), rps3 (121 aa), rps4 (158 aa), rps7 (128 aa), rps8 (97 aa), rps12 (122 aa), rps14 (75 aa), rps18 (52 aa), and rps19 (88 aa).
To assess if different proteins supported conflicting plastid phytogenies, we divided the data into functionally related groups under the assumption that interacting proteins could potentially share a common evolutionary history (e.g., de Queiroz et al. 1995 [but see Vogl et al. 2003]). The first major group was defined by photosystem (PSII + II) I and II proteins (i.e., psaX and psbX proteins) and was of total length 3498 aa and the second major group included proteins involved in transcription and translation ([T + T] i.e., ribosomal proteins and RNA polymerase subunits) and was of total length 3347 aa. We also generated data sets of only the ribosomal proteins (1623 aa), only the RNA polymerase subunits (1724 aa), the ATP synthase subunits (1138 aa), the pet proteins (671 aa), and the total photosynthetic apparatus (i.e., PSI + II, ATP synthase subunits, electron transfer [petA, B, D, and G], and ccsA–5463 aa).
A maximum likelihood (ML) approach was used to reconstruct the plastid trees and three other methods were used to infer support for nodes in these phylogenies. For the ML approach, PROML in PHYLIP V3.6b (Felsenstein 2003) was used to generate globally optimized protein trees (one random addition, JTT + Γ model). The gamma parameter value was calculated using TREE-PUZZLE (V5.1; Schmidt et al. 2002; see below). To calculate support values, we first did unweighted maximum parsimony (MP) bootstrap analysis (2000 replications) using PAUP* V4.0b8 (Swofford 2002), with the number of random-addition replicates set to 10 with tree bisection–reconnection optimization. Second, we did Bayesian analysis of the plastid data (MrBayes V3.0b4; Huelsenbeck and Ronquist 2001) using the WAG model (Whelan and Goldman 2001). Metropolis-coupled Markov chain Monte Carlo from a random starting tree was initiated in the Bayesian inference and run for 500,000 generations (except for the full genome data, for which we used 350,000 generations due to the extreme run time), with trees sampled every 100th generation. Four chains were run simultaneously, of which three were heated and one was cold. Stationarity of the log likelihoods was monitored and a consensus phylogeny was made with the “post burn-in” trees for each protein data set to determine the posterior probabilities at the different nodes. And third, quartet puzzling-maximum likelihood analysis was done with the WAG + Γ model using TREE-PUZZLE. Quartet puzzling support (QPS) values were calculated from 5000 puzzling steps.
Tree Comparisons
Trees were compared among the different data sets using the reciprocal 70% bootstrap support criterion (Mason-Gamer and Kellogg 1996). Under this criterion, if the same set of terminal taxa receives bootstrap support larger than 70% for a monophyletic relationship in a data partition and is not monophyletic with support values larger than 70% in a different data partition, then we interpret this as a topological conflict. We also tested the robustness of our findings by generating alternative topologies of the 41-protein, PS I + II, and T + T ML trees using MacClade (V4.05; Maddison and Maddison 2002). Groups of interest were positioned at alternate branch points in the ML trees and the log likelihoods of the “best” ML and the rearranged trees were calculated and compared using the one-sided Kishino–Hasegawa (K-H) test (Kishino and Hasegawa 1989; Goldman et al. 2000) implemented in TREE PUZZLE.
Results
Genome Structure and Gene Composition General Features
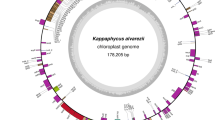
The complete circular plastid genome of Gracilaria tenuistipitata var. liui was 183,883 bp in length (Fig. 1), with a GC content of 29.1% (Table 1). The genome does not contain inverted or direct repeats and encodes only a single copy of the ribosomal RNA (rRNA) operon. There are 238 predicted genes including 204 protein-encoding genes, 29 tRNAs, 3 rRNAs, 1 tmRNA, and 1 ribonuclease gene (rnpB) (for the full list of genes, see Table 3 in the supplementary information at http://gracilaria.lbi.ic.unicamp.br/chloroplast). The coding sequences lack introns and span 84.1% of the genome. There are just two cases of overlapping genes (atpF/atpD and psbC/ psbD). As expected, codon usage reflects the low GC content of the plastid genome, with codons ending in G or C comprising only 17.5% of the total. The termination codon TAA is used in 77.5% of the ORFs (see Table 4 in the supplementary information).

The Gracilaria tenuistipitata chloroplast genome. Genes on the outside circle are transcribed in a clockwise direction, whereas those in the middle circle are transcribed counterclockwise. The inside circle indicates G + C content. Identified genes based on database comparisons were named as in Stoebe et al. (1998) and hypothetical ORFs were named as “ORF” followed by the number of codons. The color indicates functional category, photosynthesis (green), transcription and translation (blue), biosynthesis (yellow), miscellaneous (orange), hypothetical, and conserved hypothetical (gray).
Photosynthesis
Based on databank comparisons, we have identified 57 genes related to photosynthetic function in the Gracilaria plastid genome. All the ATP synthase genes are present with the exception of atpC, which was transferred to the nucleus early in the evolution of plastids (Kowallik 1997). The ATP synthase genes are arranged in two clusters. The atpB and atpE genes are adjacent, whereas atpA, D, F, G, H, and I form a second cluster. There is an overlap of a single base in the atpF and atpD genes as described for Synechococcus PCC 6301 (Cozens and Walker 1987). The genes petA, B, D, F, G, J, and N (ycf6) of the electron transfer chain are present, whereas petL and petM are missing. Twenty-seven genes for the assembly of photosystem I and II are present. The genes psbC and psbD share a 92-bp region of overlap. The genome encodes both subunits of Rubisco (rbcL and rbcS) and all 10 phycobiliprotein genes.
Transcription and Translation
The consensus sequences for −10 (TATAAT) and −35 (TTTAAA and TTGACA) promoters were searched in the intergenic regions of Gracilaria plastid genome. The −10 consensus was detected for 102 ORFs and the −35 for 70 ORFs. Only three ORFs contained the promoter TTGACA (for the full list of genes see Table 5 in the supplementary information). A set of 46 ORFs contained both the canonical −10 and −35 sequences. We searched restricted regions for the −10 (from −1 to −25) and −35 (from −1 to −100) promoter sequences. These motifs were detected for just 17 and 39 ORFs, respectively. The ribosome-binding site (RBS) was detected for only 97 ORFs using the Shine–Dalgarno canonical sequences (AAGG, AGGA, GGAG, and GAGG) to search the intergenic regions immediately upstream of all start codons (Table 5). The number of hits decreased to 61 ORFs when we restricted the search from −1 to −50. The absence of promoter regions and RBS for some ORFs is likely due to an alternative sequence being used and/or different location or may indicate an operon organization or “relay race”-type translation (Link 1996; Löffelhardt et al. 1997).
All four subunits of the eubacterial-like RNA-polymerase are encoded in the Gracilaria plastid genome. The genes rpoB, rpoC1, and rpoC2 form a cluster, whereas rpoA is part of a large cluster of ribosomal proteins. The presence of the putative transcription regulators lysR (ycf 30), ompR (ycf 27), tctD (ycf 29), and ntcA (ycf28) indicate the potential for regulation of gene expression in the plastid. The genes coding for the ATP-binding protein involved in Rubisco expression (cfxQ) and ribonuclease E (rne) were also detected.
The Gracilaria plastid has a nearly complete set of ribosomal proteins (Table 1). It lacks rps15, which is present only in green plant plastids. The Gracilaria plastid genome contains 19 genes for 30S ribosomal subunit proteins and 28 genes for 50S ribosomal subunit proteins. There is one major cluster of 29 genes, which includes 26 ribosomal protein genes, tufA, rpoA, and secY, and is flanked by dnaK. Other ribosomal protein genes are scattered throughout the genome or are in “mixed” operons like psaA–psaB–rps14. Two ORFs coding for initiation factors IF-2 (inf B) and IF-3 (inf C) and two ORFs coding for elongation factors EF-Ts (tsf) and EF-Tu (tufA) were found in the Gracilaria plastid genome but the ORF for IF-1 (infA) is missing.
The Gracilaria plastid genome contains only one set of rRNA genes organized in an operon encoding the 16S, 23S, and 5S subunits. The spacer region between 16S and 23S encodes two tRNA genes (tRNAIle and tRNAAla). Twenty-nine tRNA genes, encompassing all 20 amino acids, are found either singly or in clusters of two or three genes. The tRNA gene positions are highly conserved in the red algal plastid lineage (see Table 6 in the supplementary information). Two tRNA synthetases have been identified in Gracilaria, histidyl-tRNA synthetase (hisS or syh) and phenylalanyl-tRNA synthetase beta chain (pheT or syfB), which have homologs only in the Porphyra plastid, in cyanobacteria, and in other eubacterial genomes. The genes for tmRNA (K. Williams, personal communication) and ribonuclease P (rnpB) were detected based on secondary structure predictions.
Biosynthesis
Two unique genes involved in leucine biosynthesis, leuC and leuD, were found in the Gracilaria plastid genome. Gracilaria contains many other genes involved in the biosynthesis of amino acids (e.g., ilvB and H), fatty acids (e.g., accA, D, B), and components of the photosynthetic apparatus (e.g., chlI, moeB, pbsA). The chlorophyll biosynthesis genes chlL, chlN, and chlB were absent. The gene gltB that encodes a protein involved in nitrogen assimilation was detected, but the regulating gene glnB was absent.
Miscellaneous
Within the miscellaneous category, we detected the following genes in the Gracilaria plastid genome: (1) two homologs of bacterial genes involved in protein translocation across membranes secA and secY, (2) the chaperonin subunits groEL and dnaK, (3) the ATP-binding subunit clpC, (4) the genes involved in replication and division dnaB (this gene contained an intein of 143 amino acids) and ftsH, (5) two adjacent genes coding for cytochrome C-type biogenesis proteins (ccdA and ccs1/yfc44), (6) a gene coding for phosphoglycerate mutase (pgmA) involved in glycolysis, and (7) the gene coding for a thioredoxin (trxA) detected only in red algae.
Conserved (ycfs) and Hypothetical ORFs
We identified 33 conserved hypothetical genes in the Gracilaria plastid genome and attributed ycf numbers (as proposed in Stoebe et al. 1998) for all but 1 gene (ORF65), which shares 69% similarity with ORF58 of Porphyra purpurea. The gene coding for ycf17 is truncated in Gracilaria. We identified nine hypothetical ORFs (Table 2) varying from 83 to 446 amino acids in length. No promoter sequences were detected for those ORFs. The codon usage for these hypothetical ORFs is quite similar to the average for all the ORFs, indicating that they are expressed (Table 4). Six of the hypothetical ORFs are clustered between position 136,053 and position 144,145. The GC content of this region is 22.8%, which is lower than the average value (29.1%) for the genome.
Comparisons with Porphyra purpurea
The plastid genomes of Gracilaria and Porphyra are quite similar in gene content and share colinearity over large regions. Gracilaria lacks the duplication of the rRNA genes present as direct repeats in Porphyra as well as two genes (chlL and chlN) flanking one of these direct repeats. Open reading frames that are exclusively present between the Porphyra and Gracilaria plastid genomes are listed in Table 2. Adjacent to the single rRNA operon in Gracilaria are the unique leuD and leuC genes (GC content of this region is 32.6%). Another interesting region in Gracilaria is between 136,053 and 144,145, which includes six hypothetical proteins (ORF245, 109, 446, 220, 198, and 197) that have no homologs in Porphyra. The corresponding region in Porphyra contains two exclusive ORFs (ORF62 and 621). Gracilaria has 29 of the 37 tRNA genes present in Porphyra. The locations of all these genes are conserved between both genomes (see Table 5 in the supplementary information). Of the eight additional tRNA genes present in Porphyra, two are located at the intergenic spacer of the extra rRNA operon, and three are flanking rearrangement regions.
We identified 11 colinear regions in comparisons of the Gracilaria and Porphyra plastid genomes (Fig. 2). These 11 regions contain from 2 to 54 genes. The breaks in colinearity are due to a major inversion, four minor inversions, the deletion of the rRNA repeat, and the insertion of Gracilaria’s exclusive leuC/leuD and hypothetical ORFs. Genes coding for tRNAs are flanking 7 of the 11 breaks in colinearity. The inversion of region 5 occurred in Porphyra, breaking the conserved operon psaA–psaB–rps14. The inversions of regions 3, 7, and 8 appear to have occurred in Gracilaria, since the gene order of this region is conserved among Guillardia, Cyanidium, and Porphyra. When we align the Gracilaria and Porphyra plastid genomes, a clear “X” shape is observed (Fig. 2B), indicating inversions around the origin and/or terminus in these genomes. These X-alignments are a common feature of bacterial evolution and are evident for moderately closely related species (Eisen et al. 2000).

Comparison of Gracilaria tenuistipitata (Gt) and Porphyra purpurea (Pp) plastid genome structure. A Illustration of the backbone of both genomes with the relative position, size, and orientation of colinear regions (numbered 1 to 11). The arrow direction within each chromosome fragment indicates its relative orientation. The rRNA operons and regions exclusive to each genome are indicated (see Table 2). B The nucleotide alignment of both plastid genomes. The colinear regions are numbered as in A.
Phylogeny
Protein ML analysis of the 41-protein data set resulted in the phylogeny that is shown in Fig. 3A. This tree resolves nearly every node as indicated by the significant Bayesian posterior probabilities and robust support values in the MP bootstrap and quartet-puzzling analyses. The relationships within the chlorophyte and streptophyte lineages generally agree with well-accepted views, for example, the early divergences of the charophyte Chaetosphaeridium within the Streptophyta (Karol et al. 2001) and the prasinophyte Nephroselmis within the Chlorophyta (Steinkoetter et al. 1994). In addition, Gracilaria is resolved with strong bootstrap support as sister to the Bangiales red alga Porphyra purpurea. Three areas in the tree that lacked resolution were the position of the diatom Odontella sinensis within the clade of red algal and red algal-derived (i.e., chromist) plastids, the relative branching order of the hornwort + liverwort ([bryophytes] Anthoceros formosae, Marchantia polymorpha, respectively) and tracheophyte (Adiantum capillus–veneris, Psilotum nudum) lineages, and the relative position of the glaucophyte Cyanophora paradoxa with respect to the red and green plastid lineages. Use of the K-H test showed that the ML tree shown in Fig. 3A had a lower probability (p = 0.123) than the “best” rearranged tree that united the chromists (i.e., moving Odontella to Guillardia; difference in log likelihood units = 34.67 is, however, nonsignificant [the converse rearrangement of moving Guillardia to Odontella was overwhelmingly rejected; p < 0.000]). However, moving the bryophyte branch either inside the tracheophyte divergence or as sister to this group did not result in significantly worse trees (p = 0.363; p = 0.066, respectively). Moving Cyanophora to the base of all plastids also did not result in a significantly worse tree (p = 0.173). Interestingly, placing Mesostigma at the base of the streptophytes or the chlorophytes both resulted in significantly worse trees (p = 0.017 and p = 0.010, respectively). These data indicate that uncertainty remains about the divergence order of early land plants and Cyanophora and that ML analysis of the 41-protein data set does not convincingly resolve the issue of chromist monophyly. This tree strongly supports the position of Mesostigma at the base of all green plastids.

Phylogenetic relationships of algae and plants based on ML analysis of plastid genome data. The trees inferred from the concatenated data set of 41 proteins (A), from only PSI+II proteins (B), and from the T+T proteins (C) are shown, as well as the red lineage portions of the ML trees inferred from the full photosynthesis (full PS apparatus), ATP-synthase, pet, ribosomal protein, and RNA polymerase subunit data sets (D). The Bayesian inference was run using the WAG model. The posterior probabilities (when >95%) inferred from this analysis are shown as the thicker branches. The values shown above the branches result from an unweighted maximum parsimony bootstrap analysis, whereas the values shown below the branches result from a quartet-puzzling maximum likelihood analysis (WAG+Γ model). Only bootstrap or puzzle support >60% is shown. The trees are rooted on the branch leading to the cyanobacterial sequences. The branch lengths are proportional to the number of substitutions per site (see scales in figures).
To gain additional insights into these results, we initially partitioned the 41-protein alignment into two functional groups (photosystem I + II proteins [PSI + II, 3498 aa: psaA–C and J; psbA–F, H, L, N, and T] and transcription/translation proteins [T + T, 3347 aa: rp12, 14, 16, and 20; rps2-4, 7, 8, 12, 14, 18, and 19; rpoA, B, C1, and C2]) and did separate phylogenetic analyses with these data sets. The motivation in this approach (see de Queiroz et al. 1995) was to assess whether the two different sets of interacting and presumably coevolving proteins would support congruent plastid phylogenetic histories, in particular, with regard to the areas that were unresolved in the 41-protein tree (Fig. 1A). ML analysis of the PSI+II data set (Fig. 3B) resulted in a tree that was identical to the 41-protein tree with one important well-supported difference. The stramenopile Odontella is positioned in the PSI+II as sister to the other chromist, Guillardia theta, with support in the MP and Bayesian analyses. Use of the K-H test with the PSI+II data set showed that breaking chromist monophyly by moving either Odontella (p = 0.027) or Guillardia (p < 0.000) to the base of the Cyanidiales (as in Fig. 3A) or by moving Odontella to the branch uniting Porphyra+Gracilaria (p = 0.007) resulted in significantly worse trees. Again, however, moving the bryophyte branch to either before the tracheophyte divergence (p = 0.424) or as sister to this group (p = 0.330) did not result in significantly worse trees. Moving Cyanophora to the base of all plastids resulted in a marginally better (difference in log likelihood units = 3.63 is nonsignificant) tree than that shown in Fig. 3B and moving Mesostigma to the base either of the streptophytes (p = 0.243) or of all green plastids (p = 0.470) was not rejected by the K-H test. Analysis of the PSI+II data indicates therefore that considerable uncertainty remains about the divergence order of early land plants, Cyanophora, and Mesostigma but that the K-H test (under the WAG+Γ model) strongly supports the monophyly of chromist plastids.
ML analysis of the T+T data set (Fig. 3C) resulted in a tree that showed ambiguity with respect to the position of Cyanophora and the relative branching order of the bryophyte and tracheophyte lineages. Odontella was, however, positioned with support as sister to the Cyanidiales red algae in the T+T analyses. Use of the K-H test with these data showed surprisingly that despite bootstrap and Bayesian support for chromist polyphyly, uniting these taxa by moving either Odontella to Guillardia (p = 0.232) or vice versa (p = 0.308) did not result in significantly worse trees. Similar to the other data sets, moving Cyanophora either to the base of all green plastids or, in this case, to the base of the red plastids or rearranging the branch points of the bryophytes and tracheophytes relative to each other did not result in significantly worse trees. Placing Mesostigma at the base of the chlorophytes did, however, result in a significantly different log likelihood score (p = 0.024). Analysis of the T+T data indicates therefore that despite bootstrap support for chromist polyphyly, this result does not hold up under the K-H test and that, like the 41-protein and PSI+II data, the divergence points of early land plants and Cyanophora remain in question. Application of the reciprocal 70% bootstrap support criterion (Mason-Gamer and Kellogg 1996) showed that three well-supported areas of conflict existed between the PSI+ II and the T + T trees, the positions of Odontella and Mesostigma and the divergence of the first land plants. Despite considerable uncertainty about the branch points of some algal plastids (e.g., Cyanophora and Mesostigma), the monophyly of Gracilaria and Porphyra was robustly supported by the K-H test with all three data sets. Moving Gracilaria to either Odontella or Guillardia always resulted in significantly worse trees (p < 0.000).
We looked in more detail at the phylogeny of the red and chromist algal plastids using other functionally related subsets of the 41-protein data set. Here, the ML method was used to infer a phylogeny for the full photosynthetic protein data set (i.e., PSI+II plus ATP-synthase, ccsA, and pet proteins, 5463 aa), the ATP-synthase proteins (1138 aa), the pet proteins (671 aa), the ribosomal proteins (1623 aa), and proteins of the RNA polymerase subunits (1724 aa). The red+chromist portions of these ML trees are shown in Fig. 3D. The full photosynthesis and pet data sets supported chromist monophyly with support, whereas the ATP synthase, ribosomal proteins, and RNA polymerase data sets did not. In particular, the ribosomal protein data provided the strongest support for a specific association between Odontella and the Cyanidiales exclusive of Guillardia and Gracilaria+Porphyra. These sequences most likely account for the phylogenetic signal supporting chromist polyphyly resolved in the 41-protein tree (Fig. 3A). All the data sets, however, supported a specific evolutionary relationship between Gracilaria and Porphyra. We then compared the average WAG+Γ distance for each protein data set that was calculated with TREE-PUZZLE. This analysis showed that the PSI+II proteins (average distance = 0.193) evolve at a markedly lower rate than the T+T proteins (average distance = 0.667—ca. 3.5× slower). Within the T+T data partition, the most divergent sequences were those encoding the RNA polymerase subunits (average distance = 0.736). In comparison, the average pairwise distance in the ribosomal protein data set was 0.609.
Discussion
Genome Structure and Gene Composition
Red and green plastid genomes have many differences in organization in spite of the fact that they share a single origin through a putative cyanobacterial primary endosymbiosis (Bhattacharya and Medlin 1995). These discrepancies have accumulated during their long and separate evolutionary history. Green (land plants and green algae) plastid genomes contain around 110–118 genes, including 11 genes for the NADPH dehydrogenase complex, that are absent from nongreen lineages, and their gene content is highly conserved (Palmer 1991). In this group, the early-diverging green alga Mesostigma viride (see Fig. 3A) contains the largest known repertoire, with 135 genes (Lemieux et al. 2000). In contrast, the red lineage contains many genes that are not located on the plastid genome of green plants. Rhodophyte plastid genomes have an increased coding capacity (Reith and Munholland 1993), often containing more than twice the number of genes (232–251) common to species of the green lineage. The secondary plastids in the red lineage also encode a large number of genes (174–183), but not as many as in the primary plastids.
Most plastids from the green lineage, especially the land plants, have numerous introns. Exceptions are the ancient green algae Mesostigma viride and Nephroselmis olivacea (De Las Rivas et al. 2002). Cyanophora paradoxa (Stirewalt et al. 1995) has only one intron, an ancient group I intron in the trnL (UUA) gene that is conserved in many cyanobacteria and plastid genomes of the green lineage (Tunnel et al. 2002; Besendahl et al. 2000; Simon et al. 2003). The trnL intron is absent from the red lineage with the exception of some brown algae (Xanthophyceae and Phaeophyceae), indicating that it was likely lost in most taxa and retained only in the stramenopiles (Simon et al. 2003). The invasion and proliferation of autocatalytic introns seem to have occurred secondarily in the green lineage and are probably associated with the substantial rearrangements seen in this lineage (Turmel et al. 2002).
Typically, plastid genomes possess two unique sequence regions and two large inverted repeats (5–30 kb) containing the rRNA genes. As the number of reported genomic sequences increases, we see more and more variation in this general organization. Some angiosperm, gymnosperm, and green algal plastids do not contain the inverted repeats (Gillham 1994). In the red lineage, large repeats are absent in Gracilaria and in the Cyanidiales genera Cyanidium and Cyanidioschyzon. Porphyra purpurea has direct nonidentical repeats containing only the rRNA genes (Reith and Munholland 1993). The plastid genomes of other red algae have been mapped. The Florideophycidae species Griffithsia pacifica (Shivji et al. 1992) and Chondrus crispus (Leblanc et al. 1995) contain only one rRNA operon. In contrast, the Bangiophycidae Porphyra yezoensis encodes two operons for rRNA genes on inverted repeats (Shivji 1991), as does the cryptophyte Guillardia theta (Douglas and Penny 1999). The stramenopiles Odontella sinensis (Kowallik et al. 1995), Pylaiella littoralis (Goër et al. 1988), Olisthodiscus luteus, and Ochromonas danica (Shivji et al. 1992) contain two rRNA operons on inverted repeats. Analysis of the rDNA operons and their flanking regions in Guillardia and Porphyra suggests that the directly repeated rDNA genes of the ancestral rhodophyte were transformed into inverted repeats in Guillardia (Douglas 1998). The Bangiales, which contains the genus Porphyra, is a sister group of the monophyletic Florideophycidae (Oliveira and Bhattacharya 2000; Müller et al. 2001). Therefore, it seems reasonable to assume that the rDNA direct repeat present in Porphyra was lost secondarily in the Florideophycidae. An independent loss of the rDNA repeat could also have occurred in Cyanidium. In the absence of additional data on basal rhodophytes, it remains unclear which arrangement of large repeats was present in the ancestral rhodophyte plastid genome.
In the red lineage, there is one major cluster that includes 26 ribosomal protein genes plus tufA, rpoA, and secY. This cluster is highly conserved in gene order and composition in the red lineage and present striking resemblance to the str, S10, spc, and α operons of E. coli (Ohta et al. 1997; Stoebe and Kowallik 1999). Ohta et al. (1997) proposed a model for the evolution of these operons in bacteria, plastids, and mitochondria. The conserved arrangement of these clusters between Synechocystis and plastids supports the monophyletic origin of plastids. They also proposed a translocation of the str cluster early after the separation of the rhodophyte lineage. In the green plastid lineage, more variability is seen in the organization of these operons. There is evidence that due to the presence of numerous introns, the plastid genome of the Streptophyta underwent substantial changes in its architecture (Turmel et al. 2002). In land plant plastid genomes, the initiation factor IF-1 (infA) is present but they lack IF-2 and IF-3 (inf B, inf C, respectively [Gillham 1994]). Both inf B and infC are found only in Porphyra and Gracilaria. The cryptophyte Guillardia retains only inf B and Cyanidium only inf C (Glöckner et al. 2000). The diatom Odontella has lost all three genes (Kowallik et al. 1995). Given this distribution, we surmise that the ancestor of these plastids probably encoded all three translational initiation factors, which were differentially lost in the extant lineages.
Two unique genes involved in leucine biosynthesis were found in the Gracilaria plastid genome. These genes, leuC and leuD, have no homologs in any other plastid genome and are most similar to genes in Arabidopsis thaliana, the bacteria Chlorobium tepidum, and several Archaea species. Interestingly, leuC and leuD are located between ORFs ompR (ycf 27) and psbD, which are separated by only 206 bp in Porphyra. In Cyanidium, this region (about 1200 bp) contains a hairpin loop flanked by two short repeated units that are thought to be involved in replication (Glöckner et al. 2000). The GC content of this region is 32.6%, which is similar to the Gracilaria plastid genome GC content average of 29.1%. Interestingly, when the leuC 5′-flanking region is used to search for similarities using BLASTX (Altschul et al. 1997), the first two hits are two plasmids (3.9 and 7.2 kb) described from Gracilaria chilensis, which have a GC content of 25% (NC_002107, NC_002106). Considering the above, it seems probable that this region (∼4.6 kb), containing the leuC and leuD genes, could have been originated through plasmid integration in the plastid genome of Gracilaria.
The genes chlI, B, L, and N that are involved in chlorophyll biosynthesis are present in Porphyra, Cyanophora, and the green alga Chlorella vulgaris (Stoebe et al. 1998), indicating their presence in the plastid common ancestor. It is interesting to note that chlN and chlL in Porphyra flank one of the direct repeats and this whole region was lost in Gracilaria. The same was observed for the green lineage, where the inverted repeat of Chaetosphaeridium globosum contains a pair of extra genes (chlL and chlN) compared to the corresponding region in Marchantia cpDNA (Turmel et al. 2002).
Turmel et al. (2002) identified 12 blocks of colinear sequences when comparing the cpDNAs of the charophyte Chaetosphaeridium globosum and the bryophyte Marchantia polymorpha. We identified 11 colinear regions shared between the Gracilaria and the Porphyra plastid genomes. Seven breaks in colinearity could be due to recombination events across short repeated sequences within or near tRNA genes. The tRNA genes have been associated with cpDNA inversion in several Angiosperms (Cosner et al. 1997; Douglas 1998). The absence of tRNA genes at some break points indicates that other mechanisms must also be involved in generating inversions.
Gracilaria maintains a surprisingly ancient gene content in its plastid genome, containing one of the most complete repertoires of plastid genes known in photosynthetic eukaryotes. In this regard, Reith and Munholland (1993) concluded that the Porphyra plastid genome was more cyanobacterial-like than other algae based on the presence of a complete set of tRNAs, of genes encoding transcriptional regulators, of a larger number of genes, and of more operon-like structures similar to the ones found in cyanobacteria and the absence of introns. Based on these criteria, Gracilaria, which occupies a highly derived position in the red algal tree, in some aspects is even more cyanobacterial-like than Porphyra (e.g., the single base overlap of atpF and D and the presence of the psaA–psaB–rps14 operon). We will need, however, to get additional plastid genome data from both ancient and derived red algae to see if this is a general trend within the Bangiales–Florideophycidae lineage or whether most red algae maintain a relatively ancestral gene content and order.
Phylogeny of the Gracilaria Plastid Genome and the Red-Chromist Plastid Tree
Our concatenated protein phylogenies firmly place the plastid of Gracilaria (Florideophycidae) in the red algal lineage as sister to the bangiophyte Porphyra purpurea (Fig. 3). The close evolutionary relationship between Bangiales and florideophyte red algae has been previously proposed on the basis of morphological (e.g., type of reproductive cells [Magne 1989], association of the Golgi apparatus with the mitochondrion [Garbary and Gabrielson 1990], the presence of pit connections) and molecular phylogenetic data (Freshwater et al. 1994; Oliveira and Bhattacharya 2000; Müller et al. 2002; Yoon et al. 2002). The red algal plastids are monophyletic in all of our analyses with the exception of the highly divergent RNA polymerase and in the ATP synthase data sets.
The monophyly of the chromist plastids is recovered when the relatively more slowly evolving components of the photosynthetic apparatus such as the photosystem proteins are used in the phylogenetic analyses. The Chromista is united primarily on the basis of molecular sequence data (Fast et al. 2001; Yoon et al. 2002; Harper and Keeling 2003) and the shared character of chlorophyll c2 in a four-membrane bound plastid that is located in the lumen of the endoplasmic reticulum (Cavalier-Smith 1986; Bhattacharya et al. 2004). A previous analysis by Yoon et al. (2002) that incorporated broad taxon sampling (36 species) from red and chromist algae and used a five-gene plastid data set (5827 nt) resulted in a robust tree in which nearly all the nodes had significant bootstrap and Bayesian support and which supported the monophyly of chromist plastids (i.e., supporting a single secondary endosymbiotic origin of this plastid from an early-diverging red alga). The addition of a sixth plastid gene to the DNA analyses and expansion of the taxon sampling to include green algae and land plants further strengthened this result, however, protein analyses did not provide bootstrap support for chromist monophyly (Yoon et al. 2004). Existing plastid genome trees inferred from a more limited sampling of taxa (i.e., that did not include a haptophyte) do not support chromist monophyly (e.g., Martin et al. 2002; Maul et al. 2003; Ohta et al. 2003), leaving in question the phytogeny of this plastid clade. Our present analyses do not convincingly settle this issue but provide insights into the behavior of different plastid proteins in tree reconstruction. We suggest that the lack of support for chromist monophyly, and possibly for the lack of resolution in other key nodes of the plastid genome tree (e.g., early land plant origin), may be caused by inadequate taxon sampling in these regions (e.g., Wheeler 1992; Rannala et al. 1998; Pollock et al. 2002; Zwickl and Hillis 2002) and the high divergence rates of some plastid proteins. Importantly, the K-H test results show that despite bootstrap and/or Bayesian support to the contrary (see Fig. 3A and C), the 41-protein and T+T data sets do not robustly support the polyphyletic origin of chromist plastids.
Given large differences in divergence rates (e.g., proteins of the plastid transcription/translation apparatus evolve on average at a rate that is about 3.5× higher than photosystem proteins), a reasonable expectation is that the more slowly evolving proteins should be better markers for deep phylogenetic splits in the plastid tree. In contrast, the more highly divergent sequences would be better suited for resolving more recent phylogenetic splits. Concatenation of slowly and rapidly diverging sequences would result in the swamping of the phylogenetic signal in the former data by that in the latter. This potentially explains why the genome and T+T trees both support chromist polyphyly, whereas the relatively more slowly evolving photosystem proteins (see also Morden and Sherwood 2002) support a different topology that is consistent with chromist monophyly (see Yoon et al. 2002; Nozaki et al. 2003; Yoon et al. 2004). In this regard, the splits in the red algal lineage are likely ancient based on molecular clock analyses that date the putative chromist secondary endosymbiosis at about 1.3 billion years ago (Yoon et al. 2004) and the fossil record which shows the potential existence of sexual red algae from this time (Butterfield 2001 [but see Cavalier-Smith 2002]). Convergence to a single well-supported chromist plastid tree will, however, likely require the addition of more genes to data sets with broad taxon sampling (e.g., Yoon et al. 2002) and the addition of more taxonomic breadth to the narrowly sampled plastid genome data sets (e.g., Martin et al. 2002; Maul et al. 2003; Ohta et al. 2003). In addition, there is a need for detailed analyses of single plastid proteins to assess their contribution to phylogenetic signal within the chromist and red algal plastid clades (H.S. Yoon, J.D. Hackett, S. Heard, D. Bhattacharya, unpublished data). The result that is apparent both here and in previous analyses (e.g., Vogl et al. 2003) is that a different plastid gene(s) often supports discordant phylogenies. Despite this vexing issue, it is also clear that most of the nodes in the plastid tree are in fact well supported. This suggests that more extensive taxon sampling may be the best approach to resolve this issue. Other aspects of the red-chromist plastid subtree that were strongly supported regardless of which data set was used are the monophyly of Gracilaria and Porphyra (also found in other studies; e.g., Oliveira and Bhattacharya 2000; Müller et al. 2002) and the resolution of the Cyanidiales as a monophyletic lineage in the red algae (Yoon et al. 2002; Ohta et al. 2003; Pinto et al. 2003; Ciniglia et al. 2004).
References
SF Altschul TL Madden AA Schaffer J Zhang Z Zhang W Miller DJ Lipman (1997) ArticleTitleGapped BLAST and PSI-BLAST: A new generation of protein database search programs Nucleic Acids Res 25 3389–3402 Occurrence Handle1:CAS:528:DyaK2sXlvFyhu7w%3D Occurrence Handle9254694
JO Andersson AJ Roger (2002) ArticleTitleA cyanobacterial gene in nonphotosynthetic protists: an early chloroplast acquisition in eukaryotes? Curr Biol 12 115–119
JM Archibald MB Rogers M Toop K Ishida PJ Keeling (2003) ArticleTitleLateral gene transfer and the evolution of plastid-targeted proteins in the secondary plastid-containing alga Bigelowiella natans Proc Natl Acad Sci USA 100 7678–7683 Occurrence Handle10.1073/pnas.1230951100 Occurrence Handle1:CAS:528:DC%2BD3sXlt1Wqsrs%3D Occurrence Handle12777624
A Besendahl YL Qiu J Lee JD Palmer D Bhattacharya (2000) ArticleTitleThe cyanobacterial origin and vertical transmission of the plastid tRNA(Leu) group-I intron Curr Genet 37 12–23 Occurrence Handle10.1007/s002940050002 Occurrence Handle1:CAS:528:DC%2BD3cXitFartQ%3D%3D Occurrence Handle10672439
D Bhattacharya L Medlin (1995) ArticleTitleThe phylogeny of plastids: A review based on comparisons of small-subunit ribosomal RNA coding regions J Phycol 31 489–498 Occurrence Handle1:CAS:528:DyaK2MXos1Gls7Y%3D
D Bhattacharya HS Yoon JD Hackett (2004) ArticleTitlePhotosynthetic eukaryotes unite: endosymbiosis connects the dots BioEssays 26 50–60 Occurrence Handle10.1002/bies.10376 Occurrence Handle14696040
R Bock (2001) ArticleTitleTransgenic plastids in basic research and plant biotechnology J Mol Biol 312 425–438 Occurrence Handle10.1006/jmbi.2001.4960 Occurrence Handle1:CAS:528:DC%2BD3MXmvV2isrs%3D Occurrence Handle11563907
NJ Butterfield (2001) ArticleTitlePaleobiology of the late Mesoproterozoic (ca. 1200 ma) hunting formation, Somerset Island, Arctic Canada Precambr Res 111 235–256 Occurrence Handle10.1016/S0301-9268(01)00162-0 Occurrence Handle1:CAS:528:DC%2BD3MXlvFWnsrc%3D
T Cavalier-Smith (1986) ArticleTitleThe kingdoms of organisms Nature 324 416–417 Occurrence Handle10.1038/324416a0 Occurrence Handle1:STN:280:BiiD28rhtFU%3D Occurrence Handle2431320
T Cavalier-Smith (2002) ArticleTitleThe neomuran origin of archaebacteria, the negibacterial root of the universal tree and bacterial megaclassification Int J Syst Evol Microbiol 52 7–76
C Ciniglia HS Yoon A Pollio G Pinto D Bhattacharya (2004) ArticleTitleHidden biodiversity of the extremophilic Cyanidiales red algae Mol Ecol 13 1827–1838 Occurrence Handle1:CAS:528:DC%2BD2cXlvFelurk%3D Occurrence Handle15189206
ME Cosner RK Jansen JD Palmer SR Downie (1997) ArticleTitleThe highly rearranged chloroplast genome of Trachelium caeruleum (Campanulaceae): Multiple inversions, inverted repeat expansion and contraction, transposition, insertions/deletions, and several repeat families Curr Genet 31 419–429 Occurrence Handle10.1007/s002940050225 Occurrence Handle1:CAS:528:DyaK2sXjvFOkt70%3D Occurrence Handle9162114
AL Cozens JE Walker (1987) ArticleTitleThe organization and sequence of the genes for ATP synthase subunits in the cyanobacterium Synechococcus 6301 J Mol Biol 294 359–383 Occurrence Handle10.1016/0022-2836(87)90667-X
J Rivas ParticleDe Las JJ Lozano AR Ortiz (2002) ArticleTitleComparative analysis of chloroplast genomes: functional annotation, genome-based phytogeny, and deduced evolutionary patterns Genome Res 12 567–583 Occurrence Handle10.1101/gr.209402 Occurrence Handle11932241
AL Delcher D Harmon S Kasif O White SL Salzberg (1999) ArticleTitleImproved microbial gene identification with GLIMMER (306K, PDF format) Nucleic Acids Res 27 4636–4641 Occurrence Handle10.1093/nar/27.23.4636 Occurrence Handle1:CAS:528:DyaK1MXnvVanu7o%3D Occurrence Handle10556321
A de Queiroz MJ Donoghue J Kim (1995) ArticleTitleSeparate versus combined analysis of phylogenetic evidence Annu Rev Ecol Syst 26 657–681 Occurrence Handle10.1146/annurev.es.26.110195.003301
SE Douglas (1998) ArticleTitlePlastid evolution: origins, diversity, trends Curr Opin Genet Dev 8 655–661 Occurrence Handle10.1016/S0959-437X(98)80033-6 Occurrence Handle1:CAS:528:DyaK1MXhs1yisw%3D%3D Occurrence Handle9914199
SE Douglas SLJ Penny (1999) ArticleTitleThe plastid genome of the cryptophyte alga, Guillardia theta: Complete sequence and conserved synteny groups confirm its common ancestry with red algae J Mol Evol 48 236–244 Occurrence Handle1:CAS:528:DyaK1MXotVyqsQ%3D%3D Occurrence Handle9929392
JA Eisen JF Heidelberg O White L Salzberg (2000) ArticleTitleEvidence for symmetric chromosomal inversions around the replication origin in bacteria Genome Biol 1 0011.1–0011.9 Occurrence Handle10.1186/gb-2000-1-6-research0011
NM Fast JC Kissinger DS Roos PJ Keeling (2001) ArticleTitleNuclear-encoded, plastid-targeted genes suggest a single common origin for apicomplexan and dinoflagellate plastids Mol Biol Evol 18 418–426 Occurrence Handle1:CAS:528:DC%2BD3MXhvVKrt78%3D Occurrence Handle11230543
J Felsenstein (2002) PHYLIP (Phylogeny Inference Package) version 3.6. Department of Genetics University of Washington Seattle
W Freshwater S Fredericq BS Butler MH Hommersand MW Chase (1994) ArticleTitleA gene phylogeny of the red algae (Rhodophyta) based on plastid rbcL Proc Natl Acad Sci USA 91 7281–7285 Occurrence Handle1:CAS:528:DyaK2cXltVerurg%3D Occurrence Handle8041781
DJ Garbary PW Gabrielson (1990) Taxonomy and evolution KM Cole RG Sheath (Eds) Biology of the red algae Cambridge University Press Cambridge 477–498
J Gewolb (2002) ArticleTitlePlant scientists see big potential in tiny plastids Science 295 258–259 Occurrence Handle10.1126/science.295.5553.258 Occurrence Handle11786623
NW Gillham (1994) Organelle genes and genomes Oxford University Press New York
P Gilson GI McFadden (1995) ArticleTitleThe chlorarachniophyte: A cell with two different nuclei and two different telomeres Chromosoma 103 635–641 Occurrence Handle1:STN:280:BymD1cfjslM%3D Occurrence Handle7587586
G Glöckner A Rosenthal K Valentin (2000) ArticleTitleThe structure and gene repertoire of an ancient red algal plastid genome J Mol Evol 51 382–390 Occurrence Handle11040290
SL Goër Y Markowicz J Dalmon H Audren (1988) ArticleTitlePhysical maps of the two circular plastid DNA molecules of the brown alga Pylaiella littoralis (L.) Kjellm Curr Genet 14 155–162 Occurrence Handle10.1007/BF00569339
N Goldman JP Anderson AG Rodrigo (2000) ArticleTitleLikelihood-based tests of topologies in phylogenetics Syst Biol 49 652–670 Occurrence Handle10.1080/106351500750049752 Occurrence Handle1:STN:280:DC%2BD38zntVOjtw%3D%3D Occurrence Handle12116432
D Gordon C Abajian P Green (1998) ArticleTitleConsed: A graphical tool for sequence finishing Genome Res 8 195–202 Occurrence Handle1:CAS:528:DyaK1cXitlWksr0%3D Occurrence Handle9521923
MW Gray (1992) ArticleTitleThe endosymbiont hypothesis revisited Int Rev Cytol 141 233–357 Occurrence Handle1:STN:280:ByyD1cvkslI%3D Occurrence Handle1452433
JC Hagopian P Nyvall MC Oliveira (2002) ArticleTitlePurification of plastid DNA from an enriched rhodoplast fraction of the red alga Gracilaria tenuistipitata Plant Mol Biol Rep 20 399–406 Occurrence Handle1:CAS:528:DC%2BD3sXhtVSksrg%3D
V Hannaert E Saavedra F Duffieux JP Szikora DJ Rigden PA Michels FR Opperdoes (2003) ArticleTitlePlantlike traits associated with metabolism of Trypanosoma parasites Proc Natl Acad Sci USA 100 10677 Occurrence Handle10.1073/pnas.0335769100
JT Harper PJ Keeling (2003) ArticleTitleNucleus-encoded, plastid-targeted glyceraldehyde-3-phosphate dehydrogenase (GAPDH) indicates a single origin for chromalveolate plastids Mol Biol Evol 20 1730–1735 Occurrence Handle10.1093/molbev/msg195 Occurrence Handle1:CAS:528:DC%2BD3sXotlWjsL0%3D Occurrence Handle12885964
JP Huelsenbeck F Ronquist (2001) ArticleTitleMRBAYES: Bayesian inference of phylogenetic trees Bioinformatics 17 754–755 Occurrence Handle10.1093/bioinformatics/17.8.754 Occurrence Handle1:STN:280:DC%2BD3MvotV2isw%3D%3D Occurrence Handle11524383
KG Karol RM McCourt MT Cimino CF Delwiche (2001) ArticleTitleThe closest living relatives of land plants Science 294 2351–2353 Occurrence Handle10.1126/science.1065156 Occurrence Handle1:CAS:528:DC%2BD3MXptFGrt78%3D Occurrence Handle11743201
H Kishino M Hasegawa (1989) ArticleTitleEvaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data and the branching order in hominoidea J Mol Evol 29 170–179 Occurrence Handle1:CAS:528:DyaL1MXkvFCnsbc%3D Occurrence Handle2509717
KV Kowallik (1997) Origin and evolution of chloroplasts: Current status and future perspectives HEA Schenk (Eds) Eucariotism and symbiosis Spring-Verlag Berlin 3–23
KV Kowallik B Stoebe I Schaffran P Kroth-Pancic U Freier (1995) ArticleTitleThe chloroplast genome of a chlorophyll a+c-containing alga, Odontella sinensis Plant Mol Biol Rep 13 336–342 Occurrence Handle1:CAS:528:DyaK28XhtFWmsL4%3D
C Leblanc C Boyen SL Goër (1995) ArticleTitleOrganization of the plastid genome from the rhodophyte Chondrus crispus (Gigartinaels); Sequence and phylogeny of the 16S rRNA gene Eur J Phycol 30 133–140
C Lemieux C Otis M Turmel (2000) ArticleTitleAncestral chloroplast genome in Mesostigma viride reveals an early branch of green plant evolution Nature 403 649–652 Occurrence Handle10.1038/35001059 Occurrence Handle1:CAS:528:DC%2BD3cXht1Oqt7g%3D Occurrence Handle10688199
G Link (1996) ArticleTitleGreen life: Control of chloroplast gene transcription BioEssay 18 465–471 Occurrence Handle1:CAS:528:DyaK28XkslSntbs%3D
W Löffelhardt HJ Bohnert DA Bryant (1997) The complete sequence of the Cyanophora paradoxa cyanelle genome (Glaucocystophyceae) D Bhattacharya (Eds) Origin of algae and their plastids Springer-Verlag Wein/New York 149–162
TM Lowe SR Eddy (1997) ArticleTitletRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequences Nucleic Acids Res 25 955–964 Occurrence Handle10.1093/nar/25.5.955 Occurrence Handle1:CAS:528:DyaK2sXhvVahtrk%3D Occurrence Handle9023104
DR Maddison WP Maddison (2002) MacClade V4.05 Sinauer Associates Sunderland, MA
F Magne (1989) ArticleTitleClassification et phylogéne des Rhodophycées Crypt Algol 10 1–115
W Martin P Borst (2003) ArticleTitleSecondary loss of chloroplasts in trypanosomes Proc Natl Acad Sci USA 100 765–767 Occurrence Handle10.1073/pnas.0437776100 Occurrence Handle1:CAS:528:DC%2BD3sXhtF2hu74%3D Occurrence Handle12552110
W Martin B Stoebe V Goremykin S Hansmann M Hasegawa KV Kowallik (1998) ArticleTitleGene transfer to the nucleus and the evolution of chloroplasts Nature 393 162–165 Occurrence Handle10.1038/30234 Occurrence Handle1:CAS:528:DyaK1cXjt1ahsL0%3D Occurrence Handle11560168
W Martin T Rujan E Richly A Hansen S Cornelsen T Lins D Leister B Stoebe M Hasegawa D Penny (2002) ArticleTitleEvolutionary analysis of Arabidopsis, cyanobacterial, and chloroplast genomes reveals plastid phylogeny and thousands of cyanobacterial genes in the nucleus Proc Natl Acad Sci USA 99 12246–12251 Occurrence Handle10.1073/pnas.182432999 Occurrence Handle1:CAS:528:DC%2BD38XntlCks70%3D Occurrence Handle12218172
RJ Mason-Gamer EA Kellogg (1996) ArticleTitleTesting for phylogenetic conflict among molecular data sets in the tribe Triticeae (Gramineae) Syst Biol 45 524–545
JE Maul JW Lilly L Cui CW DePamphilis W Miller EH Harris DB Stern (2002) ArticleTitleThe Chlamydomonas reinhardtii plastid chromosome: Islands of genes in a sea of repeats Plant Cell 14 2659–2679
GI McFadden (2001) ArticleTitlePrimary and secondary endosymbiosis and the origin of plastids J Phycol 37 951 Occurrence Handle10.1046/j.1529-8817.2001.01126.x
CW Morden AR Sherwood (2002) ArticleTitleContinued evolutionary surprises among dinoflagellates Proc Natl Acad Sci USA 99 11558–11560 Occurrence Handle10.1073/pnas.192456999 Occurrence Handle1:CAS:528:DC%2BD38XntFWqs78%3D Occurrence Handle12195026
D Moreira H Le Guyader H Phillippe (2000) ArticleTitleThe origin of red algae and the evolution of chloroplasts Nature 405 69–72 Occurrence Handle10.1038/35011054 Occurrence Handle1:STN:280:DC%2BD3c3mvFemtA%3D%3D Occurrence Handle10811219
KM Muller MC Oliveira RG Sheath D Bhattacharya (2001) ArticleTitleRibosomal DNA phylogeny of the Bangiophycidae (Rhodophyta) and the origin of secondary plastids Am J Bot 88 1390–1400 Occurrence Handle1:CAS:528:DC%2BD3MXmsVyhtb8%3D
H Nozaki N Ohta M Matsuzaki O Misumi T Kuroiwa (2003) ArticleTitlePhylogeny of plastids based on cladistic analysis of gene loss inferred from complete plastid genome sequences J Mol Evol 57 377–382 Occurrence Handle10.1007/s00239-003-2486-6 Occurrence Handle1:CAS:528:DC%2BD3sXovFCgt7o%3D Occurrence Handle14708571
N Ohta N Sato H Nozaki T Kuroiwa (1997) ArticleTitleAnalysis of the cluster of ribosomal protein genes in the plastid genome of a unicellular red alga Cyanidioschyzon merolae: Translocation of the str cluster as an early event in the Rhodophyte-Chromophyte lineage of plastid evolution J Mol Evol 45 688–695 Occurrence Handle1:CAS:528:DyaK1cXktlKi Occurrence Handle9419246
N Ohta M Matsuzaki O Misumi SY Miyagishima H Nozaki K Tanaka IT Shin Y Kohara T Kuroiwa (2003) ArticleTitleComplete sequence and analysis of the plastid genome of the unicellular red alga Cyanidioschyzon merolae DNA Res 10 67–77 Occurrence Handle1:CAS:528:DC%2BD3sXjsFCqsLs%3D Occurrence Handle12755171
EC Oliveira EJ Paula EM Plastino R Petti (1995) Metodologías para el cultivo no axénico de macroalgas marinas in vitro K Alveal ME Ferrario EC Oliveira E Sar (Eds) Manual de métodos ficológicos Universidad de Concepción Concepción 429–441
MC Oliveira D Bhattacharya (2000) ArticleTitlePhylogeny of the Bangiophycidae (Rhodophyta) and the secondary endosymbiotic origin of algal plastids Am J Bot 87 482–492 Occurrence Handle10766719
JD Palmer (1991) Plastid chromosomes: Structure and evolution L Bogorad IK Vasil (Eds) The molecular biology of plastids Academic Press San Diego 5–53
JD Palmer (2003) ArticleTitleThe symbiotic birth and spread of plastids: How many times and whodunit? J Phycol 39 4–11 Occurrence Handle10.1046/j.1529-8817.2003.02185.x Occurrence Handle1:CAS:528:DC%2BD3sXhvV2hsb0%3D
G Pinto P Albertano C Ciniglia S Cozzolino A Pollio HS Yoon D Bhattacharya (2003) ArticleTitleComparative approaches to the taxonomy of the genus Galdieria merola (Cyanidiales, Rhodophyta) Crypt Algol 24 13–32
DD Pollock DJ Zwickl JA McGuire DM Hillis (2002) ArticleTitleIncreased taxon sampling is advantageous for phylogenetic inference Syst Biol 51 664–671 Occurrence Handle10.1080/10635150290102357 Occurrence Handle12228008
B Rannala JP Huelsenbeck Z Yang R Nielsen (1998) ArticleTitleTaxon sampling and the accuracy of large phytogenies Syst Biol 47 702–710 Occurrence Handle10.1080/106351598260680 Occurrence Handle1:STN:280:DC%2BD38zitleltw%3D%3D Occurrence Handle12066312
ME Reith J Munholland (1993) ArticleTitleA high-resolution gene map of the chloroplast genome of the red alga Porphyra purpurea Plant Cell 5 465–475 Occurrence Handle10.1105/tpc.5.4.465 Occurrence Handle1:CAS:528:DyaK3sXkvVyju7w%3D Occurrence Handle12271072
ME Reith J Munholland (1995) ArticleTitleComplete nucleotide sequence of the Porphyra purpurea chloroplast genome Plant Mol Biol Rep 13 333–335 Occurrence Handle1:CAS:528:DyaK28XhtFWms7c%3D
HA Schmidt K Strimmer M Vingron A Haeseler Particlevon (2002) ArticleTitleTREE-PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing Bioinformatics 18 502–504 Occurrence Handle10.1093/bioinformatics/18.3.502 Occurrence Handle1:CAS:528:DC%2BD38XivFKrsL0%3D Occurrence Handle11934758
MS Shivji (1991) ArticleTitleOrganization of the chloroplast genome in the red alga Porphyra yezoensis Curr Genet 19 49–54 Occurrence Handle10.1007/BF00362087 Occurrence Handle1:CAS:528:DyaK3MXisVWlu7Y%3D
MS Shivji N Li RA Cattolico (1992) ArticleTitleStructure and organization of rhodophyte and chromophyte plastid genomes: implications for the ancestry of plastids Mol Gen Genet 232 65–73 Occurrence Handle10.1007/BF00299138 Occurrence Handle1:CAS:528:DyaK38Xit12nsrY%3D Occurrence Handle1552904
D Simon D Fewer T Friedl D Bhattacharya (2003) ArticleTitlePhylogeny and self-splicing ability of the plastid tRNA-Leu group I intron J Mol Evol 57 710–720 Occurrence Handle10.1007/s00239-003-2533-3 Occurrence Handle1:CAS:528:DC%2BD2cXjsVemtQ%3D%3D Occurrence Handle14745540
J Steinkötter D Bhattacharya I Semmelroth C Bibeau M Melkonian (1994) ArticleTitlePrasinophytes form independent lineages within the Chlorophyta: Evidence from ribosomal RNA sequence comparisons J Phycol 30 340–345 Occurrence Handle10.1111/j.0022-3646.1994.00340.x
JW Stiller BD Hall (1997) ArticleTitleThe origin of the red algae: implications for plastid evolution Proc Natl Acad Sci USA 94 4520–4525 Occurrence Handle10.1073/pnas.94.9.4520 Occurrence Handle1:CAS:528:DyaK2sXjtV2jsbw%3D Occurrence Handle9114022
JW Stiller DC Reel JC Johnson (2003) ArticleTitleA single origin of plastids revisited: convergent evolution in organellar genome content J Phycol 39 95–105 Occurrence Handle10.1046/j.1529-8817.2003.02070.x Occurrence Handle1:CAS:528:DC%2BD3sXhvV2htr8%3D
VL Stirewalt CB Michalowski W Luffelhardt HJ Bohnert DA Bryant (1995) ArticleTitleNucleotide sequence of the cyanelle genome from Cyanophora paradoxa Plant Mol Biol Rep 13 327–332 Occurrence Handle1:CAS:528:DyaK28XhtFWms7Y%3D
B Stoebe KV Kowallik (1999) ArticleTitleGene-cluster analysis in chloroplast genomics Trends Genet 15 344–347 Occurrence Handle10.1016/S0168-9525(99)01815-6 Occurrence Handle1:CAS:528:DyaK1MXltlymsrY%3D Occurrence Handle10461201
B Stoebe W Martin KV Kowallik (1998) ArticleTitleDistribution and nomenclature of protein-coding genes in 12 sequenced chloroplast genomes Plant Mol Biol Rep 16 243–255 Occurrence Handle10.1023/A:1007568326120 Occurrence Handle1:CAS:528:DyaK1MXlslSntL0%3D
DL Swofford (2002) PAUP*: Phylogenetic analysis using parsimony (*and other methods), 4.0b8 Sinauer Associates Sunderland, MA
M Turmel C Otis C Lemieux (1999) ArticleTitleThe complete chloroplast DNA sequence of the green alga Nephroselmis olivacea: Insights into the architecture of ancestral chloroplast genomes Proc Natl Acad Sci USA 96 10248–10253 Occurrence Handle10.1073/pnas.96.18.10248 Occurrence Handle1:CAS:528:DyaK1MXlvFensr4%3D Occurrence Handle10468594
M Turmel C Otis C Lemieux (2002) ArticleTitleThe chloroplast and mitochondrial genome sequences of the charophyte Chaetosphaeridium globosum: Insights into the timing of the events that restructured organelle DNAs within the green algal lineage that led to land plants Proc Natl Acad Sci USA 99 11275–11280 Occurrence Handle10.1073/pnas.162203299 Occurrence Handle1:CAS:528:DC%2BD38XmslSmtbs%3D Occurrence Handle12161560
C Vogl J Badger P Kearney M Li M Clegg T Jiang (2003) ArticleTitleProbabilistic analysis indicates discordant gene trees in chloroplast evolution J Mol Evol 56 330–340 Occurrence Handle10.1007/s00239-002-2404-3 Occurrence Handle1:CAS:528:DC%2BD3sXhslWhsro%3D Occurrence Handle12612836
W Wheeler (1992) Extinction, sampling, and molecular phylogenetics MJ Novacek QD Wheeler (Eds) Extinction and phylogeny Columbia University Press New York 205–215
S Whelan N Goldman (2001) ArticleTitleA general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach Mol Biol Evol 18 691–699 Occurrence Handle1:CAS:528:DC%2BD3MXjtFyktr4%3D Occurrence Handle11319253
HS Yoon JP Hackett G Pinto D Bhattacharya (2002) ArticleTitleThe single, ancient origin of chromist plastids Proc Natl Acad Sci USA 99 15507–15512 Occurrence Handle10.1073/pnas.242379899 Occurrence Handle1:CAS:528:DC%2BD3sXjvVOg Occurrence Handle12438651
HS Yoon J Hackett C Ciniglia G Pinto D Bhattacharya (2004) ArticleTitleA molecular timeline for the origin of photosynthetic eukaryotes Mol Biol Evol 21 809–818 Occurrence Handle10.1093/molbev/msh075 Occurrence Handle1:CAS:528:DC%2BD2cXjsFyqsrw%3D Occurrence Handle14963099
DJ Zwickl DM Hillis (2002) ArticleTitleIncreased taxon sampling greatly reduces phylogenetic error Syst Biol 51 588–598 Occurrence Handle12228001
Acknowledgments
This work was supported by FAPESP and CNPq. D.B. was supported by a grant from the (U.S.) National Science Foundation (DEB 01-0774). J.C.H thanks the U.S. Fulbright Program, the Brazilian Fulbright Commission, and the Organization of American States for fellowships.
Author information
Authors and Affiliations
Corresponding author
Additional information
[Reviewing Editor: Dr. W. Ford Doolittle]
Rights and permissions
About this article
Cite this article
Hagopian, J.C., Reis, M., Kitajima, J.P. et al. Comparative Analysis of the Complete Plastid Genome Sequence of the Red Alga Gracilaria tenuistipitata var. liui Provides Insights into the Evolution of Rhodoplasts and Their Relationship to Other Plastids. J Mol Evol 59, 464–477 (2004). https://doi.org/10.1007/s00239-004-2638-3
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/s00239-004-2638-3