
Abstract
The [3 + 2] cycloaddition reaction of N-(p-methylphenacyl)benzothiazolium ylide (NBY) and 1-nitro-2-(p-methoxyphenyl) ethane (NME), experimentally investigated by Yan et al., was studied theoretically. They reported that the reaction proceeds via a stepwise mechanism and produces two diastereomeric adducts in a 4:1 ratio. For the study of the reaction, two regioselective attacks were considered between the reagents and their theoretical parameters were calculated. In excellent agreement with the experimental outcomes, the Fukui and Parr functions reactivity indices analysis as well as the energetic results indicated that among the two studied regioselective attacks, one which leads to the formation of two experimentally reported adducts, is more favorable than the other. The molecular mechanism of the studied reactions was characterized using the IRC, QTAIM and Wiberg bond indices analyses and the results suggested that two diastereomeric adducts are generated via two different mechanisms. The major adduct is produced via a two-stage one-step mechanism without the formation of any stable intermediate, whereas the minor one is generated through a stepwise mechanism along with the formation of a stable zwitterionic intermediate. The analysis of global electron density transfer showed that the reactions are polar and electron density fluxes from NBY toward NME. It was found from molecular electrostatic potential map that at the more favorable transition state, approach of reactants locates the oppositely charged regions over each other resulting in attractive forces between the two fragments. Analysis of the frontier molecular orbitals indicated that the HOMO orbital of NBY is also a frontier effective-for-reaction molecular orbital.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
One of the most useful reactions for the synthesis of homocyclic as well as heterocyclic compounds is cycloaddition reactions [1]. Many of the heterocyclic compounds can be easily synthesized by using [3 + 2] cycloaddition reaction (which is also called 1,3-dipolar cycloaddition) between a three-atom component (or 1,3-dipole) and an ethylene derivative (or dipolarophile). This reaction, which often takes place in a highly regio- and stereoselective fashion, can be used as a powerful method for the synthesis of various five-membered nitrogen-, oxygen- and sulfur-containing cyclic compounds [2].
The mechanistic aspects of the cycloaddition reactions have been the subject of the most interesting controversies. Woodward and Hoffmann believed that a pericyclic reaction is a concerted reaction in which all bonds are formed or broken through a cyclic transition state [3]. In 1986, Dewar et al. introduced the two-stage mechanisms as asynchronous concerted, in which the changes in bonding take place in two stages. In other words, some of the bonding changes occur in the first half of the reaction pathway while the other changes occur in the second one [4]. After that in 1995, Houk and his coworker believed that an asynchronous concerted process takes place via two-stage mechanism, in which two new bonds are formed in separate but overlapping processes [5]. Further studies on those cycloaddition processes, which occur in a high asynchronous transition state but in a single kinetic step, revealed that two new σ bonds are formed between two fragments in a nonconcerted fashion [6]. According to the Domingo’s definition, when a reaction takes place through a one-step nonconcerted process, the reaction can be considered as a two-stage process. In this mechanism, the formation of two new bonds between two fragments is not concerted, and the formation of the second σ bond begins after much progress in the formation of the first one [7].
Among the fused five-membered heterocycles, pyrrolo[2,1-b]thiazoles are one of the nitrogen and sulfur-containing compounds, possessing remarkable biological activities. These heterocyclic compounds are well known as structural building blocks of many biologically active molecules and can be used as anti-breast cancer, antitumor, anti-inflammatory, antibiotic, anticonvulsant, hepatoprotective and antidiabetic agents [8, 9]. Owing to the importance of pyrrolo[2,1-b]thiazoles, the development of efficient synthetic procedures to build such heterocyclic structures is of interest [8, 10, 11].
One of the well-known methods for the synthesis of five-membered nitrogen-containing heterocycles is [3 + 2] cycloaddition of azomethine ylides and various dipolarophiles. In this way, in 2017, Yan et al. reported an intermolecular [3 + 2] cycloaddition reaction between N-phenacylbenzothiazolium bromidese and nitroalkenes under basic conditions for the synthesis of tetrahydrobenzo[d]pyrrolo[2,1-b]thiazoles (Scheme 1) [10].

Synthesis of tetrahydrobenzo[d]pyrrolo[2,1-b]thiazoles using a [3 + 2] cycloaddition [10]
Based on the proposed mechanism by Yan et al., initial deprotonation of N-phenacylbenzothiazolium bromidese (A) by triethylamine generates a benzothiazolium ylide (B) or its resonance form (B′). By nucleophilic attack of ylide (B) on nitroalkene, a zwitterionic intermediate (C) is generated, which undergoes a ring closure to produce two diastereomeric tetrahydrobenzo[d]pyrrolo[2,1-b]thiazoles (D) and (D′) (Scheme 2).

The proposed mechanism for the synthesis of tetrahydrobenzo[d]pyrrolo[2,1-b]thiazoles (D) and (D′) via a [3 + 2] cycloaddition reaction between N-phenacylbenzothiazolium bromidese and nitroalkenes under basic conditions
Herein, in continuation of our previous theoretical studies on various organic compounds [12, 13], the [3 + 2] cycloaddition of N-(p-methylphenacyl)benzothiazolium ylide (NBY, which generates from deprotonation of N-(4-methylphenacyl)benzothiazolium bromide) and 1-nitro-2-(p-methoxyphenyl) ethene (NME), experimentally explored by Yan and co-workers [10] (see Scheme 3), is theoretically analyzed at the density functional theory (DFT) levels to elucidate the regioselectivities and molecular mechanism of the reaction.

Reaction paths involved in the [3 + 2] cycloaddition reaction of NBY toward NME
The important aims of the present research are:
-
Analysis of the global and local reactivity indices at the ground state of the reagents involved in the [3 + 2] cycloaddition of NBY and NME.
-
Potential energy surface exploration of reaction paths involved in the studied reaction.
-
To study the regioselectivity and stereoselectivity of the reaction.
-
To elucidate the global electron density transfer (GEDT) at transition states.
-
To study the molecular mechanism and investigation of the reaction synchronicity.
2 Computational details
The geometry of the reactants and products was optimized using wB97XD/6-311G** and M062X/6-311G methods [14, 15]. Solvent effects of ethanol in the optimizations were considered using the conductor-like polarizable continuum model (CPCM) [16]. The transition states were determined by using the Berny algorithm or the synchronous transit-guided quasi-Newton (STQN) procedure [17, 18]. The frequency calculations were carried out on the optimized structures to verify that all transition states have one imaginary frequency and all reactants and products have only real frequency values. The optimized geometries of the transition states were verified by using intrinsic reaction coordinates (IRC) calculations [19, 20].
NBO population analysis was applied to compute the atomic charges and the Wiberg bond indices of the reactants, transition states and products [21, 22].
QTAIM analysis was carried out using the AIM2000 program [23].
The global nucleophilicity index N was computed using the following equation:
where EHOMO(TCE), the HOMO energy of tetracyanoethylene (TCE), is considered as the reference and EHOMO(Nu) denotes the HOMO energy of nucleophile [24]:
The global electrophilicity index ω was calculated using the following equation from the chemical hardness η and electronic chemical potential μ [25].
Both η and μ quantities were computed from the frontier molecular orbitals energies using the following equations [26, 27]:
All calculations were performed using the Gaussian 09 software [28].
3 Results and discussion
3.1 Analysis of the global reactivity indices of the reagents
Conceptual density functional theory (CDFT) is one of the powerful tools to study the reactivity as well as regioselectivity in organic transformations [29, 30]. This theory can also predict satisfactorily the polar character of a reaction. Thus, at the first step of the work, the CDFT global reactivity indices were computed to assess the reactivity of the reactants as well as the polar character of the reaction. Table 1 illustrates the wB97XD/6-311G** and M062X/6-311G** computed CDFT global reactivity indices, i.e., the chemical potential (μ), chemical hardness (η), global nucleophilicity (N) and global electrophilicity (ω) of NBY, NME in both gas and ethanol phases.
An analysis of the results given in Table 1 indicates that the electronic chemical potential of NME is more negative than that of NBY. Therefore, along a polar [3 + 2] cycloaddition reaction between NME and NBY electron density is expected to flow from NBY, as nucleophile, toward NME, as electrophile. Also, the values of nucleophilicity N and electrophilicity ω indices reveal that NBY is a stronger nucleophile than NME. On the other hand, NME shows an electrophilicity power more than NBY. Thus, we can expect that the relevant reaction is polar in character and probably exhibits a low activation barrier.
3.2 Study of the regioselectivity of the reaction
Both electronic and steric effects can control regioselectivity in a reaction. Electronic effects are considered when the reactants are separated from each other and can be analyzed at the reactive sites while steric effects become important when the reactants approach to each other. To study the regioselectivity in the reaction, four possible reactive channels were considered between the reactants, NME and NBY, to form four possible cycloadducts P-I through P-IV (Scheme 3). As portrayed in scheme 3, all reactive channels can take place through one of the two possible Cγ–Cε and Cγ–Cδ regioisomeric routs, which are characterized by Cγ–Cε and Cγ–Cδ attacks, respectively. Within the Cγ–Cε attack, two diastereomeric adducts, namely P-I and P-II, are formed, while Cγ–Cδ attack leads to the formation of two different diastereomeric adducts P-III and P-IV. As mentioned in Sect. 1, Yan et al. reported experimentally that the reaction proceeds through Cγ–Cε regioisomeric attack to form P-I and P-II adducts in a 4:1 ratio [10].
In polar reactions between non-symmetrical reactants, when electrophile and nucleophile approach to each other, the most favorable interaction occurs between the most nucleophilic center of nucleophile and the most electrophilic center of electrophile. Thus, the calculation of local reactivity associated with the reactants can help us to predict the best interaction between two species. Fukui functions reactivity indices are one of the powerful tools to elucidate the local reactivity of the reactants. According to a model established by Yang and Mortier, the Fukui functions can be obtained using Mulliken population analysis of an atom, k, in a molecule [31]:
The local electrophilicity \(\omega_{k}\) and nucleophilicity \(N_{k}\), which are widely used to predict the reactivity and regioselectivity in various polar processes are computed using following equations [32, 33]:
Herein, the local electrophilicity \(\omega_{k}\) and nucleophilicity \(N_{k}\) indices at the reactive sites of the reagents involved in the cycloaddition reaction were computed using NBO, Hirshfeld and Mulliken population analyses. Since, in the studied reaction, NBY and NME act as nucleophile and electrophile, respectively, the local nucleophilicity (\(N_{k}\)) of the former and the local electrophilicity (\(\omega_{k}\)) of the later were computed. The results are presented in Table 2.
As given in Table 2, the Cε atom of NME possessing higher value of local electrophilicity than the Cδ atom, which indicates NME is more electrophilically activated at the Cε atom. On the other hand, all three population analyses clearly indicate that NBY is more nucleophilically activated at the Cγ atom than the Cα one. According to the Fukui function analysis, it can be concluded that along the nucleophilic attack of NBY on NME, the most favorable nucleophile–electrophile interaction will occur between the most electrophilic center of NME, Cε atom, and the most nucleophilic center of NBY, Cγ atom, to proceed the reaction within the Cε–Cγ regioselectivity (paths 1 and 2 in Scheme 3). This regioselectivity prediction which leads to the formation of P-I and P-II cycloadducts is in excellent agreement with the experimental results [10].
Another approach to investigate the local reactivity and regioselectivity in polar reactions is based on nucleophilic \(P_{k}^{ - }\) and electrophilic \(P_{k}^{ + }\) Parr functions, which are obtained from the excess of spin electron density reached via the electron transfer process between two reagents involved in polar reactions [34, 35]. Since NME and NBY act as electrophile and nucleophile, respectively, the electrophilic \(P_{k}^{ + }\) Parr functions of NME and the nucleophilic \(P_{k}^{ - }\) Parr functions of NBY were computed. Table 3 summarizes the computed Parr functions for the reagents.
As given in Table 3, the Cε atom of NME with higher value of \(P_{k}^{ + }\) is more electrophilic than the Cδ atom. On the other hand, the Cγ atom of NBY is more nucleophilic than the Cα and Nβ atoms. So again, it is clear that the most nucleophilic/electrophilic interaction occurs between the Cε atom of NME as the most reactive site of the electrophile, and the Cγ atom of NBY as the most reactive site of the nucleophile. Accordingly, the reaction proceeds through the Cε–Cγ regioselective attack to produce either P-I or P-II adducts, which is in complete agreement with the experimental outcomes [10].
3.3 Potential energy surface (PES) analysis of the reactive channels
To examine the aforementioned local reactivity indices, the transition states associated with the four reactive channels between NME and NBY were computed by using wB97XD/6-311G** method. Also, analysis of the IRC profiles relevant to the transition states TS-I, TS-III and TS-IV involved in the three reactive channels in ethanol clearly revealed that the corresponding cycloaddition reactions proceed via a one-step mechanism without the formation of any stable intermediate. On the other hand, the IRC analysis of TS-II (which from now on is called TS-II-1) in ethanol indicated that the relevant reactive channel (path 2) proceeds through a stepwise mechanism along with the formation of a stable zwitterionic intermediate, which from now on is called Int-II. This intermediate then experiences a rapid intramolecular ring-closure reaction to yield the corresponding cycloadduct P-II through transition state TS-II-2. As mentioned in Sect. 1, Yan et al. proposed that the reaction proceeds via a stepwise mechanism with the formation of a zwitterionic intermediate (Scheme 2) [10]. As mentioned above, the IRC analyses showed that only the reactive channels 2 in ethanol involves the formation of a stable intermediate. It should be here noted that all reactive channels in the gas phase proceed via a one-step mechanism without the formation of any stable intermediate.
To have more details about the PES of the reaction, the free energies of the reactants, transition states, intermediate and products and also the relative free energies associated with the transition states, intermediate and products (ΔG) involved in the four reactive channels were computed. Table 4 summarizes the calculated free energies in both gas and ethanol phases. Also, the relative free energies were plotted to evaluate the reactive channels in a more comparable manner. The results are depicted in Fig. 1.

Relative free energy diagram in the presence of ethanol at 25 °C for the species involved in the four reactive channels between NME and NBY
An overview of Fig. 1 indicates that all reactive channels are energetically possible because the corresponding products are more stable than the reactants. It is clear that the Cγ–Cε regioselective attack is more favorable both kinetically and thermodynamically than the Cγ–Cδ one, which is in excellent agreement with the experimental outcomes [10]. In addition, P-II, generated in a pathway releasing a greater amount of energy, ~ 140 kJ mol−1, is the most stable cycloadduct. On the other hand, P-I, which is generated in a pathway that has a low activation barrier, ~ 20 kJ mol−1, can be considered as a product under kinetically controlled conditions. As mentioned in Sect. 1, Yan et al. experimentally reported the formation of P-I and P-II in a 4:1 ratio [10]. In accordance with the kinetic principles, the formation rate of each product, P, can be calculated as k[NBY][NME], where k denotes the second-order rate constant. The rate constant can be calculated from Eyring equation:
where KB denotes the Boltzmann constant, T is the Kelvin temperature, h is the Plank’s constant, ΔG‡ is the activation free energy, and R is the universal constant of the gases [36].
Using kI to kIV, the reaction rate constants associated with the four reactive channels, the Cγ–Cε to Cγ–Cδ regioselectivity can be estimated as:
where kII is the rate constant for the first step (the rate-determining step) of the reactive channel 2.
Such a very high Cγ–Cε regioselectivity, in complete agreement with the experimental outcomes [10], indicates that only both P-I and P-II adducts are produced over the course of a Cγ–Cε regiospecific reaction between NME and NBY. These results are completely consistent with those obtained from Fukui and Parr function analyses (see Fukui and Parr functions analyses section).
Similarly, the calculated concentration ratio of I-P to II-P in the reaction mixture can be estimated as:
This computed stereoselectivity for the formation of I-P rather than II-P is not in complete agreement with the experimental outcomes (I-P:II-P = 4:1) [10].
The dielectric constant of the medium influences the rate of the polar reactions. If the transition state is more polar than the reactants, the reaction accelerates when the dielectric constant of the medium is increased. On the other hand, if the polarity of the reactants is more than the transition state, the reaction rate is reduced by increasing the dielectric constant of the medium. Since, in the studied reaction, the polarity of the reactants (especially of NBY as 1,3-dipole) is more than that of the transition state, the rate of all reactive channels in ethanol is smaller than that in the gas phase (Table 4, compare the activation barriers associated with the transition states). Ethanol as a polar solvent with dielectric constant of 24.5 lowers the energy of the reagents more than the transition states and increases the activation barriers. In addition, the zwitterionic intermediate Int-II is only detected in the presence of ethanol and not in the gas phase, which can be attributed to the stabilizing effects of the solvent molecules.
3.4 Thermodynamic and kinetic studies of four other alternative reactive channels
As mentioned in Sect. 1, Yan et al. reported the synthesis of two tetrahydrobenzo[d]pyrrolo[2,1-b]thiazoles using a [3 + 2] cycloaddition [10]. They prepared the single crystal of two major (D, Ar = p-MeC6H4, Ar′ = C6H5) and minor (D′, Ar = p-FC6H4, Ar′ = p-MeC6H4) cycloadducts (Scheme 2), and successfully determined the molecular structures using X-ray diffraction method. The results indicated that the both cycloadducts have an R configuration at the Cγ center, which correlate with the P-I and P-II geometries. Thus, it appears that these geometries are more stable than the alternative geometries possessing opposite configuration at the Cγ center. However, four alternative reactive channels can be considered between NBY and NME in which the Cγ center has an opposite configuration (S) in the generated cycloadducts (P-I′ to P-IV′). Scheme 4 depicts these reactive channels.

Four alternative reaction paths between NBY and NME
At the next step, the geometries of the corresponding transition states (TS-I′ to TS-IV′) and products (P-I′ to P-IV′) were optimized in both gas and ethanol phases and their free energies were extracted. The results are presented Table 5.
Also, the relative free energies for all original and alternative reactive channels (paths 1 to 4 and 1′ to 4′, respectively) were plotted to evaluate the reactive channels in a more comparable manner. The results are depicted in Fig. 2.

Relative free energy diagram in the presence of ethanol at 25 °C for the species involved in the eight reactive channels between NME and NBY
According to the results presented in Table 5 and Fig. 2, it is clear that all alternative reactive channels (paths 1′ to 4′) are both thermodynamically and kinetically more unfavorable than the original reactive channels (paths 1 to 4). In facts, all cycloadducts obtained from the reactive channels 1′ to 4′ have higher energy than those obtained from reactive channels 1 to 4. In addition, all transition states corresponding to the reactive channels 1′ to 4′ are more unstable relative to those of 1′ to 4′ paths. Among the eight transition states, TS-I which leads to the P-I adduct, is the most favorable transition state. This is in complete agreement with the experimental outcomes. The IRC analyses on TS-II-1′ show that the reactive channel 2′ in ethanol proceeds through a stepwise mechanism along with the formation of a stable zwitterionic intermediate Int-II′. This intermediate then experiences a rapid intramolecular ring-closure reaction to generate the corresponding cycloadduct P-II′ through transition state TS-II-2′. This sequence is completely identical to that of the reactive channel 2.
In the continuation of the present work, the reactive channels 1 to 4 are studied because such pathways are energetically more favorable than 1′ to 4′ ones and also produce the experimentally reported products (P-I and P-II).
3.5 Study of charge transfer in the transition states
The polar character of a cycloaddition reaction can be evaluated using analysis of the magnitude of global electron density transfer (GEDT) between two interacting fragments in the corresponding transition state [37]. The magnitude of the GEDT can be considered as a relative criterion to determine the polarity of a reaction. To investigate the charge transfer, numerical amounts of the GEDT were computed for the intermediate and transition states associated with the Cγ–Cε regioselective attack (TS-I, TS-II-1, Int-II and TS-II-2). The molecular electrostatic potential (MESP) map of the reactants, intermediate and transition states were also computed in order to display the charge transfer in a comparable manner. Figure 3 illustrates the computed values of the GEDTs along with the MESP maps. In the MESP map, the red and blue colors indicate the region with higher and lower electron density, respectively.

The molecular electrostatic potential MESP maps of the reactants, intermediate and transition states, and the computed values of the global electron density transfer (GEDT) for the intermediate and transition states associated with the Cγ–Cε regioselective attack. The H and L abbreviations refer to the regions with high and low electron density, respectively. The GEDT values corresponding to the gas phase are given in parentheses
As given in Fig. 3, a very large value of GEDT found at the transition states, which fluxes from the NBY fragment toward the NME one, 0384 e (for TS-I), 0.398 e (for TS-II-1) and 0.674 e (for TS-II-2), shows a high polar character for the Cγ–Cε regioselective attack. A comparison between the intensity of the red color around the oxygen atom of NBY (which has been specified by H abbreviation in Fig. 3) and that in TS-I and TS-II clearly indicates that the electron density fluxes from NBY fragment toward the NME one. In addition, the magnitude of GEDT, and consequently the polarity of the reactions, is slightly more in ethanol than in the gas phase.
The high value of GEDT for Int-II, 0.804 e, clearly suggests a high degree of charge separation as well as the zwitterionic character of this intermediate. According to the MESP of Int-II, it is clear that the negative charge obtained from nucleophilic attack of NBY on NME is delocalized over the nitro group. On the other hand, the obtained positive charge on the NBY fragment is delocalized over the benzothiazol moiety.
It is clear from Fig. 3 that at the energetically more favorable transition state, TS-I, approach of reactants locates the oppositely charged regions over each other resulting in attractive forces between two fragments. On the other hand, at the energetically less favorable transition state, TS-II-1, regions with the same charge are forced to be located over each other leading to repulsive forces between two fragments. Therefore, due to the electrostatic attractive forces between two interacting fragments, formation of TS-I is more favorable than formation of TS-II-1, which is in complete agreement with the experimental outcomes [10].
3.6 Study of the interaction between the frontier molecular orbitals of the reactants
When a cycloaddition reaction takes place between two species, the interaction between those frontier molecular orbitals, namely the HOMO of one (donor) and the LUMO of the other (acceptor), is a determining factor and often controls the total orbital interaction between two species. In the studied reaction, NBY and NME act as electron donor and acceptor, respectively. Thus, it can be concluded that the HOMO of NBY can interact with the LUMO of NME. However, recently it was found that sometimes the HOMO–LUMO interactions fail to describe the bond formation correctly, because two factors can determine the interaction between the frontier orbitals: the HOMO–LUMO energy difference of the reagents and the relative contribution of the active sites in the molecular orbital. The HOMO–LUMO interaction considers only the first factor. Thus, a new concept called frontier effective-for-reaction molecular orbital (FERMO) has emerged to describe the interaction of the frontier molecular orbitals. This concept considers one of the highest occupied molecular orbitals with a large contribution in the reactive atoms. For many reactions, the FERMO can work better than HOMO [38]. To determine the FERMO orbital of NBY as electron donor, the relative contribution of its active sites, Cα, Nβ and Cγ atoms, was computed in the three highest occupied MOs, namely HOMO, HOMO-1 and HOMO-2. The results are depicted in Fig. 4.

The LUMO of NME and the three highest occupied MOs of NBY along with the relative contribution of the active sites
An analysis of the relative contribution of the active sites in the three highest occupied MOs presented in Fig. 4 clearly indicates that the HOMO of NBY is also a FERMO orbital, because the relative contributions corresponding to the active sites in HOMO, 8.90%, 5.03% and 38.64%, respectively, for Cα, Nβ and Cγ atoms, are more than those in the other occupied MOs. The high contribution of the Cγ atom in the FERMO orbital, 38.64%, relative to the other sites, Cα and Nβ, clearly confirms that NBY is nucleophilically activated at the Cγ atom. This is in complete agreement with the local reactivity indices obtained from Fukui and Parr function analysis.
3.7 Study of the synchronicity and the molecular mechanism of the reactions
A one-step cycloaddition reaction may take place in a synchronous or asynchronous fashion, in which the bond changes occur, respectively, in one or two stages but through one transition state. Since, in the studied reaction, the IRC analysis ruled out the formation of any stable intermediate for the reactive channels 1, 3 and 4, it can be concluded that these reactive channels should take place via a synchronous or asynchronous mechanism. On the other hand, it was found from IRC analysis that the reactive channel 2 proceeds through a stepwise mechanism yielding a stable zwitterionic intermediate, Int-II. To study the synchronicity of the transition states, the Wiberg bond indices, corresponding to those bonds that participate directly in the reaction, were calculated using NBO analysis. For this purpose, the Wiberg bond indices, Bi, associated with the reagents, intermediate and transition states were calculated and then, relative change in each bond index, ΔBi, was calculated using the following equation:
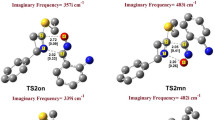
Figure 5 depicts the wB97XD/6-311G** optimized structure of the transition states and intermediate involved in the studied reaction in the presence of ethanol along with the values of ΔBi. Also, the imaginary frequency of TSs and the corresponding displacement vectors are shown in Fig. 5. The optimized structures of the reactants and products and their Wiberg bond indices are given in Fig. A1 of supplementary materials.

The wB97XD/6-311G** optimized structure of the intermediate and transition states at the presence of ethanol along with the TS imaginary frequencies as well as the changes in the Wiberg bond indices ΔBi
As given in Fig. 5, it is apparent that there is an imaginary frequency for each transition state with a displacement vector in the direction of bond formation between the two fragments, which indicates that all TSs have been computed correctly. In addition, analysis of the ΔBi values indicates that the one-step reactive channels (paths 1, 3 and 4) are asynchronous. It is clear that the degree of progress of two new σ bonds between the two reactants is not equivalent in the corresponding transition states. For instance, the value of ΔBi for the Cγ–Cε and Cα–Cδ bonds in TS-I is + 0.38 and + 0.08, respectively, which reveals that on going from the reactants to the TS, the reaction progresses mainly through the formation of the Cγ–Cε bond. On the other hand, when the formation of the Cγ–Cε bond is very advanced, the Cα–Cδ bond begins to form. This trend is in complete agreement with the results of the local Fukui and Parr function analysis, because the most favorable interaction was found to be between the Cγ and Cε atoms of two fragments. This behavior reveals that this [3 + 2] cycloaddition reactive channel takes place via a two-stage one-step mechanism in which the formation of the second σ bond, Cα–Cδ, begins when the first one, Cγ–Cε, is mainly formed. For the transition state associated with Cγ–Cε regioisomeric attack, the order of Cγ–Cε bond is 0.38 at TS-I and 0.40 at TS-II-1, while the order of Cα–Cδ bond is 0.08 (TS-I) and 0.06 (TS-II-1). This is indicative of a higher degree of asynchronicity for TS-II-1 compared to TS-I. However, this high degree of asynchronicity leads to a stepwise mechanism with the formation of a stable zwitterionic intermediate, Int-II. A comparison between the absolute value of ΔBi for the Cγ–Cε and Cε–Cδ bonds, 0.91 and 0.71, implies that these bonds experience significant changes during the conversion of TS-II-1 to Int-II. In addition, it is clear that on going from Int-II toward TS-II-2, the Cα–Cδ bond begins to form and its ΔBi value increases from + 0.04 to + 0.18, whereas the Cγ–Cε and Cε–Cδ bonds do not experience any significant variation. During the conversion of TS-II-2 to the P-II adduct, this trend continues and the ΔBi value of the Cα–Cδ bond increases from + 0.18 to + 0.97. Among the studied transition states, TS-III and TS-IV are more synchronous compared to the others in which the two new bonds are formed in overlapping processes. In these transition states, the Cγ–Cδ bond begins to form when the Cα–Cε bond has formed partially.
The IRC profiles associated with the first reactive channel (paths 1) is shown in part A of Fig. 6. It is clear that the reaction proceeds via a one-step mechanism without the formation of any stable intermediate. However, there is a shoulder-like region on the pathway from TS-I to P-I, but we were unable to locate any minimum energy geometry on this region of the potential energy surface. This indicates that the probable intermediate is very unstable in ethanol, in which quickly experiences a ring closure without any activation barrier to afford the corresponding adduct, P-II.

The IRC profile (A), the variations of the Wiberg bond indices Bi (B) and the variations of the BCP electron densities ρi (C) during the reactive channel 1 at the presence of ethanol
To evaluate the bond formation during the Cγ–Cε regioselective attack, the Wiberg bond index corresponding to the two new Cγ–Cε and Cα–Cδ bonds between two fragments was calculated for a series of points on the IRC profile. The results are given in Table 6. Also, the corresponding diagram is depicted in part B of Fig. 6. An overview over the results clearly indicates that the formation of two new bonds is asynchronous. Actually, the Cγ–Cε bond is formed faster than the Cα–Cδ one. For instance, at point P7, where the formation of Cγ–Cε bond is very advanced, Bi = 0.867, the second bond, Cα–Cδ, is slightly formed, Bi = 0.141. Thus, it can be concluded that the reaction takes place via a two-stage one-step mechanism, in which two new bonds are formed in separate but overlapping processes. In other words, the reaction proceeds through an asynchronous one-step mechanism and the formation of the Cα–Cδ bond begins after much progress in the formation of the Cγ–Cε one.
An appropriate criterion to evaluate various interactions has been established by Espinoza in terms of the ratio of potential to kinetic electron energy density, \(\frac{\varvec\mid{{\varvec{V}}\mid}}{\varvec{G}}\), at the corresponding bond critical point (BCP) [39]. According to this method, pure closed-shell interactions, including non-covalent, ionic, and hydrogen bonding, suggest \(\frac{\varvec\mid{{\varvec{V}}\mid}}{\varvec{G}}\) < 1, while pure shared-shell interactions (covalent) indicate \(\frac{\varvec\mid{{\varvec{V}}\mid}}{\varvec{G}}\) > 2. A borderline interaction can be characterized using a \(\frac{\varvec\mid{{\varvec{V}}\mid}}{\varvec{G}}\) value between 1 and 2. The calculated values of ρ and \(\frac{\varvec\mid{{\varvec{V}}\mid}}{\varvec{G}}\) for the Cγ–Cε and Cα–Cδ BCPs obtained from QTAIM analysis, corresponding to a series of points on the IRC profile, are given in Table 6. The diagrams of changes in the electron density (ρ) along the IRC profile are also depicted in part C of Fig. 6.
Analysis of the topological QTAIM descriptors presented in Table 6 and part C of Fig. 6 indicates that, similar to the Wiberg bond indices, during the first half of the reaction, the electron density along the Cγ–Cε bond increases faster than it does along the Cα–Cδ bond. Once again, it is clear that the former bond is formed faster than the latter. Also, the values of \(\frac{\varvec\mid{{\varvec{V}}\mid}}{\varvec{G}}\) are increased during the reaction coordinate and this increasing trend is in parallel with the electron densities. Also, most of the BCPs between the interacting atoms show \(\frac{\varvec\mid{{\varvec{V}}}\mid}{\varvec{G}}\geq 1\) along the reaction coordinate which is indicative of the covalent nature of the interactions.
4 Conclusion
By using the computational methods at the wB97XD/6-311G** and M062X/6-311G** levels of theory a [3 + 2] cycloaddition reaction between N-(p-methylphenacyl)benzothiazolium ylide (NBY) and 1-nitro-2-(p-methoxyphenyl) ethene (NME) experimentally studied by Yan et al., has been investigated. They reported that the studied reaction is regioselective and stereoselective with the formation of two cycloadducts in a 4:1 ratio.
In this study, two Cγ–Cε and Cγ–Cδ regioselective attacks were considered between NBY and NME, and studied theoretically. Analysis of the global CDFT reactivity indices indicated that NBY and NME act as donor (nucleophile) and acceptor (electrophile), respectively. The Fukui and Parr functions reactivity indices analysis of the reactants confirmed the experimentally observed regioselectivity. Also, the energetic results indicated that in complete agreement with the experimental outcomes, the Cγ–Cε regioselective attack is kinetically more favorable than the Cγ–Cδ one.
The IRC, QTAIM and Wiberg bond indices analyses clearly indicated that the studied reactions occur via two different mechanisms. The major cycloadduct is generated through a two-stage one-step mechanism without the formation of any stable intermediate, whereas the minor one is generated through a stepwise mechanism along with the formation of a stable zwitterionic intermediate. In the two-stage one-step mechanism two new bonds (Cγ–Cε and Cγ–Cδ) are formed in separate but overlapping processes.
The global electron density transfer (GEDT) analysis suggested that the reactions are polar and electron density fluxes from NBY toward NME. It was found from MESP map that at the more favorable transition state, approach of reactants locates the oppositely charged regions over each other resulting in attractive forces between two reactants.
Analysis of the frontier molecular orbitals suggested that the HOMO orbital of NBY also can be considered as a frontier effective-for-reaction molecular orbital (FERMO).
References
Carruthers W (1990) Cycloaddition reactions in organic synthesis. Pergamon, Oxford
Padwa A, Pearson WH (2002) Synthetic applications of 1,3-dipolar cycloaddition chemistry toward heterocycles and natural products. Wiley, New York
Woodward RB, Hoffmann R (1965) J Am Chem Soc 87:395–397
Dewar MJS, Olivella S, Stewart JJP (1986) J Am Chem Soc 108:5771–5779
Houk KN, Gonza´lez J, Li Y (1995) Acc Chem Res 28:81–90
Berski S, Andre´s J, Silvi B, Domingo LR (2006) J Phys Chem A 110:13939–13947
Domingo LR, Sae´z JA, Zaragoza´ RJ, Arno M (2008) J Org Chem 73:8791–8799
Soares MIL, Brito AF, Laranjo M, Paixão JA, Botelho MF, Pinho e Melo TMVD (2013) Eur J Med Chem 60:254–262
Shen YM, Lv P-Ch, Zhang M-Zh, Xiao H-Q, Deng L-P, Zhu H-L, Qi Ch-Z (2011) Monatshe Chem Chem Mon 142:521–528
Jin G, Sun J, Yang R-Y, Yan Ch-G (2017) Sci Rep 7:46470
Shen G-L, Sun J, Yan Ch-G (2015) Org Biomol Chem 13:10929–10938
Soleymani M (2018) Monatshe Chem Chem Mon 149:2183–2193
Soleymani M (2019) Theoretical study on the [4+2] cycloaddition of 1,3-dimethylindole with 2,6-dimethylquinone. Struct Chem. https://doi.org/10.1007/s11224-018-1259-1
Chai J-D, Head-Gordon M (2008) Phys Chem Chem Phys 10:6615–6620
Zhao Y, Truhlar DG (2006) J Phys Chem 110:5121–5129
Barone V, Cossi M (1998) J Phys Chem A 102:1995–2001
Schlegel HB (1982) J Comput Chem 3:214–218
Peng C, Ayala PY, Schlegel HB, Frisch MJ (1996) J Comput Chem 17:49–56
Gonzalez C, Schlegel HB (1989) J Chem Phys 90:2154–2161
Gonzalez C, Schlegel HB (1990) J Phys Chem 94:5523–5527
Reed AE, Curtiss LA, Weinhold F (1988) Chem Rev 88:899–926
Carpenter JE, Weinhold FJ (1988) J Mol Struct 169:41–62
Biegler-Konig F, Schonbohm J, Bayles D (2001) J Comp Chem 22:545–559
Domingo LR, Perez P, Ortega DE (2013) J Org Chem 78:2462–2471
Parr RG, Szentpaly LV, Liu S (1999) J Am Chem Soc 121:1922–1924
Parr RG, Pearson RG (1983) J Am Chem Soc 105:7512–7516
Parr RG, Yang W (1989) Density functional theory of atoms and molecules. Oxford University Press, New York
Frisch MJ, Trucks GW, Schlege HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Peralta JE Jr., Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2010) Gaussian 09, Revision E.01. Gaussian, Inc., Wallingford
Geerlings P, De Proft F, Langenaeker W (2003) Chem Rev 103:1793–1874
Ess DH, Jones GO, Houk KN (2006) Adv Synth Catal 348:2337–2361
Yang W, Mortier WJ (1986) J Am Chem Soc 108:5708–5711
Domingo LR, Aurell MJ, Pêrez P, Contreras R (2002) J Phys Chem A 106:6871–6875
Pêrez P, Domingo LR, Duque-Noreňa M, Chamorro E (2009) J Mol Struct THEOCHEM 895:86–91
Domingo LR, Pérez P, Sáez JA (2013) RSC Adv 3:1486–1494
Chamorro E, Pérez P, Domingo LR (2013) Chem Phys Lett 582:141–143
Eyring H (1935) J Chem Phys 3:107–115
Domingo LR (2014) RSC Adv 4:32415–32428
Da Silva RR, Ramalho TC, Santos JM, Figueroa-Villar JD (2006) J Phys Chem A 110:1031–1040
Espinosa E, Alkorta I, Elguero J, Molins E (2002) J Chem Phys 117:5529–5542
Acknowledgements
I am thankful to the Research Council and Office of Graduate Studies of the Ayatollah Boroujerdi University for their financial support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Soleymani, M. A density functional theory study on the [3 + 2] cycloaddition of N-(p-methylphenacyl)benzothiazolium ylide and 1-nitro-2-(p-methoxyphenyl) ethene: the formation of two diastereomeric adducts via two different mechanisms. Theor Chem Acc 138, 87 (2019). https://doi.org/10.1007/s00214-019-2477-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-019-2477-3