
Abstract
Dye-sensitized solar cells have become in one important and promising technology in the photovoltaic field. The ability for a sensitizer to harvest light photons and inject the excited electrons into a photoanode, typically a metal oxide, determines the performance and operation range of the solar cell. Metal complexes with 1,3-dithiole-2-thione-4,5-dithiolate (dmit) ligands, which are an important class of functional materials, have received extensive attention due to their intriguing chemical and physical properties. The electronic and molecular properties of isolated and adsorbed nickel complexes with dmit ligands have been investigated using first-principles calculations based on the density functional theory (DFT). Adsorption energies of metal complexes supported on the anatase TiO2(101) surface were calculated for three different configurations, linked by sulfur atom of Sthione, Sthiole–Sthiolate, and planar. The most stable adsorption configurations found in this study are the Sthiole–Sthiolate and the planar forms for the nickel complex. TD-DFT molecular calculations reveal that the lowest energy transition in ultraviolet visible near-infrared (UV–Vis-NIR) mainly corresponds to the HOMO–LUMO π–π* excitation for the nickel complex. The effect of the TiO2 (101) surface on the absorption spectra of the nickel complex is practically limited to a red shift of about 0.1–0.3 eV. The analysis of the density of states for the dmit/TiO2 (101) system shows that the LUMO of the metal complex lies at the edge of the TiO2 conduction band indicating, therefore, that electron injection from the complex excited state into the semiconductor surface is unlikely.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The sensitization of wide band-gap semiconductors lays at the heart of dye-sensitized solar cells (DSSCs), a rising technology for solar energy harvesting that offers some advantages over classical Si-based devices [1, 2]. A key strength of DSSCs is the separation of electron generation and transport processes in two distinct materials. This allows the disconnected optimization of the dye for photon absorption and of a wide band-gap semiconductor for electron–hole separation and collection [3].
Typically, metal oxides like zinc oxide (ZnO) [4, 5], stannic oxide (SnO2) [6,7,8], and titanium dioxide (TiO2) [2] have been used as the semiconductor material. However, different experimental results have shown that TiO2 is preferable over either ZnO or SnO2. Titanium dioxide is non-toxic, highly abundant, and provides a mesoporous structure for both organic and inorganic dye adsorption.
The role of the dye is to absorb the incoming photons and to transfer the excited electron to the conduction band of the semiconductor. Thus, an efficient dye should (a) be strongly adsorbed at the semiconductor surface; (b) show intense absorption in the visible and near-infrared regions of the electromagnetic spectrum; (c) be stable enough as to be capable of multiple oxidation–reduction cycles; and (d) be stable enough in its oxidized form as to be reduced by the electrolyte and his lowest excited state should be higher than the semiconductor conduction band edge.
Basically, photosensitizer dyes are either pure organic compounds or metal-based organometallic complexes. Metal-free organic sensitizers are cheaper, easy to modify structurally to tune the dye properties; in some cases, they are environmentally benign and non-toxic and have high molar absorption coefficients. However, they also show important stability and efficiency problems. Many metal complex-based dyes have been proposed. Of these, ruthenium (II) polypyridyl complexes have been shown to be the best so far [9]. However, the low abundance of the metal, its high cost, and toxicity impose severe limitations on its practical and widespread use. Thus, transition metal complexes based on iron (Fe), nickel (Ni), cobalt (Co), palladium (Pd), platinum (Pt), and zinc (Zn), among others, have been proposed as alternatives in the design of photovoltaic sensitizers [2, 3, 10,11,12,13,14].
In this regard, square-planar complexes with sulfur-containing ligands that absorb in the near-infrared region (NIR) of the spectrum have attracted special interest and have been examined both experimentally and theoretically [12, 15,16,17,18,19]. Islam et al. [15] were the first to explore the application of a series of square-planar diimine–dithiolate complexes as sensitizers. They synthesized and characterized a series of platinum-based polypyridyl complexes with dithiolate ligands that were also anchored to nanocrystalline TiO2 in photoelectrochemical cells. The intense charge-transfer band in these complexes was shown to be tunable by changing the dithiolate ligands. Geary et al. [16] prepared and examined a family of Pt(II)(diimine)(dithiolate) complexes, analyzing the influence of 3,3′-, 4,4′-, and 5,5′-bipyridyl substituents on their electronic properties. All synthesized complexes were attached to a TiO2 substrate and tested as solar cells sensitizers with the 3,3′-disubstituted bipyridyl complex showing the highest photovoltaic performance.
In a later study [17], the superior performance of the 3,3′- bipyridyl complex was rationalized by using density functional theory calculations based on a hybrid functional that suggested that the longer-lived charge-separated state for this complex on TiO2 was related to the non-planar geometry of the complex, reducing the electronic coupling between ligands. Lazarides et al. [18] have attempted to increase the light absorption properties of Pt(II)(diimine)(dithiolate) chromophores by combining them with boron-dipyrromethene, a strongly absorbing dye, in a dual chromophore system. By using time-dependent DFT calculations, the authors show that the many paths for electron transfer that exist in these systems result in unexpected routes for excited-state relaxation and loss of the desired properties of the excited charge-transfer state. Despite the intense work developed on examining the potential of Pt(II)(diimine)(dithiolate) complexes as sensitizers for DSSC cells, only the paper by Linfoot et al. [12] has studied some Ni(II)(diimine)(dithiolate) dyes in relation with their use as dyes in a DSSC cell. The authors characterized the complexes using electrochemical, spectroscopic, and computational techniques and assigned intense visible absorptions to ligand-to-ligand charge-transfer transitions that would suggest appropriate charge separation for using on a photoelectrochemical device. However, low photocurrents were found when the complex was adsorbed on a TiO2 film, a problem that was linked to a short-lived excited state of the Ni(II) complex.
Because of their unique properties related to applications in fields as diverse as conducting and superconducting materials, nonlinear optics, catalysis, and dyes, metal dithiolene complexes, R2M(dmit)2, R = PyMe, NEt4, NMe4, NPr4, NBu4, and dmit = 1,3-dithiole-2-thione-4,5-dithiolate) have been extensively studied for more than forty years [20]. These applications result from an interplay of different properties, including highly delocalized frontier orbitals that allow direct electron transfer through the ligand π orbitals. For this reason, these complexes are considered promising candidates for photochemical devices [21].
Here, we present a study of the structural and spectroscopic properties of the model dithiolene complex [(CH3)2][Ni(dmit)2] by combining DFT and TD-DFT calculations. First, we analyze the properties of the isolated complex using the B3LYP functional and an atom-centered basis set. Second, the geometric and electronic properties of the complex adsorbed on a model TiO2 (101) anatase surface have been examined by using plane-wave calculations that include both the use of a Hubbard correction to properly localize the metal d electrons and an approximate functional to improve the description of the dispersion forces on the DFT calculations. Finally, we theoretically examine the performance of the model [(CH3)2][Ni(dmit)2] complex as a sensitizer.
2 Computational details
For the isolated [CH3]2[Ni(dmit)2] complex, DFT calculations have been performed using the Gaussian 09 quantum chemical package [22]. Equilibrium geometry and electronic properties were determined by employing the hybrid Becke three-parameter functional with the Lee, Yang, and Parr (B3LYP) exchange correlation functional [23, 24], with CEP-121G [25,26,27] effective core potentials and basis sets for Ni and S atoms and 6–31++G basis set for C and H atoms. Given that some charge-transfer character was found in the first excited state, the CAM-B3LYP hybrid functional, that includes long-range corrections, was also employed [28]. TD-DFT single-point energy calculations were performed on optimized geometries.
To determine the geometric and electronic properties of the TiO2 (101) surface and TiO2 (101) surface with the adsorbed nickel complex, we performed periodic DFT calculations using the Vienna ab initio Simulation Package (VASP) [29,30,31]. The projector augmented wave (PAW) method was used, and the cutoff energy was set to 400 eV for slab and adsorption calculations. The generalized gradient approximation (GGA) functional was used (Perdew-Burke–Ernzerhof, PBE) [32]. In order to better render the anatase band gap, usually underestimated in plain GGA DFT calculations, a Hubbard-type on-site Coulomb correction term was used as implemented by Dudarev et al. [33]. The GGA + U procedure was applied on the transition metal d electrons, being the \( U_{\text{eff}} \) values employed in this work 4.5 eV and 5.5 eV for 3d levels of Ti and Ni, respectively [34, 35]. Optical spectra were obtained from the frequency-dependent dielectrical functions as proposed by Gajdoš et al. [36].
Because GGA functionals neglect attractive long-range contributions, computed adsorption energies are generally underestimated [37]. To include the van der Waals corrections into the density functional approach (vdW-DF) and obtain a more accurate description, the method proposed by Tkatchenko and Scheffler was employed in this work [38].
The slab model of anatase surface was obtained by appropriately cutting the most stable TiO2 (101) surface and is represented by 96 [TiO2] units arranged according to anatase crystalline structure. The model 5 × 3 supercell consisted of two O-Ti–O trilayers, 144 atoms each, where the bottom layer was fixed. The orthorhombic supercell has, thus, dimensions: a = 31.254, b = 15.288 and c = 35.916 Å, including a vacuum space of 20 Å in the c direction. All calculations were performed at the Γ point.
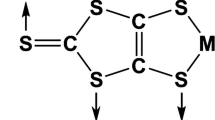
Adsorption of [CH3]2[Ni(dmit)2] complex on the TiO2 (101) surface was done in three different adsorption configurations: linked by Sthione, linked by Sthiole–Sthiolate (bridge), and plane (Fig. 1).

Schematic structure of adsorption form Sthione, Sthiole–Sthiolate
Adsorption energies (\( E_{\text{ADS }} \)) for the optimized metal complexes on the TiO2(101) surface were calculated using
where \( E_{{\left( {{\text{TiO}}_{2} } \right) + \left( {{\text{Ni}}_{\text{dmit}} } \right)}} \) is the energy of (Ni-dmit) complex adsorbed on the TiO2 (101) surface, \( E_{{{\text{Ni}}_{\text{dmit}} }} \) and \( E_{{\left( {{\text{TiO}}_{2} } \right)}} \) are the energies of the isolated Ni-dmit complex and clean TiO2 (101) surface, respectively. With this definition, negative adsorption energies represent bound states stable with respect to desorption.
3 Results and discussion
3.1 Structure and electronic properties of [CH3]2[Ni(dmit)2] complex
Figure 2 shows the optimized structure of the square-planar complex [CH3]2[Ni(dmit)2]. Table 1 presents the geometric parameters obtained at the B3LYP and PBE + U levels, in comparison with the experimental structure. We report only the relevant bond lengths and bond angles.

Optimized structure of [CH3]2[Ni(dmit)2] complex. Atoms colors code: Ni, gray; C, black; S, yellow; H, white
From the data shown in Table 1, a general agreement between calculated and experimental values is observed. Optimized bond distances are systematically overestimated, and the values obtained from PBE + U calculations in general are in better agreement than those estimated with either the B3LYP or the CAM-B3LYP functionals, except for the C=C double bond. Ni–S bond lengths are very similar to each other and in agreement with the experimental results. In contrast, the S=C bond is significantly overestimated. The disagreement found can be related to the fact that experimental data derive from solid crystal structure diffraction experiments in which packing forces may alter the geometry of individual molecules.
The calculated harmonic vibrational frequencies and band assignments for the nickel complex are presented in Table 2.
The calculated peaks associated with the C–H stretch modes of the [CH3] groups were found at 3073/3098/3001 cm−1 for B3LYP, CAM-B3LYP and PBE + U, respectively. The bands at 1331/1374/1297 cm−1 and 993/1063/950 cm−1 were assigned to C=C and C=S stretching modes, respectively, and were compatible with other published results [40]. Valade et al. [41] also reported the C=C peak at 1430 cm−1 and listed two peaks at 455 cm−1 and 310 cm−1 and both were assigned as Ni–S vibration. The bands at 496/509/490 cm−1 are characteristic of the fundamental vibrations of the thiocarbonate group (–S–(C=Sthione)-S–). The C=S stretching vibration is the characteristic vibration in the IR spectra of DMIT complexes. According to various authors, several peaks appear in the 1050–995 cm−1 range, making it difficult to assign as C=S [42, 43]. The results show the same tendency for different levels of calculations in describing the vibrational properties.
Considering now the electronic properties of metal complexes, we first start analyzing the HOMO and LUMO Kohn–Sham frontier orbitals of [CH3]2[Ni(dmit)2]. As shown in Fig. 3, the HOMO is of π-character and mainly corresponds to the C2S2 2− unit of dmit ligand and Ni(II) d orbital center. The LUMO is mainly contributed from the thiole ring of dmit ligand without metal participation. The same profile was observed by Fan et al. [45].

Kohn–Sham frontier orbitals of isolated [CH3]2[Ni(dmit)2] HOMO (left); LUMO (right)
Absorption electronic spectra were obtained from TD-DFT calculations performed at the optimized ground-state geometries (Fig. 4). Calculated oscillator strengths, transition energies, and wave function for the most relevant transitions of electronic absorption bands are listed in Table 3. The influence of the solvent environment on the absorption spectra was not considered in our calculations. Only transitions with significant oscillator strengths are presented. Fifty singlet electronic-excited states were included.

Gas phase UV–Vis absorption spectrum of [CH3]2[Ni(dmit)2] computed at the TDDFT/B3LYP and TDDFT/CAM-B3LYP levels of theory
As shown in both Table 3 and Fig. 4, the theoretical description of the UV–Vis absorption spectrum of [CH3]2[Ni(dmit)2] complex is, quantitatively, quite different for the two DFT functionals tested. The first absorption appears at a wavelength of ~800 nm when the B3LYP functional is used. However, the CAM-B3LYP functional offers a different picture, with a first, quite intense band appearing at ~590 nm, in much good agreement with the experiment [46, 47]. Both functionals assign this excitation to a HOMO–LUMO π → π* transition, but with significant contribution of the HOMO-1 → LUMO + 1 transition in the case of the CAM-B3LYP functional. At shorter wavelengths, the general features of the CAM-B3LYP computed spectrum also agree in a better way with the experimental data. There are some excitations that give rise to an absorption band around 380 nm assigned to transitions involving molecular orbitals arising from the interaction between d metal orbitals with p orbitals of dmit moieties [48]. However, in the experimental spectrum the band is found at 400 nm and shows a higher intensity than the 600 nm band, pointing to some defects still present in the model. In the high region energy, however, there is a much better agreement with the experiment, noting a prominent band at 4.79 eV (259 nm) that involves intra- and inter-ligand π → π* excited determinants.
3.2 Adsorption energies, structural, and electronic properties of [CH3]2[Ni(dmit)2] supported on the TiO2 (101) surface
Three different types of adsorptions onto TiO2 surface configurations were considered: linked by Sthione (in this case, the methyl group has been displaced to a nearby Sthiole site to allow Sthione to interact with the TiO2 surface), linked by Sthiole–Sthiolate, and planar. The optimized geometry of these structures is shown in Fig. 5, and the values of the adsorption energies are summarized in Table 4.

Side-view optimized geometries for [CH3]2[Ni(dmit)2] adsorbed on TiO2 (101) anatase surface
Table 4 shows that adsorption modes Sthiole–Sthiolate and planar of Ni complex are moderately exothermic, while binding through the Sthione atom is endothermic. Two main ingredients contribute to this different behavior. First, the stability of a complex with a bidentate ligand is invariably greater than the corresponding complex of monodentate (Sthione) ligands. Second, there is a large contribution from the dispersion forces for the Sthiole–Sthiolate and planar modes, which is practically absent in the Sthione mode. In fact, the more efficient Van der Waals interaction in the case of planar adsorption makes it the preferred mode. There are no significant variations on the internal geometry of the Ni complex after adsorption on TiO2 (101) surface.
To analyze the electronic structure, density of states (DOS) was calculated for the optimized structure of clean TiO2 (101) surface, isolated [CH3]2[Ni(dmit)2] complex, and [CH3]2[Ni(dmit)2] complex adsorbed on TiO2 (101) surface. The corresponding DOS plots for the isolated [CH3]2[Ni(dmit)2] complex and the TiO2 surface are reported in Fig. 6. The DOS of clean TiO2(101) shows a broad, filled valence band and a broad, empty conduction band, separated by a gap of 2.48 eV, which is consistent with previous theoretical works [49]. Inspection of Fig. 6 shows that the HOMO of the [CH3]2[Ni(dmit)2] complex appears ~0.98 eV above the TiO2 valence band edge, lying within the band gap. In its turn, the LUMO level lies just below the conduction band edge.

Top: Calculated DOS for the isolated [CH3]2[Ni(dmit)2] complex and clean TiO2 (101) surface; bottom: after adsorption. The zero of the energy is set at the Fermi level of the complex
The performance of DSSC is associated with alignment of the metal complex HOMO–LUMO levels with respect to the TiO2 band edges; this property is the key for an efficient electron injection. The LUMO of metal complex has to be higher in energy than the conduction band edge of the semiconductor, so the electron transfer occurs between LUMO and the TiO2 conduction band. Therefore, charge injection times depend upon the electronic overlap between the TiO2 and dye orbital. The analysis of DOS shows that the metal complex LUMO position relative the TiO2 (101) surface is energetically unfavorable for electron injection on TiO2 conduction band. After adsorption, the DOS, bottom of Fig. 6, is found to be quite similar to that for the isolated components. The HOMO–LUMO gap of nickel complex decreases slightly, while the LUMO level is somewhat pushed into the conduction band of the titania surface. As can be seen, some new occupied states appear at the TiO2 band gap after adsorption of [CH3]2[Ni(dmit)2] complex. These bands are contributed almost exclusively from the Ni complex orbitals. We also observe that metal complexes have a strong overlap with the conduction band. On the other hand, it is worth noting that the presence of [CH3]2[Ni(dmit)2] affects the magnitude of the band gap. In any case, the calculated DOS for the different modes of adsorption is quite similar.
Finally, the UV–Vis absorption spectra for the [CH3]2[Ni(dmit)2] nickel complex adsorbed on the TiO2(101) anatase surface in the planar configuration were obtained. The imaginary part of the dielectric function, ε 2(ω), computed from PBE + U calculations is reported in Fig. 7. In order to make the analysis easier, the optical spectrum of the isolated nickel complex was also computed and plotted in Fig. 7.

Optical absorption spectra of isolated [CH3]2[Ni(dmit)2] complex and adsorbed onto the anatase TiO2 (101) surface, planar mode, computed at the PBE + U level of theory
We start with the spectrum of the isolated nickel complex. The lowest energy peak appears at 0.87 eV that according to the DOS analysis is assigned to a HOMO–LUMO transition. This is followed by a small peak at 1.95 eV, and several absorptions at energies higher than 3 eV. If we compare this spectrum with that obtained from TDDFT B3LYP calculations (Fig. 4), a general lowering of the transition energies is observed. Beyond the specified technical differences in the setup calculations: plane-waves vs. contracted Gaussian basis sets and theoretical framework, the main responsible for this lowering is the exchange–correlation functional used in the calculations [50]. While hybrid exchange–correlation functionals use to overestimate HOMO–LUMO gaps, plane GGA functionals clearly underestimate them, which leads to a shift in the spectra [51, 52]. Notice that although a Hubbard-type correction is present in the PBE calculations, the U term is only applied to Ni atoms, while the lowest transitions mainly are of π → π* character, primordially involving carbon and sulfur ligand atoms.
After deposition of the nickel complex on the TiO2(101) surface, the optical spectrum shows a low energy strong absorption at 0.73 eV that corresponds to the HOMO–LUMO excitation of the free complex. Below 3 eV, the observed peaks are quite similar to those of the free complex but at lower energies, indicating that the interaction with the titanium oxide surface induces a general red shift of the electronic bands in agreement with the behavior reported for CdSe clusters supported on TiO2(110) rutile surface [51]. At energies higher than 3 eV, the spectrum shows complex features resulting from the superposition of the electronic absorption of both the nickel complex and the TiO2(101) surface [51]. This assignment is consistent with the DOS plots computed for [CH3]2[Ni(dmit)2] nickel complex adsorbed on the TiO2(101) anatase surface reported at the bottom of Fig. 6. In any case, it is worth to remark the absence of significant electronic absorptions in the visible spectrum window (1.6–3.1 eV), indicating the unsuitability of this type of nickel complex as DSSC sensitizer. This situation, however, may be different if the complex is chemically bound to the TiO2 surface, a possibility that will be the subject of a future work.
4 Conclusions
Density functional calculations have been carried out to explore the structural, electronic, and optical properties of [CH3]2[Ni(dmit)2] complex, both isolated and supported on TiO2(101) anatase surfaces. With this aim, two different theoretical frameworks have been undertaken: molecular B3LYP calculations with a contracted Gaussian type basis set and periodic PBE + U slab calculations using plane-waves. From a structural point of view, both approaches give geometrical parameters and vibrational frequencies in agreement with experimental data. TDDFT/B3LYP calculations of [CH3]2[Ni(dmit)2] isolated complex show an intense and low energy absorption at 1.51 eV mainly arising from a HOMO–LUMO π → π* excitation. The HOMO isosurface clearly shows the electron density being localized on C2S2 2− moieties of dmit ligand and Ni (II) d orbital. In contrast, the LUMO is mainly composed by the dmit ligand thiole ring without nickel contribution.
For the deposition of [CH3]2[Ni(dmit)2] on the TiO2(101) surface, three different adsorption configurations were considered. Our calculations indicate that the most stable adsorption mode corresponds to a planar form where the dmit π system interacts with the TiO2 surface, mainly through long-range dispersion forces. Partial DOS plots show that the LUMO of [CH3]2[Ni(dmit)2] lies at the conduction band edge of TiO2 which is unfavorable for a hypothetic electron injection onto the semiconductor surface. On the other hand, the HOMO of the complex also falls within the TiO2 band gap, making it that the lowest (and most intense) electronic absorption of the nickel complex supported on the TiO2 is similar to that of the unsupported complex. The effect of the TiO2(101) surface on the absorption spectra of the nickel complex is practically limited to a red shift of about 0.1–0.3 eV. The absence of significant electronic absorption in the visible spectrum region allows us to discard this complex as sensitizer in DSSC.
References
Nazeeruddin MK, Baranoff E, Grätzel M (2011) Dye-sensitized solar cells: a brief overview. Sol Energy 85:1172–1178
Hagfeldt A, Boschloo G, Sun L, Kloo L, Pettersson H (2010) Dye-sensitized solar cells. Chem Rev 110:6595–6663
Robertson N (2006) Optimizing dyes for dye-sensitized solar cells. Angew Chem Int Edit 45:2338–2345
Lupan O, Guérin VM, Tiginyanu IM, Ursaki VV, Chow L, Heinrich H, Pauporté T (2010) Well-aligned arrays of vertically oriented ZnO nanowires electrodeposited on ITO-coated glass and their integration in dye sensitized solar cells. J Photoch Photobio A 211:65–73
Sakai N, Miyasaka T, Murakami TN (2013) Efficiency enhancement of ZnO-based dye-sensitized solar cells by low-temperature TiCl4 treatment and dye optimization. J Phys Chem C 117:10949–10956
Han DW, Heo JH, Kwak DJ, Han CH, Sung YM (2009) Texture, morphology and photovoltaic characteristics of nanoporous F:SnO2 films. J Electr Eng Technol 4:93–97
Ferrere S, Zaban A, Gregg BA (1997) Dye sensitization of nanocrystalline Tin oxide by perylene derivatives. J Phys Chem B 101:4490–4493
Niu H, Zhang S, Wang R, Guo Z, Shang X, Gan W, Qin S, Wan L, Xu J (2014) Dye-sensitized solar cells employing a multifunctionalized hierarchical SnO2 nanoflower structure passivated by TiO2 nanogranulum. J Phys Chem C 118:3504–3513
Nazeeruddin MK, De Angelis F, Fantacci S, Selloni A, Viscardi G, Liska P, Ito S, Takeru B, Grätzel M (2005) Combined experimental and DFT-TDDFT computational study of photoelectrochemical cell ruthenium sensitizers. J Am Chem Soc 127:16835–16847
Yella A, Lee HW, Tsao HN, Yi C, Chandiran AK, Nazeeruddin MK, Diau EWG, Yeh CY, Zakeeruddin SM, Grätzel M (2011) Porphyrin-sensitized solar cells with cobalt (II/III)–based redox electrolyte exceed 12 percent efficiency. Sci 334:629–634
Diwan K, Chauhan R, Singh SK, Singh B, Drew MGB, Bahadur L, Singh N (2014) Light harvesting properties of some new heteroleptic dithiocarbimate-diamine/diimine complexes of Ni, Pd and Pt studied as photosensitizer in dye-sensitized TiO2 solar cells. New J Chem 38:97–108
Linfoot CL, Richardson P, McCall KL, Durrant JR, Morandeira A, Robertson N (2011) A nickel-complex sensitiser for dye-sensitised solar cells. Sol Energy 85:1195–1203
Duchanois T, Thibaud E, Cebrián C, Lui L, Monari A, Baley M, Assfeld X, Haacke S, Gross PC (2015) An iron-based photosensitizer with extended excited-state lifetime: photophysical and photovoltaic properties. Eur J Inorg Chem 14:2469–2477
Duchanois T, Etienne T, Beley M, Assfeld X, Perpète EA, Monari A, Gros PC (2014) Heteroleptic pyridyl-carbene iron complexes with tuneable electronic properties. Eur J Inorg Chem 23:3747–3753
Islam A, Sugihara H, Hara K, Singh LP, Katoh R, Yanagida M, Takahashi Y, Murata S, Arakawa H, Fujihashi G (2001) Dye sensitization of nanocrystalline titanium dioxide with square planar platinum(II) diimine dithiolate complexes. Inorg Chem 40:5371–5380
Geary EAM, Yellowlees LJ, Jack LA, Oswald IDH, Parsons S, Hirata N, Durrant JR, Robertson N (2005) Synthesis, structure, and properties of [Pt(II)(diimine)(dithiolate)] dyes with 3,3‘-, 4,4‘-, and 5,5‘-disubstituted bipyridyl: applications in dye-sensitized solar cells. Inorg Chem 44:242–250
Geary EAM, McCall KL, Turner A, Murray PR, McInnes EJL, Jack LA, Yellowlees LJ, Robertson N (2008) Spectroscopic, electrochemical and computational study of Pt-diimine-dithiolene complexes: rationalising the properties of solar cell dyes. Dalton T:3701–3708
Lazarides T, McCormick TM, Wilson KC, Lee S, McCamant DW, Eisenberg R (2011) Sensitizing the sensitizer: the synthesis and photophysical study of bodipy−Pt(II)(diimine)(dithiolate) conjugates. J Am Chem Soc 133:350–364
Browning C, Hudson JM, Reinheimer EW, Kuo FL, McDougaldJr RN, Rabaâ H, Pan H, Bacsa J, Wang X, Dunbar KR, Shepherd ND, Omary MA (2014) Synthesis, spectroscopic properties, and photoconductivity of black absorbers consisting of Pt(bipyridine)(dithiolate) charge transfer complexes in the presence and absence of nitrofluorenone acceptors. J Am Chem Soc 136:16185–16200
Kato R (2004) Conducting metal dithiolene complexes: structural and electronic properties. Chem Rev 104:5319–5346
Zarkadoulas A, Koutsouri E, Mitsopoulou CA (2012) A perspective on solar energy conversion and water photosplitting by dithiolene complexes. Coord Chem Rev 256:2424–2434
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery Jr JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski V G, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ, (2010) Gaussian Inc., Wallingford
Lee C, Yang W, Parr RG (1988) Development of the Colle–Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37:785
Becke AD (1988) Density-functional exchange-energy approximation with correct asymptotic behavior. Phys Rev A 38:3098
Stevens WJ, Basch H, Krauss M (1984) Compact effective potentials and efficient shared-exponent basis sets for the first- and second-row atoms. J Chem Phys 81:6026
Stevens WJ, Krauss M, Basch H, Jasien PG (1992) Relativistic compact effective potentials and efficient, shared-exponent basis sets for the third-, fourth-, and fifth-row atoms. Can J Chem 70:612–630
Cundari TR, Stevens WJ (1993) Effective core potential methods for the lanthanides. J Chem Phys 98:5555
Yanai T, Tew DP, Handy NC (2004) A new hybrid exchange-correlation functional using the coulomb-attenuating method (CAM-B3LYP). Chem Phys Lett 393:51
Kresse G, Hafner J (1993) Ab initio molecular dynamics for liquid metals. Phys Rev B 47:558
Kresse G, Furthmüller J (1996) Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys Rev B 54:11169
Kresse G, Furthmüller J (1996) Efficiency of ab initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp Mat Sci 6:15–50
Perdew JP, Burke K, Ernzerhof M (1996) Generalized gradient approximation made simple. Phys Rev Lett 77:3865
Dudarev SL, Botton GA, Savrasov SY, Humphreys CJ, Sutton AP (1998) Electron-energy-loss spectra and the structural stability of nickel oxide: an LSDA + U study. Phys Rev B 57:1505
Calzado CJ, Hernández NC, Sanz JF (2008) Effect of on-site Coulomb repulsion term U on the band-gap states of the reduced rutile (110) TiO2 surface. Phys Rev B 77:045118
Cao C, Hill S, Cheng HP (2008) Strongly correlated electrons in the [Ni(hmp)(ROH)X]4 single molecule magnet: a DFT + U study. Phys Rev Lett 100:167206
Gajdoš M, Hummer K, Kresse G, Furthmüller J, Bechstedt F (2006) Linear optical properties in the projector-augmented wave methodology. Phys Rev B 73:045112
Klimeš J, Bowler DR, Michaelides A (2011) Van der Waals density functionals applied to solids. Phys Rev B 83:195131
Breitzer JG, Rauchfuss TB (2000) Studies on α-C3S5 2− (dmit2−) and its dinuclear Ni(II) complex: spectroscopic and structural characterization. Polyhedron 19:1283–2391
Bousseau M, Valade L, Legros JP, Cassoux P, Garbauskas M, Interrante LV (1986) Highly conducting charge-transfer compounds of tetrathiafulvalene and transition metal-”dmit” complexes. J Am Chem Soc 108:1908–1916
Ferreira GB, Comerlato NM, Wardell JL, Hollauer E (2004) Vibrational spectra of bis(dmit) complexes of main group metals: IR, Raman and ab initio calculations. J Braz Chem Soc 15:951–963
Valade L, Legros JP, Cassoux P, Kubel F (1986) Synthesis, electrical conductivity, and crystal structure of (AsPh4)0.25 [Ni(dmit)2]. Mol Cryst Liq Cryst 140:335–351
Singh JD, Singh HB (1993) Synthesis and characterization of [M(dmit)2]2− and [M(dmt)2]2− complexes (M = Se(II) or Te(II); dmit = 4,5-dimercapto-1,3-dithiole-2-thione and dmt = 4,5-dimercapto-1,2-dithiole-3-thione). Polyhedron 12:2849–2856
Liu G, Fang Q, Xu W, Chen H, Wang C (2004) Vibration assignment of carbon–sulfur bond in 2-thione-1,3-dithiole-4,5-dithiolate derivatives. Spectrochim Acta A 60:541–550
Bernardo da Cruz AG, Wardell JL, Rocco AM (2006) The decomposition kinetics of [Et4N]2[M(dmit)2] (M = Ni, Pd) in a nitrogen atmosphere using thermogravimetry. Thermochim Acta 443:217–224
Fan HL, Ren Q, Wang XQ, Li TB, Sun J, Zhang GH, Xu D, Yu G, Sun ZH (2009) Investigation on third-order optical nonlinearities of two organometallic Dmit2- complexes using Z-scan technique. Natural Sci 01:136–141
da Cruz AGB, Wardell JL, Rangel MVD, Simão RA, Rocco AM (2007) Preparation and characterization of a polypyrrole hybrid film with [Ni(dmit)2]2−, bis(1,3-dithiole-2-thione-4,5-dithiolate)nickellate(II). Synthetic Met 157:80–90
Ferreira GB, Hollauer E, Comerlato NM, Wardell JL (2006) An experimental and theoretical study of the electronic spectra of tetraethylammonium [bis(1,3-dithiole-2-thione-4,5-dithiolato)zincate(II)], [NEt4]2[Zn(dmit)2], and tetraethylammonium [bis(1,3-dithiole-2-one-4,5-dithiolato)zincate(II)], [NEt4]2[Zn(dmio)2]. Inorg Chim Acta 359:1239–1247
da Cruz AGB, Wardell JL, Simão RA, Rocco AM (2007) Preparation, structure and electrochemistry of a polypyrrole hybrid film with [Pd(dmit)2]2−, bis(1,3-dithiole-2-thione-4,5-dithiolate)palladate(II). Electrochim Acta 52:1239–1247
Arroyo-de Dompablo ME, Morales-García A, Taravillo M (2011) DFT + U calculations of crystal lattice, electronic structure, and phase stability under pressure of TiO2 polymorphs. J Chem Phys 135:054503–054511
Nadler R, Sanz JF (2013) Simulating the optical properties of CdSe clusters using the RT-TDDFT approach. Theor Chem Acc 132:1342
Nadler R, Sanz JF (2015) Effect of capping ligands and TiO2 supporting on the optical properties of a (CdSe)13. Cluster, J Phys Chem A 119:1218–1227
Amaya Suárez J, Plata JJ, Marquez AM, Sanz JF (2016) Structural, electronic and optical properties of copper, silver and gold sulfide: a DFT study. Theor Chem Acc 135:70
Acknowledgements
The first author would like to acknowledge fellowships granted by CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior) and CNPq to fund her stay at the University of Seville. Computations were done at the University of Seville facilities funded by Spanish Ministerio de Economía y Competitividad, Grant CTQ2015-64669-P, Junta de Andalucía, Grant P12-FQM-1595, and European FEDER.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published as part of the special collection of articles derived from the 10th Congress on Electronic Structure: Principles and Applications (ESPA-2016).
Rights and permissions
About this article

Cite this article
Paes, L.W.C., Suárez, J.A., Márquez, A.M. et al. First-principles study of nickel complex with 1,3-dithiole-2-thione-4,5-dithiolate ligands as model photosensitizers. Theor Chem Acc 136, 71 (2017). https://doi.org/10.1007/s00214-017-2098-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-017-2098-7