
Abstract
Structural, electronic and optical properties of copper, silver and gold sulfides have been analyzed using density functional theory (DFT). A systematic study of all these properties using different functionals has been carried out and compared to the performance of more expensive hybrids functionals (HSE06). On-site Coulomb repulsion terms (DFT + U) and the inclusion of London dispersion forces (B86b-vdW) seem to be crucial for the good description of these solids, even improving the predictions of hybrids functionals in some cases. This article describes how the inclusion of some corrections in GGA functional makes possible a balanced description of the properties of this group of complex materials and makes affordable the future investigation of more complex systems that contain these solids within a reasonable computational cost.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Copper, silver and gold sulfides belong to I–VI semiconductor materials. These compounds have shown interesting properties for various applications such as light captors in photovoltaic solar cells. Specially, copper and silver sulfides have been identified as efficient photon absorbers of solar light, not only in thin films [1, 2], but also forming quantum dots [3, 4]. Rather than by its electronic properties, the suitability of gold sulfide as sensitizer is limited because it is a metastable compound [5]. Band gap values for gold sulfide are controversial with theoretical estimations varying in the 1.3–2.6 eV range [6]. Different authors, however, have reported the synthesis and characterization of Au2S nanoparticles of 2–5 nm in diameter with direct band gaps in the 1.8–2.5 eV range [7, 8]. These values are close to the band gap values of cadmium and lead chalcogenides that have been widely used in photovoltaics [9–12]. The crystal structure and electronic properties of these compounds have been examined in previous theoretical studies using different methodologies. For copper sulfide, Zhang et al. [13] studied the electronic structure of the antifluorite structure of Cu2S using a variety of functionals. Also, Xu et al. [14] studied the crystal and electronic structure of several copper sulfide polymorphs using hybrid density functional calculations. In the case of silver sulfide, Kashida et al. [15] carried out a study of the electronic structure of some polymorphs using the LDA functional. In the same context, Ben Nasr et al. used the modified Becke Johnson potential coupled with the LDA functional (MBJLDA) in order to analyze their electronic and optical properties [16]. More recently, Lin et al. [17] have analyzed the electronic structure of Ag2S by using the Perdew-Burke-Ernzerhof (PBE) functional. Although they obtain a band gap of 1.0 eV, quite close the to the experimental value, their computed b cell parameter is largely in error by about 1.0 Å. This is a typical case where the right answer to a question (here the electronic structure represented by the band gap value) is obtained by the wrong reasons, a poorly represented geometrical structure. Finally, to the best of our knowledge, only the DFT calculations carried out by Pryadchenko et al. [18] to interpret the X-ray spectroscopic features have been reported for gold sulfide.
From a computational point of view, it has been well established that to render appropriately the electronic structure, in particular band gaps in metal oxides, hybrid exchange–correlation functionals are compulsory although this approach results prohibitive for large systems on a routinely basis. Alternatively, a less demanding computational approach makes use of a Hubbard-like term, U, to account for the strong on-site Coulomb interactions. The choice of U is a subtle point and usually its value is selected by fitting a given property [19], from linear-response calculations [20, 21], or even on a self-consistent basis [22, 23]. On the other hand, the effect of long-range dispersive forces has been recognized to play an important role in describing the structure of molecular solids, although its impact in the properties of covalent and ionic solids has been often neglected. Recently, however, the quantitative role of such interactions in the binding has been receiving much attention [24, 25].
In the present work, we report a systematic theoretical DFT study of the crystal structure, electronic and optical properties of room-temperature-stable polymorphs of copper, silver and gold sulfides. As we will show, the structural and electronic properties of these sulfides exhibit both the low band gap and long-range dispersion difficulties inherent to the DFT approach. To tackle these issues, calculations using a hybrid exchange–correlation functional, the DFT + U approach, and a functional that includes dispersion forces effects have been performed.
2 Computational details
Periodic three-dimensional (3D) calculations were carried out using the VASP 5.3 code [26–28], with the projector-augmented wave method (PAW) [29, 30]. In these calculations, the energy was computed using the generalized gradient approximation (GGA) of DFT, in particular the exchange–correlation functional proposed by Perdew, Burke and Ernzerhof (PBE) [31]. To obtain a more appropriate description of the electronic properties of the sulfides here considered, we also performed reference calculations using the hybrid functional proposed by Heyd, Scuseria and Ernzerhof (HSE06) [32, 33]. It is well known that functionals incorporating some exact exchange are superior, in particular, in the estimation of the band gaps. However, the cost of such calculations still is prohibitive for large systems when a plane-wave basis set is used. For this reason, we also performed PBE calculations incorporating an on-site Coulomb repulsion U term. Finally, to include the effects due to London dispersion forces, a series of calculations with the functional proposed by Michaelides et al. [34] (B86b-vdw) and with the DFT-D3 method of Grimme with Becke-Johnson damping [35] (HSE06+D3) were also done. In all cases, electronic states were expanded using a plane-wave basis set with a cutoff energy of 350 eV. Forces on the ions were calculated through the Hellman–Feynman theorem, including the Harris-Foulkes correction to forces [36]. Iterative relaxation of the atomic positions and lattice parameters was stopped when the forces on the atoms were <0.01 eV/Å.
The Brillouin zone was sampled using the Monkhorst–Pack set of k points [37]. The size of the mesh depends on the size of the unit cell and the functional used. For anilite (Cu7S4), a 4 × 4 × 4 mesh was selected when using the PBE, PBE + U and B86b-vdw functionals, while a 2 × 2 × 2 set was used when calculations were performed with the HSE06 hybrid functional. In the case of acanthite (α-Ag2S), an 8x4x4 mesh was selected for GGA-pure functionals, while a 4 × 2 × 2 grid was used for the hybrid HSE06. Finally, for Au2S, a 4 × 4 × 4 mesh was selected for both the GGA-pure and GGA-hybrid-functional calculations. Relative energies were converged to less than 0.01 eV with these sets of k points.
DFT + U calculations were performed for anilite and acanthite, where the U parameter was applied on the Cu-3d and Ag-4d states, respectively. The Hubbard U term was added to the plain PBE functional using the rotationally invariant approach proposed by Dudarev et al. [38], in which the Coulomb U and exchange J parameters are combined into a single parameter U eff = U − J. Optical spectra were obtained from the frequency-dependent dielectrical functions as proposed by Gajdoš et al. [39].
3 Results and discussion
3.1 Anilite (Cu7S4)
Several polymorphs of copper sulfide, of general formula Cu x S (1 ≤ x ≤ 2), have been characterized. In these structures, the location of Cu atoms in the close-packed S lattice is not well identified and their positions change as a function of the composition (x). In a copper-rich environment, the experimentally identified stable compounds of Cu x S are chalcocite (Cu2S) [40, 41], djurleite (Cu1.94S) [41], digenite (Cu1.8S) [42] and anilite (Cu1.75S) [43], all of which are p-type semiconductor due to the presence of holes in their valence bands. All of them have band gap values of about 1.1 eV [14]. It has been established that the stoichiometric polymorph chalcocite is not stable under Cu-rich conditions because of the spontaneous formation of Cu vacancies, while the non-stoichiometric polymorph anilite is the most stable one [14]. Anilite is a bluish gray solid that belongs to the orthorhombic crystal system with space group Pnma. The unit cell, shown in Fig. 1, contains four formula units (28 Cu and 16 S) with 7 of the 28 copper atoms in a +2 oxidation state [43]. The presence of this amount of Cu2+ cations makes the anilite a heavily hole-doped material.

Crystal structure of anilite. Atom colors: Cu, brown; S, yellow
Starting with the DFT calculations, we first refer to the electronic and structural properties. Plain PBE calculations give optimized cell parameters of a = 7.91 Å, b = 8.00 Å and c = 10.89 Å, in reasonable agreement with the experimental data reported in Table 1. In contrast, the estimated band gap was 0.5 eV far from the 1.2 eV experimentally reported. This underestimation of the band gap is a general issue in plain GGA DFT calculations [44]. Like transition metal oxides, copper sulfide shows a complex electronic structure, hard to describe because of the strongly correlated nature of the Cu-3d electrons. This issue can be solved, at least partially, by using hybrid functionals or perturbative methodologies such as GW [45, 46]. In the present case, the HSE06 hybrid exchange–correlation functional leads to a much better band gap of 1.4 eV. Though the improvement is noticeable, the computational cost of this approach when is associated to a plane-wave basis set, makes it unsuitable for being systematically applied to larger systems with low symmetry. That is why we explored a DFT + U approximation that incorporates a Hubbard-like term to account for the strong on-site Coulomb interactions for the 3d levels. As mentioned in the introduction, the choice of the U parameter is a delicate point: the U parameter has to be large enough to properly localize the 3d electrons without introducing undesired artifacts. In fact, the optimal U parameter can be different for different properties. In order to choose the appropriate value of the parameter U, we studied how a set of properties changes when the value of U is modified. We chose to study the cell parameters (a, b, c), the value of band gap (E g) and the energy of formation (E form) due to the availability of experimental data in the bibliography.
In the case of parameter a, it can be seen in Fig. 2a that for a U value between 0 and 3 eV the theoretical value is almost constant, but it decreases rapidly when it is increased. A value of U = 4 eV gives a theoretical value of a = 7.89 Å which is in perfect agreement with the experimental one. The parameter b (see Fig. 2b) is strongly dependent on the U value: the larger the U value, the smaller the parameter b; the experimental value being attained between U = 7 and U = 8 eV. Finally, concerning the parameter c, we found that the experimental value cannot be reached even with values as large as 8 eV. As observed in Fig. 2c, the parameter c is almost constant with an average value of 10.87 Å. The dependence of the band gap on the value of U is depicted in Fig. 2d. It can be observed that there is an almost linear dependence, and the band gap steadily increases with the value of U. The experimental band gap is reached between U = 7 and U = 8 eV. Finally, we analyze the effect of the U parameter on the energetics of the system. The value reported in the literature for the energy of formation of Cu2S is −0.824 eV [47]. The evolution of the theoretical energy as a function of U is plotted in Fig. 2e. As can be observed, it decreases when U increases, reaching the experimental value when U = 7 eV. In summary, after comparing the results, the value U = 7 eV seems to be the most appropriate for rendering this set of properties and therefore was chosen for the following PBE + U calculations. This value of U is similar to data previously reported for other copper oxides [48].

Anilite: cell parameters (a, b, c), band gap (E g) and formation energy (E form) as a function of U. Horizontal lines refer to the corresponding experimental values
Structural data obtained from optimizations using PBE, PBE + U and HSE06 functionals are reported in Table 1. As can be seen, the HSE06 approach gives crystal parameters in excellent agreement with experiment, while PBE + U calculations appear to be a good compromise. On the other hand, in order to check the effect of long-range interaction on the computed structure, optimizations performed using the B86b-vdW functional were also carried out. In contrast to what we will see in the case of silver sulfide, the correction does not seem to improve the plain PBE answer.
Density of states (DOS) and band structure using the PBE + U approach were calculated and reported in Fig. 3. Three different bands can be observed in the DOS plot. The first one, with energy values about −13.0 eV, corresponds mainly to S-3s states with minor contributions of Cu-4s and Cu-3d. The second, the valence band, located approximately between −6.3 and 0.5 eV, is dominated by Cu-3d with minor contributions of S-3p orbitals. Finally, the conduction band can be assigned to both copper and sulfur levels. As shown in the DOS plot, the Fermi level does not match with the maximum energy value of the valence band, which is consistent with the non-stoichiometry of the material and the presence of a large amount of holes. The nature of the band gap might be established examining the band structure. It can be observed that anilite is a direct band gap semiconductor at the Γ point in good agreement with previous studies [14]. Finally, the calculated absorption spectrum is reported in Fig. 4. The lowest energy band, very intense (see inset for a full view of the spectrum) and centered at 0.2 eV, arises from electron transitions involving the Cu 3d band and the holes of the material, i.e., excitations within the valence band. The absorption observed beyond 2.3 eV is assigned to excitations toward the conduction band.

Total and partial densities of states and band structure for anilite calculated with the PBE + U approach. Fermi level is set at zero

Absorption spectra of anilite, acanthite and gold sulfide
3.2 Acanthite (α-Ag2S)
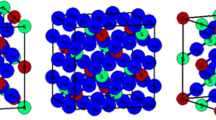
Below its melting point (1115 K), Ag2S exists in two polymorphic forms α-Ag2S and β-Ag2S. The low-temperature form, α-Ag2S, also called acanthite, belongs to the monoclinic crystal system and is essentially stoichiometric, whereas the high-temperature form, β-Ag2S or argentite, can dissolve excess Ag and S and belongs to the cubic crystal system. α-Ag2S is a thorn-shaped, black solid with space group P21/c. The unit cell, Fig. 5a, contains four formula units (8 Ag and 4 S) [49].

Crystal structure of acanthite. a Unit cell; b supercell showing the laminar arrangement. Atom colors: Ag, silver; S, yellow
Bulk geometry optimizations were performed with different functionals, and the results are summarized in Table 2. As can be seen, the PBE calculations produce cell parameters noticeably larger than the experiment, in particular for b that is overestimated by 0.85 Å. Hybrid HSE06 calculations improve the description, but the cell parameters still are overestimated, which is at variance with that found in the case of anilite, where the agreement with the experiment was much better. Yet, the laminar arrangement of acanthite, clearly observed in Fig. 5b, suggests the contribution of van der Waals interactions to the stability of the structure. Indeed, including the effect of London dispersion forces, through the B86b-vdW functional, leads to a dramatic lowering of the b parameter value, with an overall geometrical description now in good agreement with the experiment. The same is observed if the dispersion forces corrections are added to the HSE06 functional through the approach developed by Grimme (HSE06+D3) with a similar overall agreement for the geometrical cell parameters. While the calculated band gap falls short of the experimental value when the B86b-vdW functional is used, the HSE06+D3 approach results in much better agreement. However, in this case, the calculations are computationally highly expensive compared to the B86b-vdW functional (about 70 times) and cannot be proposed at this point as a practical approach to study larger systems (like Ag2S nanoparticles).
Considering now the electronic properties, Fig. 6 shows plots of DOS and band structure obtained for α-Ag2S using the B86b-vdW functional. As in the case of anilite, three regions might be differentiated. The lowest band, located around −13.0 eV, is derived almost completely from the S-3s levels. The valence band extends from −6.0 eV to the Fermi level, has a major contribution of the Ag-4d levels at the center of the band and higher mixing of Ag-4d and S-3p states both at the top and at the bottom of the band. Finally, the conduction band is contributed by both silver and sulfur electronic states. From the examination of the band structure, it results that α-Ag2S is a direct gap semiconductor at the Γ point, in agreement with other data available in the literature [15, 16], with an estimated band gap of 0.7 eV that, not surprisingly, is smaller than the experimental value, 1.1 eV [4]. Adding the on-site Coulomb correction to the B86b-vdW functional and reoptimizing the geometry does not improves the situation. The value of the b cell parameter decreases from 6.93 Å when no U correction is applied to 6.33 Å when U = 6.0 eV. At the same time, as illustrated in Fig. 7, the band gap shows an almost invariant behavior for values of U raising up to 6 eV.

Total and partial densities of states and band structure for acanthite calculated with B86b-vdW. Fermi level is set at zero

Silver sulfide band gap (E g) as a function of U value using the B86b-vdW + U functional. Horizontal line experimental band gap
However, if the cell parameters are not reoptimized from those obtained with the B86b-vdW functional, the computed band gap increases linearly with the value of the U parameter and the experimental gap of 1.1 eV is recovered for U = 3.0 eV, as illustrated also in Fig. 7. Analysis of the evolution of the DOS with increasing U parameter shows that, as expected, the occupied Ag 4d levels are displaced toward lower energies, resulting in lower mixing with Ag 5s and S 3p states at the top of the valence band. The bottom of the conduction band shows a higher contribution of S 3p and Ag 5s states and is only indirectly affected by the increasing U parameter through their mixing with Ag 4d states. This causes the erratic behavior of the band gap with U when the cell parameters are reoptimized. Although this strategy is not the most desirable from the theoretical viewpoint, one may assume that, unless proved otherwise, it should work in a similar way in Ag2S-related systems. Of course this approach should be tested against the much more expensive HSE06+D3, if possible.
Finally, the calculated absorption spectrum, obtained with the B86b-vdW functional, is also reported in Fig. 4. It can be observed that acanthite is optically active in a wide range of the visible spectrum with a maximum value of absorbance about 2.7 eV, which agree with experimental results [50].
3.3 Gold sulfide (Au2S)
Au2S presents only one polymorph, a brownish-black, metastable cuprite-type compound belonging to the cubic crystal system with space group Pn3m whose unit cell contains two formula units (4 Au and 2 S, see Fig. 8) [5]. Structural properties obtained with PBE, B86b-vdW and HSE06 functionals are reported in Table 3. In contrast with copper and silver sulfides, we found out that changing the functional does not affect significantly the computed cell parameter that is systematically overestimated. With respect to the band gap, as far as we know, experimental data for bulk gold sulfide have not been reported. The optical band gap of colloidal gold sulfide nanoparticles is estimated to be 1.8 ± 0.2 eV [7], which falls within the theoretically proposed range of 1.3–2.6 eV [6]. The value computed at the HSE06 level is 3.0 eV, clearly overestimated. In contrast, PBE plain calculations lead to a band gap of 1.94 eV that falls within the proposed range. Hence, we can conclude that in this case the PBE functional is well adapted to describe this sulfide.

Crystal structure of Au2S. Atom colors: Au, blue; S, yellow
Total and partial DOS as well as the band structure of gold sulfide calculated with the PBE functional are reported in Fig. 9. As in the case of copper and silver sulfides, three regions can be observed. The first one is centered at −13.0 eV and again mainly corresponds to S-3s states. The valence states, localized between −6.5 and Fermi level, are dominated by Au-5d states, incorporating small contributions of Au-6s and S-3p levels. Finally, the conduction band, beyond ~2 eV, can be assigned to both gold and sulfur levels. The band structure shows that Au2S is a direct band gap semiconductor at the Γ point. The absorption spectrum reported in Fig. 4 shows that Au2S is optically active in the visible spectrum, with a maximum value of absorbance about 4.0 eV, which justifies its brownish-black color.

Total and partial densities of states and band structure for Au2S calculated with PBE. Fermi level is set at zero
4 Conclusions
In conclusion, the results exposed in the preceding paragraphs allow us to rationalize the trends observed in the description by different DFT functionals of the geometric, electronic structure and optical properties of copper, silver, and gold sulfides.
Concerning the copper sulfide, anilite, it has been found that plain DFT calculations produce optimized cell parameters in reasonable agreement with available experimental data. In contrast, the computed band gap is quite below the experimental reported data. A commonly used strategy to deal with the electronic structure of highly correlated materials like those transition metal chalcogenides at a reasonable computational cost is the DFT + U approximation. In the case of anilite, a value of U = 7 eV is found to correctly describe both the experimental b cell parameter and the band gap. However, the a cell parameter is slightly underestimated and the c cell parameter is found to be insensitive to the choice of U in the range U = 0–8 eV. Computationally more expensive, the HSE06 hybrid density functional has also been employed in this system, resulting in excellent agreement for geometrical parameters, although it slightly overestimates the band gap. Finally, the inclusion of the long-range London forces through the use of a B86b-vdW functional basically has no effect on the computed cell parameters and band gap compared to plain PBE calculation. Thus, in this case the HSE06 functional produces geometrical and electronic structure in substantially good agreement with experimental available data while the PBE + U approximation seems to be a good compromise solution. Anilite is found to have a very intense band centered at 0.2 eV that arises from electronic excitations of the Cu 3d electrons within the valence band. Another absorption band is observed beyond 2.3 eV corresponding to electronic excitations to the conduction band.
For the silver sulfide, acanthite, the PBE calculations produce cell parameters that are definitely longer than the experimental data. This situation is more apparent for the b cell parameter that is overestimated by 0.85 Å. At difference of what was found for the copper sulfide, the use of a hybrid HSE06 approximation does not fully solve this issue: the cell parameters are still quite longer than the experimental values. The inclusion of the London dispersion forces proves to be crucial in this case, dramatically improving the geometrical description of the system resulting in geometrical parameters that are now in good agreement with the experiment. Concerning the electronic structure of this system, to correctly reproduce the experimental band gap, it is necessary to add the on-site correction but keeping the cell parameters obtained with the B86b-vdW functional. Acanthite is found to be a direct gap semiconductor and is optically active in the visible region, showing a broad absorption band centered at about 2.7 eV.
Finally, in the case of gold sulfide, it is found that the geometrical cell parameters are quite insensitive to the functional used, being systematically overestimated. Concerning the band gap, the HSE06 hybrid functional produces a value that is clearly larger than other theoretical data, while the plain PBE answer is the one that best fits the available experimental value. As in the case of acanthite, Au2S is found to be a direct gap semiconductor that is active in the visible spectrum showing a broad absorption that extends above 1 eV and reach a maximum at about 4 eV.
Thus, to summarize, the correct description of the geometrical and electronic structure of these materials seems to be a complex problem that requires diverse solutions. None of the tested functionals performs satisfactorily for all three systems. The highly electron deficient copper sulfide, anilite, is best described by the HSE06 functional with the PBE + U as a good compromise solution. Both silver sulfide and gold sulfide are essentially stoichiometric materials with filled metal d-shells. In these cases, the inclusion of on-site Coulomb effects in the metal d–shell does not improve the computed geometric structure. While the plain PBE approximation seems to be the best theoretical description of the electronic and geometric structure of Au2S, in the case of acanthite, its laminar structure results from the formation of planes of strong Ag–S bonds glued together by weaker van der Waals forces. Thus, in this case, the inclusion of the dispersion forces through the use of a B86b-vdW functional is mandatory to adequately describe the geometric structure of this material.
References
Reijnen L, Meester B, Goossens A, Schoonman J (2003) Chem Vapor Depos 9:15–20
Shen H, Jiao X, Oron D, Li J, Lin H (2013) J Power Sources 240:8–13
Lin MC, Lee MW (2011) Electrochem Commun 13:1376–1378
Tubtimtae A, Wu KL, Tung HY, Lee MW, Wang GJ (2010) Electrochem Commun 12:1158–1160
Ishikawa K, Isonaga T, Wakita S, Suzuki Y (1995) Solid State Ionics 79:60–66
Averitt RD, Sarkar D, Halas NJ (1997) Phys Rev Lett 78:4217–4220
Morris T, Copeland H, Szulczewski G (2002) Langmuir 18:535
Kuo C-L, Huang MH (2008) J Phys Chem C 112:11661
Schaller RD, Klimov VI (2004) Phys Rev Lett 92:186601
Peter LM, Riley DJ, Tull EZ, Wijayantha KGU (2002) Chem Commun 10:1030–1031
Robel I, Subramanian V, Kuno M, Kamat PV (2006) J Am Chem Soc 128:2385–2393
Plass R, Serge P, Krüger J, Grätzel M (2002) J Phys Chem B 106:7578–7580
Zhang Y, Wang Y, Xi L, Qiu R, Shi X, Zhang P, Zhang W (2014) J Chem Phys 140:074702
Xu Q, Huang B, Zhao Y, Yan Y, Noufi R, Wei S (2012) Appl Phys Lett 100:061906
Kashida S, Watanabe N, Hasegawa T, Iida H, Mori M, Savrasov S (2003) Solid State Ionics 158:167–175
Ben Nasr T, Maghraoui-Meherzi H, Ben Abdallah H, Bennaceur R (2013) Solid State Sci 26:65–71
Lin S, Feng Y, Wen X, Zhang P, Woo S, Shrestha S, Conibeer G, Huang S (2015) J Phys Chem C 119:867–872
Pryadchenko VV, Srabionyan VV, Avakyan LA, Van Bokhoven JA, Bugaev LA (2012) J Phys Chem C 116:25790–25796
Plata JJ, Márquez AM, Sanz JF (2012) J Chem Phys 136:041101
Fabris S, de Gironcoli S, Baroni S, Vicario G, Balducci G (2005) Phys Rev B 72:237102
Cococcioni M, de Gironcoli S (2005) Phys Rev B 71:035105
Kulik HJ, Cococcioni M, Scherlis DA, Marzari N (2006) Phys Rev Lett 97:103001
Hsu H, Umemoto K, Cococcioni M, Wentzcovitch R (2009) Phys Rev B 79:125124
Zhang GX, Tkatchenko A, Paier J, Appel H, Scheffler M (2011) Phys Rev Lett 107:245501
Bucǩo T, Lebeg̀ue S, Hafner J, Ángyań JG (2013) J Chem Theory Comput 9:4293–4299
Kresse G, Furthmuller J (1996) Phys Rev B 54:11169–11186
Kresse G, Furthmuller J (1996) Comput Mater Sci 6:15–50
Kresse G, Hafner J (1993) Phys Rev B 47:558–561
Kresse G, Joubert D (1999) Phys Rev B 59:1758–1775
Blöchl PE (1994) Phys Rev B 50:17953–17979
Perdew JP, Burke K, Ernzerhof M (1996) Phys Rev Lett 77:3865–3868
Heyd J, Scuseria GE, Ernzerhof M (2003) J Chem Phys 118:8207–8215
Heyd J, Scuseria GE, Ernzerhof M (2006) J Chem Phys 124:219906
Klimes J, Bowler DR, Michaelides A (2011) Phys Rev B 83:195131
Grimme S, Ehrilch S, Goerigk L (2011) J Comput Chem 32:1456
Harris J (1985) Phys Rev B 31:1770–1779
Monkhorst HJ, Pack JD (1976) Phys Rev B 13:5188–5192
Dudarev SL, Botton GA, Savrasov SY, Humphreys CJ, Sutton AP (1998) Phys Rev B 57:1505–1509
Gajdoš M, Hummer K, Kresse G, Furthmüller J, Bechstedt F (2006) Phys Rev B 73:045112
Potter RW (1977) Econ Geol 150:1524–1542
Evans HT (1979) Science 203:356–358
Will G, Hinze A, Rahman A, Abdelrahman M (2002) Eur J Mineral 14:591–598
Koto K, Morimoto N (1970) Am Mineral 55:106–117
Huang P, Carter EA (2008) Annu Rev Phys Chem 59:216–290
Hedin L (1965) Phys Rev 139:796
Aryasetiawan F, Gunnarsson O (1998) Rep Prog Phys 61:237–312
Lide DR (ed) (2003) CRC handbook of chemistry and physics. CRC Press, Florida
Heinemann M, Eifert B, Heiliger C (2013) Phys Rev B 87:115111
Sadanaga R, Sueno S (1967) Mineral J Jpn 5:124–143
Wang M, Wang Y, Tang A, Xu L, Yanbing H, Feng T (2012) Mater Lett 88:108–111
Acknowledgments
This work was funded by the Ministerio de Economía y Competitividad (Spain), the EU FEDER program, and the Junta de Andalucía, Grants CTQ2015-64669-P and P12-FQM-1595.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published as part of the special collection of articles “CHITEL 2015 - Torino - Italy”.
Rights and permissions
About this article

Cite this article
Suárez, J.A., Plata, J.J., Márquez, A.M. et al. Structural, electronic and optical properties of copper, silver and gold sulfide: a DFT study. Theor Chem Acc 135, 70 (2016). https://doi.org/10.1007/s00214-016-1832-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-016-1832-x